
Multiple Introductions and Distinct Genetic Groups of Canada Goldenrod (Solidago canadensis) in China Revealed by Genomic Single-Nucleotide Polymorphisms

{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
2.1. Genetic Clusters Revealed by SNP Data
2.2. Phylogenetic Relationships among Native and Invasive Canada Goldenrod
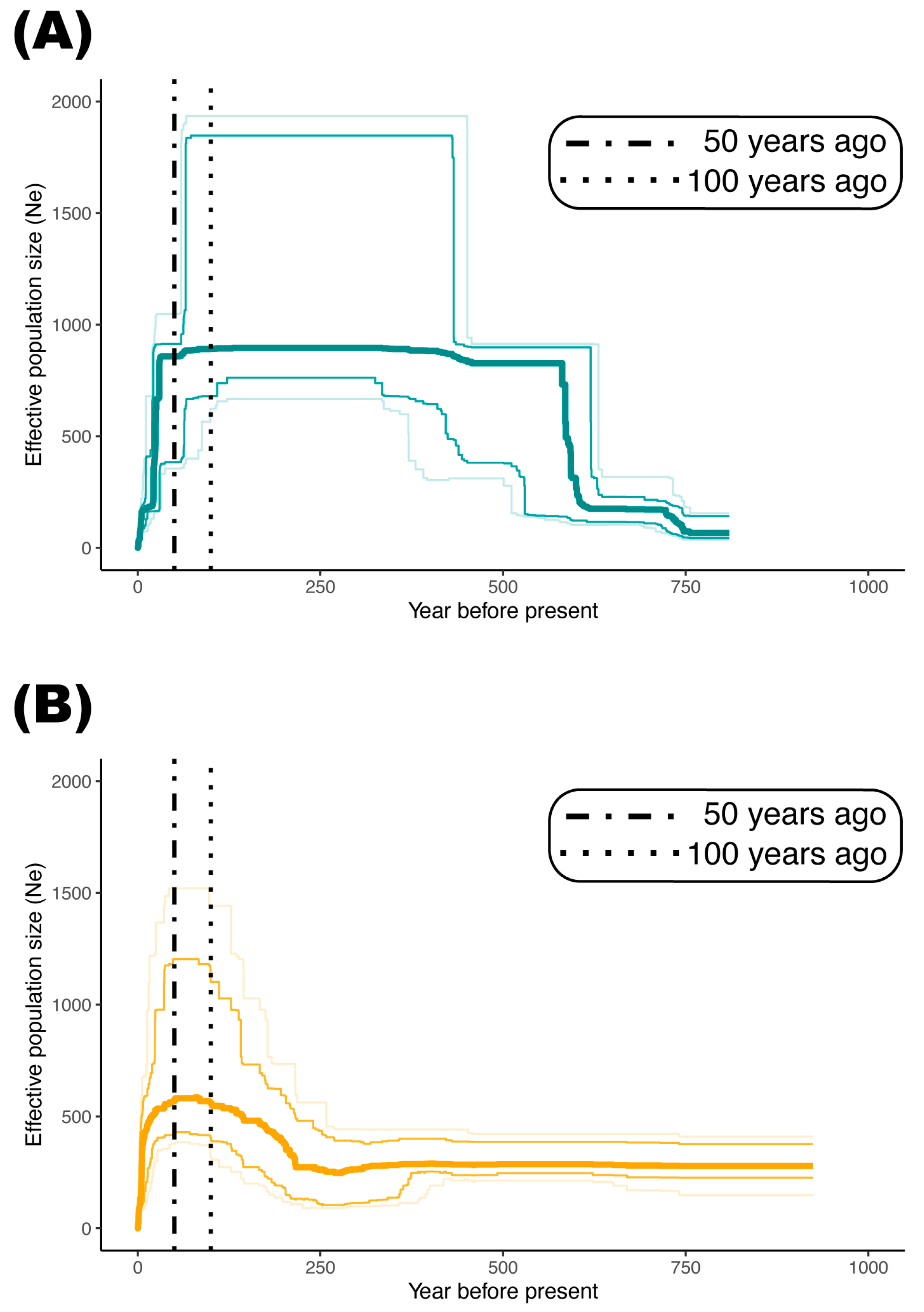
2.3. Demography of Two Genetic Groups
3. Discussion
4. Materials and Methods
4.1. Sample Collection
4.2. GBS and SNP Calling
4.3. Analysis of Population Structure
4.4. Reconstruction of Phylogeny
4.5. Demography Inference
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Butchart, S.H.M.; Walpole, M.; Collen, B.; van Strien, A.; Scharlemann, J.P.W.; Almond, R.E.A.; Baillie, J.E.M.; Bomhard, B.; Brown, C.; Bruno, J.; et al. Global biodiversity: Indicators of recent declines. Science 2010, 328, 1164–1168. [Google Scholar] [CrossRef] [PubMed]
- Simberloff, D.; Martin, J.-L.; Genovesi, P.; Maris, V.; Wardle, D.A.; Aronson, J.; Courchamp, F.; Galil, B.; García-Berthou, E.; Pascal, M.; et al. Impacts of biological invasions: What’s what and the way forward. Trends Ecol. Evol. 2013, 28, 58–66. [Google Scholar] [CrossRef]
- Lewis, S.L.; Maslin, M.A. Defining the Anthropocene. Nature 2015, 519, 171–180. [Google Scholar] [CrossRef] [PubMed]
- Facon, B.; Genton, B.; Shykoff, J.; Jarne, P.; Estoup, A.; David, P. A general eco-evolutionary framework for understanding bioinvasions. Trends Ecol. Evol. 2006, 21, 130–135. [Google Scholar] [CrossRef] [PubMed]
- Catford, J.A.; Jansson, R.; Nilsson, C. Reducing redundancy in invasion ecology by integrating hypotheses into a single theoretical framework. Divers. Distrib. 2009, 15, 22–40. [Google Scholar] [CrossRef]
- MacDougall, A.S.; Gilbert, B.; Levine, J.M. Plant invasions and the niche. J. Ecol. 2009, 97, 609–615. [Google Scholar] [CrossRef]
- Blackburn, T.M.; Pyšek, P.; Bacher, S.; Carlton, J.T.; Duncan, R.P.; Jarošík, V.; Wilson, J.R.U.; Richardson, D.M. A proposed unified framework for biological invasions. Trends Ecol. Evol. 2011, 26, 333–339. [Google Scholar] [CrossRef]
- Pearson, D.E.; Ortega, Y.K.; Eren, Ö.; Hierro, J.L. Community assembly theory as a framework for biological invasions. Trends Ecol. Evol. 2018, 33, 313–325. [Google Scholar] [CrossRef]
- Yu, W.; Li, S. Modern coexistence theory as a framework for invasion ecology. Biodivers. Sci. 2020, 28, 1362–1375. [Google Scholar] [CrossRef]
- Frankham, R. Conservation genetics. Annu. Rev. Genet. 1995, 29, 305–327. [Google Scholar] [CrossRef]
- Allendorf, F.W.; Lundquist, L.L. Introduction: Population biology, evolution, and control of invasive species. Conserv. Biol. 2003, 17, 24–30. [Google Scholar] [CrossRef]
- Estoup, A.; Ravigné, V.; Hufbauer, R.; Vitalis, R.; Gautier, M.; Facon, B. Is there a genetic paradox of biological invasion? Annu. Rev. Ecol. Evol. Syst. 2016, 47, 51–72. [Google Scholar] [CrossRef]
- Schrieber, K.; Lachmuth, S. the genetic paradox of invasions revisited: The potential role of inbreeding × environment interactions in invasion success. Biol. Rev. 2017, 92, 939–952. [Google Scholar] [CrossRef] [PubMed]
- Kolbe, J.J.; Glor, R.E.; Rodríguez Schettino, L.; Lara, A.C.; Larson, A.; Losos, J.B. Genetic variation increases during biological invasion by a Cuban lizard. Nature 2004, 431, 177–181. [Google Scholar] [CrossRef] [PubMed]
- Dlugosch, K.M.; Parker, I.M. Founding events in species invasions: Genetic variation, adaptive evolution, and the role of multiple introductions. Mol. Ecol. 2008, 17, 431–449. [Google Scholar] [CrossRef]
- Uller, T.; Leimu, R. Founder events predict changes in genetic diversity during human-mediated range expansions. Glob. Chang. Biol. 2011, 17, 3478–3485. [Google Scholar] [CrossRef]
- Keller, S.R.; Taylor, D.R. Genomic admixture increases fitness during a biological invasion. J. Evol. Biol. 2010, 23, 1720–1731. [Google Scholar] [CrossRef]
- Verhoeven, K.J.F.; Macel, M.; Wolfe, L.M.; Biere, A. Population admixture, biological invasions and the balance between local adaptation and inbreeding depression. Proc. R. Soc. B Biol. Sci. 2011, 278, 2–8. [Google Scholar] [CrossRef]
- Rius, M.; Darling, J.A. How important is intraspecific genetic admixture to the success of colonising populations? Trends Ecol. Evol. 2014, 29, 233–242. [Google Scholar] [CrossRef]
- Bock, D.G.; Caseys, C.; Cousens, R.D.; Hahn, M.A.; Heredia, S.M.; Hübner, S.; Turner, K.G.; Whitney, K.D.; Rieseberg, L.H. What we still don’t know about invasion genetics. Mol. Ecol. 2015, 24, 2277–2297. [Google Scholar] [CrossRef]
- Dlugosch, K.M.; Anderson, S.R.; Braasch, J.; Cang, F.A.; Gillette, H.D. The devil is in the details: Genetic variation in introduced populations and its contributions to invasion. Mol. Ecol. 2015, 24, 2095–2111. [Google Scholar] [CrossRef] [PubMed]
- van Boheemen, L.A.; Lombaert, E.; Nurkowski, K.A.; Gauffre, B.; Rieseberg, L.H.; Hodgins, K.A. Multiple introductions, admixture and bridgehead invasion characterize the introduction history of Ambrosia artemisiifolia in Europe and Australia. Mol. Ecol. 2017, 26, 5421–5434. [Google Scholar] [CrossRef] [PubMed]
- Qiao, H.; Liu, W.; Zhang, Y.; Zhang, Y.Y.; Li, Q.Q. Genetic admixture accelerates invasion via provisioning rapid adaptive evolution. Mol. Ecol. 2019, 28, 4012–4027. [Google Scholar] [CrossRef]
- Blumenfeld, A.J.; Eyer, P.-A.; Husseneder, C.; Mo, J.; Johnson, L.N.L.; Wang, C.; Kenneth Grace, J.; Chouvenc, T.; Wang, S.; Vargo, E.L. Bridgehead effect and multiple introductions shape the global invasion history of a termite. Commun. Biol. 2021, 4, 196. [Google Scholar] [CrossRef] [PubMed]
- Werner, P.A.; Bradbury, I.K.; Gross, R.S. The biology of Canadian weeds.: 45. Solidago Canadensis L. Can. J. Plant Sci. 1980, 60, 1393–1409. [Google Scholar] [CrossRef]
- Weber, E. Current and potential ranges of three exotic goldenrods (Solidago) in Europe. Conserv. Biol. 2001, 15, 122–128. [Google Scholar] [CrossRef]
- Dong, M.; Lu, J.; Zhang, W.; Chen, J.; Li, B. Canada goldenrod (Solidago Canadensis): An invasive alien weed rapidly spreading in China. Acta Phytotaxon. Sin. 2006, 44, 72–85. [Google Scholar] [CrossRef]
- Yang, R.; Zan, S.; Tang, J.; Chen, X. Invasion mechanisms of Solidago canadensis L.: A review. Acta Ecol. Sin. 2011, 31, 1185–1194. [Google Scholar]
- Zhang, S.; Zhu, W.; Wang, B.; Tang, J.; Chen, X. Secondary metabolites from the invasive Solidago canadensis L. accumulation in soil and contribution to inhibition of soil pathogen Pythium ultimum. Appl. Soil Ecol. 2011, 48, 280–286. [Google Scholar] [CrossRef]
- Li, J.; Du, L.; Guan, W.; Yu, F.-H.; van Kleunen, M. Latitudinal and longitudinal clines of phenotypic plasticity in the invasive herb Solidago canadensis in China. Oecologia 2016, 182, 755–764. [Google Scholar] [CrossRef]
- Jin, H.; Yuan, Y.; Gao, F.; Oduor, A.M.O.; Li, J. The invasive plant Solidago canadensis exhibits partial local adaptation to low salinity at germination but not at later life-history stages. Am. J. Bot. 2020, 107, 599–606. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Xie, Y. Invasive Alien Species in China; China Forestry Publishing House: Beijing, China, 2002; p. 170. [Google Scholar]
- Xu, H.; Qiang, S.; Genovesi, P.; Ding, H.; Wu, J.; Meng, L.; Han, Z.; Miao, J.; Hu, B.; Guo, J.; et al. An inventory of invasive alien species in China. NeoBiota 2012, 15, 1–26. [Google Scholar] [CrossRef]
- Zhao, S.Y.; Sun, S.G.; Dai, C.; Gituru, R.W.; Chen, J.M.; Wang, Q.F. Genetic variation and structure in native and invasive Solidago canadensis populations. Weed Res. 2015, 55, 163–172. [Google Scholar] [CrossRef]
- Liu, Y.; Huang, W.; Yang, Q.; Zheng, Y.L.; Li, S.P.; Wu, H.; Ju, R.; Sun, Y.; Ding, J. Research advances of plant invasion ecology over the past 10 years. Biodivers. Sci. 2022, 30, 22438. [Google Scholar] [CrossRef]
- Parvizi, E.; Dhami, M.K.; Yan, J.; McGaughran, A. Population genomic insights into invasion success in a polyphagous agricultural pest, Halyomorpha halys. Mol. Ecol. 2023, 32, 138–151. [Google Scholar] [CrossRef]
- Deschepper, P.; Vanbergen, S.; Zhang, Y.; Li, Z.; Hassani, I.M.; Patel, N.A.; Rasolofoarivao, H.; Singh, S.; Wee, S.L.; De Meyer, M.; et al. Bactrocera dorsalis in the Indian Ocean: A tale of two invasions. Evol. Appl. 2023, 16, 48–61. [Google Scholar] [CrossRef] [PubMed]
- Andrews, K.R.; Good, J.M.; Miller, M.R.; Luikart, G.; Hohenlohe, P.A. Harnessing the power of RADseq for ecological and evolutionary genomics. Nat. Rev. Genet. 2016, 17, 81–92. [Google Scholar] [CrossRef] [PubMed]
- Leaché, A.D.; Oaks, J.R. The utility of single nucleotide polymorphism (SNP) data in phylogenetics. Annu. Rev. Ecol. Evol. Syst. 2017, 48, 69–84. [Google Scholar] [CrossRef]
- Huang, H.; Guo, S. Review on ecological studies on three invasive species of Eurapean genus Solidago. Guangxi Sci. 2004, 11, 69–74. [Google Scholar]
- Moran, E.V.; Reid, A.; Levine, J.M. Population genetics and adaptation to climate along elevation gradients in invasive Solidago canadensis. PLoS ONE 2017, 12, e0185539. [Google Scholar] [CrossRef]
- Eckert, S.; Herden, J.; Stift, M.; Durka, W.; van Kleunen, M.; Joshi, J. Traces of genetic but not epigenetic adaptation in the invasive goldenrod Solidago canadensis despite the absence of population structure. Front. Ecol. Evol. 2022, 10, 856453. [Google Scholar] [CrossRef]
- Huang, H.; Guo, S. Study on reproductive biology of the invasive plant Solidago canadensis. Acta Ecol. Sin. 2005, 25, 2795–2803. [Google Scholar]
- Bradley, B.A.; Blumenthal, D.M.; Wilcove, D.S.; Ziska, L.H. Predicting plant invasions in an era of global change. Trends Ecol. Evol. 2010, 25, 310–318. [Google Scholar] [CrossRef] [PubMed]
- Wu, H.; Qiang, S. Bio-Ecological characteristics of Solidago canadensis L. and its control. Weed Sci. 2005, 1, 52–56. [Google Scholar]
- Ma, L.; Yang, H.; Du, X.; Liu, C. A summary of experiments on controlling Solidago canadensis with different chemicals. Weed Sci. 2007, 2, 56–57. [Google Scholar]
- Jiang, H.; Fang, F.; Guo, S. Influences of parasitism by Cuscuta japonica plants on eco-physiological characteristics of Solidago canadensis. Acta Ecol. Sin. 2008, 1, 399–406. [Google Scholar]
- Yang, B.; Du, L.; Li, J. Effects of Cuscuta australis parasitism on the growth, reproduction and defense of Solidago canadensis. Chin. J. Appl. Ecol. 2015, 26, 3309–3314. [Google Scholar]
- Alexander, J.M.; Poll, M.; Dietz, H.; Edwards, P.J. Contrasting patterns of genetic variation and structure in plant invasions of mountains. Divers. Distrib. 2009, 15, 502–512. [Google Scholar] [CrossRef]
- Zimmerman, S.J.; Aldridge, C.L.; Oyler-McCance, S.J. An empirical comparison of population genetic analyses using microsatellite and SNP data for a species of conservation concern. BMC Genom. 2020, 21, 382. [Google Scholar] [CrossRef]
- Friedman, J.; Barrett, S.C.H. High outcrossing in the annual colonizing species Ambrosia artemisiifolia (Asteraceae). Ann. Bot. 2008, 101, 1303–1309. [Google Scholar] [CrossRef]
- Martin, M.D.; Zimmer, E.A.; Olsen, M.T.; Foote, A.D.; Gilbert, M.T.P.; Brush, G.S. Herbarium specimens reveal a historical shift in phylogeographic structure of common ragweed during native range disturbance. Mol. Ecol. 2014, 23, 1701–1716. [Google Scholar] [CrossRef] [PubMed]
- Verhoeven, K.J.F.; Jansen, J.J.; van Dijk, P.J.; Biere, A. Stress-induced DNA methylation changes and their heritability in asexual dandelions. New Phytol. 2010, 185, 1108–1118. [Google Scholar] [CrossRef]
- Del Tredici, P. The introduction of Japanese knotweed, Reynoutria japonica, into North America. J. Torrey Bot. Soc. 2017, 144, 406–416. [Google Scholar] [CrossRef]
- Rapp, R.A.; Wendel, J.F. Epigenetics and plant evolution. New Phytol. 2005, 168, 81–91. [Google Scholar] [CrossRef]
- Pimpinelli, S.; Piacentini, L. Environmental change and the evolution of genomes: Transposable elements as translators of phenotypic plasticity into genotypic variability. Funct. Ecol. 2020, 34, 428–441. [Google Scholar] [CrossRef]
- Ashe, A.; Colot, V.; Oldroyd, B.P. How does epigenetics influence the course of evolution? Philos. Trans. R. Soc. B Biol. Sci. 2021, 376, 20200111. [Google Scholar] [CrossRef]
- Jeon, J.; Kwon, S.; Lee, Y.-H. Histone acetylation in fungal pathogens of plants. Plant Pathol. J. 2014, 30, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Couzin, J. Small RNAs make big splash. Science 2002, 298, 2296–2297. [Google Scholar] [CrossRef]
- Yu, Y.; Zhang, Y.; Chen, X.; Chen, Y. Plant noncoding RNAs: Hidden players in development and stress responses. Annu. Rev. Cell Dev. Biol. 2019, 35, 407–431. [Google Scholar] [CrossRef]
- Qin, Z.; Chen, J.; Jin, L.; Duns, G.J.; Ouyang, P. Differential expression of miRNAs under salt stress in Spartina alterniflora leaf tissues. J. Nanosci. Nanotechnol. 2015, 15, 1554–1561. [Google Scholar] [CrossRef]
- Xu, C.; Ge, Y.; Wang, J. Molecular basis underlying the successful invasion of hexaploid cytotypes of Solidago canadensis L.: Insights from integrated gene and miRNA expression profiling. Ecol. Evol. 2019, 9, 4820–4852. [Google Scholar] [CrossRef]
- Wan, J.; Oduor, A.M.O.; Pouteau, R.; Wang, B.; Chen, L.; Yang, B.; Yu, F.; Li, J. Can polyploidy confer invasive plants with a wider climatic tolerance? A test using Solidago canadensis. Ecol. Evol. 2020, 10, 5617–5630. [Google Scholar] [CrossRef] [PubMed]
- Elshire, R.J.; Glaubitz, J.C.; Sun, Q.; Poland, J.A.; Kawamoto, K.; Buckler, E.S.; Mitchell, S.E. A robust, simple genotyping-by-sequencing (GBS) approach for high diversity species. PLoS ONE 2011, 6, e19379. [Google Scholar] [CrossRef] [PubMed]
- Poland, J.A.; Brown, P.J.; Sorrells, M.E.; Jannink, J.-L. Development of high-density genetic maps for barley and wheat using a novel two-enzyme genotyping-by-sequencing approach. PLoS ONE 2012, 7, e32253. [Google Scholar] [CrossRef]
- Chen, S.; Zhou, Y.; Chen, Y.; Gu, J. Fastp: An ultra-fast all-in-one fastq preprocessor. Bioinformatics 2018, 34, i884–i890. [Google Scholar] [CrossRef]
- Rochette, N.C.; Catchen, J.M. Deriving genotypes from RAD-Seq short-read data using stacks. Nat. Protoc. 2017, 12, 2640–2659. [Google Scholar] [CrossRef]
- Li, H.; Durbin, R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 2009, 25, 1754–1760. [Google Scholar] [CrossRef]
- Van der Auwera, G.A.; Carneiro, M.O.; Hartl, C.; Poplin, R.; del Angel, G.; Levy-Moonshine, A.; Jordan, T.; Shakir, K.; Roazen, D.; Thibault, J.; et al. From fastq data to high-confidence variant calls: The genome analysis toolkit best practices pipeline. Curr. Protoc. Bioinforma. 2013, 43, 11.10.1–11.10.33. [Google Scholar] [CrossRef]
- Danecek, P.; Auton, A.; Abecasis, G.; Albers, C.A.; Banks, E.; DePristo, M.A.; Handsaker, R.E.; Lunter, G.; Marth, G.T.; Sherry, S.T.; et al. The variant call format and VCFtools. Bioinformatics 2011, 27, 2156–2158. [Google Scholar] [CrossRef]
- Foll, M.; Gaggiotti, O. A genome-scan method to identify selected loci appropriate for both dominant and codominant markers: A Bayesian perspective. Genetics 2008, 180, 977–993. [Google Scholar] [CrossRef]
- Purcell, S.; Neale, B.; Todd-Brown, K.; Thomas, L.; Ferreira, M.A.R.; Bender, D.; Maller, J.; Sklar, P.; de Bakker, P.I.W.; Daly, M.J.; et al. PLINK: A tool set for whole-genome association and population-based linkage analyses. Am. J. Hum. Genet. 2007, 81, 559–575. [Google Scholar] [CrossRef] [PubMed]
- Alexander, D.H.; Novembre, J.; Lange, K. Fast model-based estimation of ancestry in unrelated individuals. Genome Res. 2009, 19, 1655–1664. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.-H.; Qin, C.; Wang, L.; Jiao, C.; Hong, H.; Tian, Y.; Li, Y.; Xing, G.; Wang, J.; Gu, Y.; et al. Genome-wide signatures of the geographic expansion and breeding of soybean. Sci. China Life Sci. 2023, 66, 350–365. [Google Scholar] [CrossRef] [PubMed]
- Saitou, N.; Nei, M. The neighbor-joining method: A new method for reconstructing phylogenetic trees. Mol. Biol. Evol. 1987, 4, 406–425. [Google Scholar] [CrossRef] [PubMed]
- Tamura, K.; Stecher, G.; Kumar, S. MEGA11: Molecular evolutionary genetics analysis version 11. Mol. Biol. Evol. 2021, 38, 3022–3027. [Google Scholar] [CrossRef]
- Hoang, D.T.; Chernomor, O.; von Haeseler, A.; Minh, B.Q.; Vinh, L.S. UFBoot2: Improving the ultrafast bootstrap approximation. Mol. Biol. Evol. 2018, 35, 518–522. [Google Scholar] [CrossRef]
- Minh, B.Q.; Schmidt, H.A.; Chernomor, O.; Schrempf, D.; Woodhams, M.D.; von Haeseler, A.; Lanfear, R. IQ-TREE 2: New models and efficient methods for phylogenetic inference in the genomic era. Mol. Biol. Evol. 2020, 37, 1530–1534. [Google Scholar] [CrossRef]
- Kalyaanamoorthy, S.; Minh, B.Q.; Wong, T.K.F.; von Haeseler, A.; Jermiin, L.S. ModelFinder: Fast model selection for accurate phylogenetic estimates. Nat. Methods 2017, 14, 587–589. [Google Scholar] [CrossRef]
- Liu, X.; Fu, Y.-X. Stairway Plot 2: Demographic history inference with folded SNP frequency spectra. Genome Biol. 2020, 21, 1–9. [Google Scholar] [CrossRef]
- Ossowski, S.; Schneeberger, K.; Lucas-Lledó, J.I.; Warthmann, N.; Clark, R.M.; Shaw, R.G.; Weigel, D.; Lynch, M. The rate and molecular spectrum of spontaneous mutations in Arabidopsis thaliana. Science 2010, 327, 92–94. [Google Scholar] [CrossRef]



Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lin, H.; Chen, L.; Li, J. Multiple Introductions and Distinct Genetic Groups of Canada Goldenrod (Solidago canadensis) in China Revealed by Genomic Single-Nucleotide Polymorphisms. Plants 2023, 12, 1734. https://doi.org/10.3390/plants12091734
Lin H, Chen L, Li J. Multiple Introductions and Distinct Genetic Groups of Canada Goldenrod (Solidago canadensis) in China Revealed by Genomic Single-Nucleotide Polymorphisms. Plants. 2023; 12(9):1734. https://doi.org/10.3390/plants12091734
Chicago/Turabian StyleLin, Hanyang, Luxi Chen, and Junmin Li. 2023. "Multiple Introductions and Distinct Genetic Groups of Canada Goldenrod (Solidago canadensis) in China Revealed by Genomic Single-Nucleotide Polymorphisms" Plants 12, no. 9: 1734. https://doi.org/10.3390/plants12091734