
LSPpred Suite: Tools for Leaderless Secretory Protein Prediction in Plants
 , , , ,
, , , , 

Abstract
:1. Introduction
2. Methods
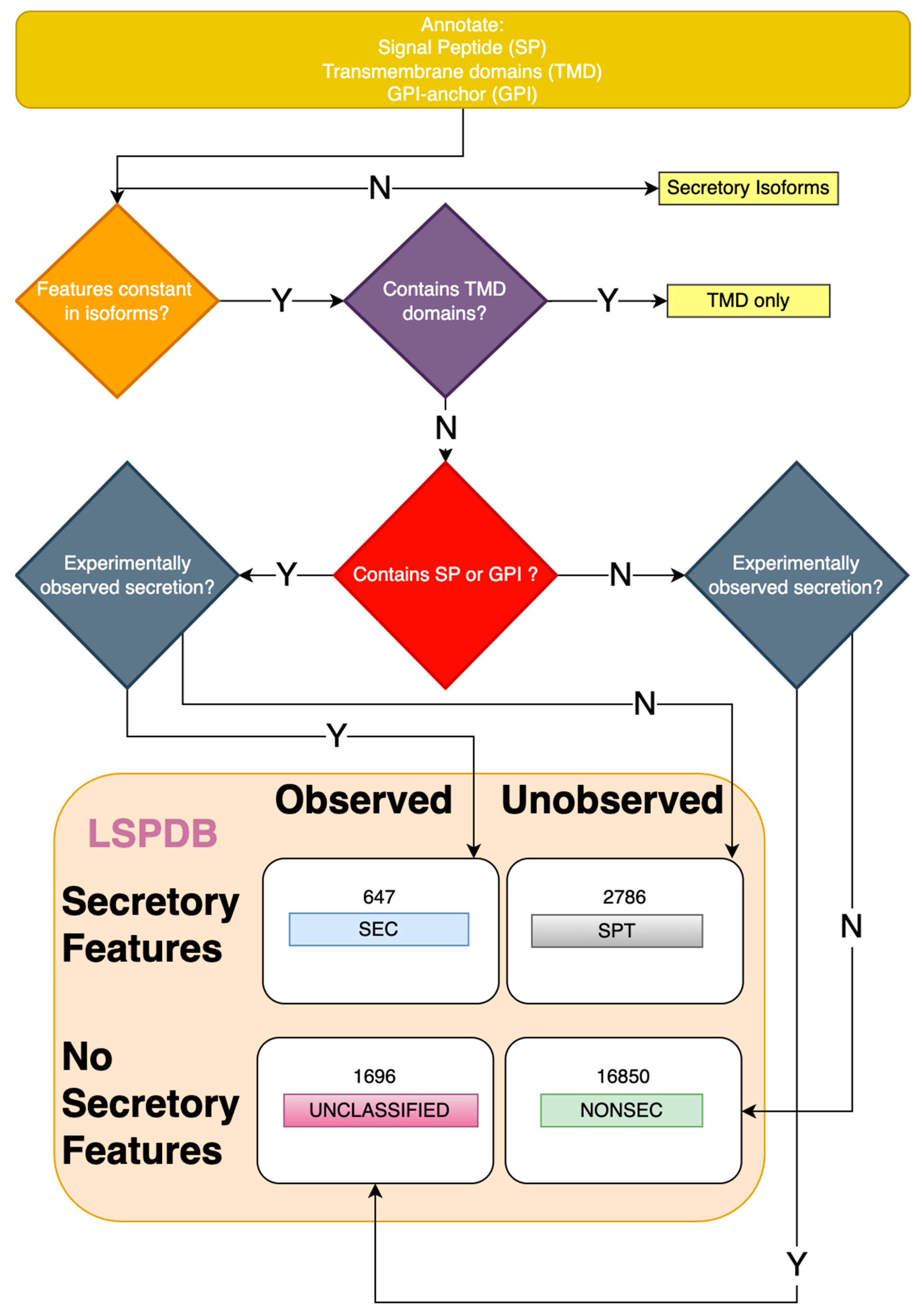
2.1. Generation of a Leaderless Secretory Protein Database (LSPDB)
2.2. Input Data for Classifiers
2.3. Training Random Forest Models on Unbalanced Data
2.4. Metrics for Preferred Classifier Model Selection
3. Results
3.1. Performance and Relevant Features of the Preferred LSPpred Model
3.2. Testing on Plant LSPs
3.3. An Independent Test of LSPpred and SPLpred Using Experimental Data
4. Discussion
4.1. Evaluation and Limitations
4.2. Insights into Feature Importance
4.3. Availability and Recommendations for Use
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Armenteros, J.J.A.; Sønderby, C.K.; Sønderby, S.K.; Nielsen, H.; Winther, O. DeepLoc: Prediction of protein subcellular localization using deep learning. Bioinformatics 2017, 33, 3387–3395. [Google Scholar] [CrossRef] [PubMed]
- Bellucci, M.; de Marchis, F.; Pompa, A. The endoplasmic reticulum is a hub to sort proteins toward unconventional traffic pathways and endosymbiotic organelles. J. Exp. Bot. 2017, 69, 7–20. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ding, Y.; Robinson, D.G.; Jiang, L. Unconventional protein secretion (UPS) pathways in plants. Curr. Opin. Cell Biol. 2014, 29, 107–115. [Google Scholar] [CrossRef] [PubMed]
- Miura, N.; Ueda, M. Evaluation of Unconventional Protein Secretion by Saccharomyces cerevisiae and other Fungi. Cells 2018, 7, 128. [Google Scholar] [CrossRef] [Green Version]
- Dimou, E.; Nickel, W. Unconventional mechanisms of eukaryotic protein secretion. Curr. Biol. 2018, 28, R406–R410. [Google Scholar] [CrossRef] [Green Version]
- Rabouille, C. Pathways of Unconventional Protein Secretion. Trends Cell Biol. 2016, 27, 230–240. [Google Scholar] [CrossRef]
- Cui, Y.; Gao, J.; He, Y.; Jiang, L. Plant extracellular vesicles. Protoplasma 2019, 257, 3–12. [Google Scholar] [CrossRef]
- Filaquier, A.; Marin, P.; Parmentier, M.-L.; Villeneuve, J. Roads and hubs of unconventional protein secretion. Curr. Opin. Cell Biol. 2022, 75, 102072. [Google Scholar] [CrossRef]
- Nickel, W.; Rabouille, C. Unconventional protein secretion: Diversity and consensus. Semin. Cell Dev. Biol. 2018, 83, 1–2. [Google Scholar] [CrossRef]
- Kim, J.; Gee, H.Y.; Lee, M.G. Unconventional protein secretion—New insights into the pathogenesis and therapeutic targets of human diseases. J. Cell Sci. 2018, 131, jcs213686. [Google Scholar] [CrossRef] [Green Version]
- Goring, D.R.; Di Sansebastiano, G.P. Protein and membrane trafficking routes in plants: Conventional or unconventional? J. Exp. Bot. 2017, 69, 1–5. [Google Scholar] [CrossRef] [Green Version]
- Nielsen, H.; Petsalaki, E.I.; Zhao, L.; Stühler, K. Predicting eukaryotic protein secretion without signals. Biochim. Biophys. Acta BBA Proteins Proteom. 2018, 1867, 140174. [Google Scholar] [CrossRef] [Green Version]
- Bendtsen, J.D.; Kiemer, L.; Fausbøll, A.; Brunak, S. Non-classical protein secretion in bacteria. BMC Microbiol. 2005, 5, 58. [Google Scholar] [CrossRef] [Green Version]
- Green, E.R.; Mecsas, J. Bacterial Secretion Systems: An Overview. Microbiol. Spectr. 2016, 4, 1. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Yu, S.; Xie, R.; Li, J.; Leier, A.; Marquez-Lago, T.T.; Akutsu, T.; Smith, A.I.; Ge, Z.; Wang, J.; et al. PeNGaRoo, a combined gradient boosting and ensemble learning framework for predicting non-classical secreted proteins. Bioinformatics 2019, 36, 704–712. [Google Scholar] [CrossRef]
- Davis, D.J.; Kang, B.-H.; Heringer, A.S.; Wilkop, T.E.; Drakakaki, G. Unconventional Protein Secretion in Plants. In Unconventional Protein Secretion: Methods and Protocols; Pompa, A., de Marchis, F., Eds.; Springer: New York, NY, USA, 2016; pp. 47–63. [Google Scholar] [CrossRef]
- Robinson, D.G.; Ding, Y.; Jiang, L. Unconventional protein secretion in plants: A critical assessment. Protoplasma 2015, 253, 31–43. [Google Scholar] [CrossRef]
- Ding, Y.; Wang, J.; Wang, J.; Stierhof, Y.-D.; Robinson, D.G.; Jiang, L. Unconventional protein secretion. Trends Plant Sci. 2012, 17, 606–615. [Google Scholar] [CrossRef]
- Agrawal, G.K.; Jwa, N.-S.; Lebrun, M.-H.; Job, D.; Rakwal, R. Plant secretome: Unlocking secrets of the secreted proteins. Proteomics 2009, 10, 799–827. [Google Scholar] [CrossRef]
- Krause, C.; Richter, S.; Knöll, C.; Jürgens, G. Plant secretome—From cellular process to biological activity. Biochim. Biophys. Acta BBA Proteins Proteom. 2013, 1834, 2429–2441. [Google Scholar] [CrossRef]
- Bendtsen, J.D.; Jensen, L.J.; Blom, N.; von Heijne, G.; Brunak, S. Feature-based prediction of non-classical and leaderless protein secretion. Protein Eng. Des. Sel. PEDS 2004, 17, 349–356. [Google Scholar] [CrossRef] [Green Version]
- Lonsdale, A.; Davis, M.J.; Doblin, M.S.; Bacic, A. Better Than Nothing? Limitations of the Prediction Tool SecretomeP in the Search for Leaderless Secretory Proteins (LSPs) in Plants. Front. Plant Sci. 2016, 7, 1451. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheng, F.-Y.; Blackburn, K.; Lin, Y.-M.; Goshe, M.B.; Williamson, J.D. Absolute Protein Quantification by LC/MSE for Global Analysis of Salicylic Acid-Induced Plant Protein Secretion Responses. J. Proteome Res. 2008, 8, 82–93. [Google Scholar] [CrossRef] [PubMed]
- Pinedo, M.; Regente, M.; Elizalde, M.; Quiroga, I.Y.; Pagnussat, L.A.; Jorrin-Novo, J.; Maldonado, A.; de la Canal, L. Extracellular sunflower proteins: Evidence on non-classical secretion of a jacalin-related lectin. Protein Pept. Lett. 2012, 19, 270–276. [Google Scholar] [CrossRef] [PubMed]
- Del Rio, M.; de la Canal, L.; Pinedo, M.; Regente, M. Internalization of a sunflower mannose-binding lectin into phytopathogenic fungal cells induces cytotoxicity. J. Plant Physiol. 2018, 221, 22–31. [Google Scholar] [CrossRef]
- Wang, J.; Ding, Y.; Wang, J.; Hillmer, S.; Miao, Y.; Lo, S.W.; Wang, X.; Robinson, D.G.; Jiang, L. EXPO, an Exocyst-Positive Organelle Distinct from Multivesicular Endosomes and Autophagosomes, Mediates Cytosol to Cell Wall Exocytosis in Arabidopsis and Tobacco Cells. Plant Cell 2010, 22, 4009–4030. [Google Scholar] [CrossRef] [Green Version]
- Zhang, H.; Li, J. From Cytosol to the Apoplast: The Hygromycin Phosphotransferase (HYGR) Model in Arabidopsis. In Unconventional Protein Secretion: Methods and Protocols; Pompa, A., de Marchis, F., Eds.; Springer: New York, NY, USA, 2016; pp. 81–90. [Google Scholar] [CrossRef]
- Zhang, Y.; Giboulot, A.; Zivy, M.; Valot, B.; Jamet, E.; Albenne, C. Combining various strategies to increase the coverage of the plant cell wall glycoproteome. Phytochemistry 2011, 72, 1109–1123. [Google Scholar] [CrossRef] [Green Version]
- Viotti, C. ER to Golgi-Dependent Protein Secretion: The Conventional Pathway. In Unconventional Protein Secretion: Methods and Protocols; Pompa, A., de Marchis, F., Eds.; Springer: New York, NY, USA, 2016; pp. 3–29. [Google Scholar] [CrossRef]
- Li, W.; Godzik, A. Cd-hit: A fast program for clustering and comparing large sets of protein or nucleotide sequences. Bioinformatics 2006, 22, 1658–1659. [Google Scholar] [CrossRef] [Green Version]
- Ofer, D.; Linial, M. ProFET: Feature Engineering Captures High-Level Protein Functions. Bioinformatics 2015, 31, 3429–3436. [Google Scholar] [CrossRef] [Green Version]
- Larrañaga, P.; Calvo, B.; Santana, R.; Bielza, C.; Galdiano, J.; Inza, I.; Lozano, J.A.; Armañanzas, R.; Santafé, G.; Pérez, A.; et al. Machine learning in bioinformatics. Briefings Bioinform. 2006, 7, 86–112. [Google Scholar] [CrossRef] [Green Version]
- Pedregosa, F.; Varoquaux, G.; Gramfort, A.; Michel, V.; Thirion, B.; Grisel, O.; Blondel, M.; Prettenhofer, P.; Weiss, R.; Dubourg, V.; et al. Scikit-Learn: Machine Learning in Python. J. Mach. Learn. Res. JMLR 2011, 12, 2825–2830. [Google Scholar]
- Lemaître, G.; Nogueira, F.; Aridas, C.K. Imbalanced-Learn: A Python Toolbox to Tackle the Curse of Imbalanced Datasets in Machine Learning. J. Mach. Learn. Res. JMLR 2017, 18, 1–5. [Google Scholar]
- Garg, A.; Raghava, G.P.S. A machine learning based method for the prediction of secretory proteins using amino acid composition, their order and similarity-search. Silico Biol. 2008, 8, 129–140. [Google Scholar]
- Almagro Armenteros, J.J.; Tsirigos, K.D.; Sønderby, C.K.; Petersen, T.N.; Winther, O.; Brunak, S.; Von Heijne, G.; Nielsen, H. SignalP 5.0 improves signal peptide predictions using deep neural networks. Nat. Biotechnol. 2019, 37, 420–423. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sperschneider, J.; Dodds, P.N.; Singh, K.B.; Taylor, J.M. ApoplastP: Prediction of Effectors and Plant Proteins in the Apoplast Using Machine Learning. New Phytol. 2018, 217, 1764–1778. [Google Scholar] [CrossRef] [Green Version]
- Sperschneider, J.; Dodds, P.N. EffectorP 3.0: Prediction of Apoplastic and Cytoplasmic Effectors in Fungi and Oomycetes. Mol. Plant-Microbe Interact. 2022, 35, 146–156. [Google Scholar] [CrossRef] [PubMed]
- Zhao, L.; Poschmann, G.; Waldera-Lupa, D.; Rafiee, N.; Kollmann, M.; Stühler, K. OutCyte: A novel tool for predicting unconventional protein secretion. Sci. Rep. 2019, 9, 19448. [Google Scholar] [CrossRef] [Green Version]
- Ras-Carmona, A.; Gomez-Perosanz, M.; Reche, P.A. Prediction of unconventional protein secretion by exosomes. BMC Bioinform. 2021, 22, 333. [Google Scholar] [CrossRef]
- Ceballos-Laita, L.; Gutierrez-Carbonell, E.; Takahashi, D.; Lonsdale, A.; Abadía, A.; Doblin, M.S.; Bacic, A.; Uemura, M.; Abadía, J.; López-Millán, A.F. Effects of Excess Manganese on the Xylem Sap Protein Profile of Tomato (Solanum lycopersicum) as Revealed by Shotgun Proteomic Analysis. Int. J. Mol. Sci. 2020, 21, 8863. [Google Scholar] [CrossRef]
- Ho, T.C.; Blackburn, R.K.; Goshe, M.B.; Williamson, J.D. Identification of multiple proteins whose interaction with mannitol dehydrogenase is induced by salicylic acid: Implications for unconventional secretion. Proteomics 2021, 22, e2100091. [Google Scholar] [CrossRef]
- Dubchak, I.; Muchnik, I.; Holbrook, S.R.; Kim, S.H. Prediction of Protein Folding Class Using Global Description of Amino Acid Sequence. Proc. Natl. Acad. Sci. USA 1995, 92, 8700–8704. [Google Scholar] [CrossRef] [Green Version]
- Pierleoni, A.; Martelli, P.L.; Casadio, R. PredGPI: A GPI-anchor predictor. BMC Bioinform. 2008, 9, 392. [Google Scholar] [CrossRef] [Green Version]
- Basu, U.; Francis, J.L.; Whittal, R.M.; Stephens, J.L.; Wang, Y.; Zaiane, O.R.; Goebel, R.; Muench, D.G.; Good, A.G.; Taylor, G.J. Extracellular Proteomes of Arabidopsis thaliana and Brassica napus Roots: Analysis and Comparison by MudPIT and LC-MS/MS. Plant Soil 2006, 286, 357–376. [Google Scholar] [CrossRef]
- Nguyen-Kim, H.; Clemente, H.S.; Balliau, T.; Zivy, M.; Dunand, C.; Albenne, C.; Jamet, E. Arabidopsis thaliana root cell wall proteomics: Increasing the proteome coverage using a combinatorial peptide ligand library and description of unexpected Hyp in peroxidase amino acid sequences. Proteomics 2015, 16, 491–503. [Google Scholar] [CrossRef]
- Hervé, V.; Duruflé, H.; Clemente, H.S.; Albenne, C.; Balliau, T.; Zivy, M.; Dunand, C.; Jamet, E. An enlarged cell wall proteome of Arabidopsis thaliana rosettes. Proteomics 2016, 16, 3183–3187. [Google Scholar] [CrossRef]
- Chen, Y.; Ye, D.; Held, M.A.; Cannon, M.C.; Ray, T.; Saha, P.; Frye, A.N.; Mort, A.J.; Kieliszewski, M.J. Identification of the Abundant Hydroxyproline-Rich Glycoproteins in the Root Walls of Wild-Type Arabidopsis, an ext3 Mutant Line, and Its Phenotypic Revertant. Plants 2015, 4, 85–111. [Google Scholar] [CrossRef] [Green Version]
- Boudart, G.; Jamet, E.; Rossignol, M.; Lafitte, C.; Borderies, G.; Jauneau, A.; Esquerré-Tugayé, M.-T.; Pont-Lezica, R. Cell wall proteins in apoplastic fluids of Arabidopsis thaliana rosettes: Identification by mass spectrometry and bioinformatics. Proteomics 2004, 5, 212–221. [Google Scholar] [CrossRef] [Green Version]
- Haslam, R.P.; Downie, A.L.; Raveton, M.; Gallardo, K.; Job, D.; Pallett, K.E.; John, P.; Parry, M.A.J.; Coleman, J.O.D. The assessment of enriched apoplastic extracts using proteomic approaches. Ann. Appl. Biol. 2003, 143, 81–91. [Google Scholar] [CrossRef]
- Trentin, A.R.; Pivato, M.; Mehdi, S.M.M.; Barnabas, L.E.; Giaretta, S.; Fabrega-Prats, M.; Prasad, D.; Arrigoni, G.; Masi, A. Proteome readjustments in the apoplastic space of Arabidopsis thaliana ggt1 mutant leaves exposed to UV-B radiation. Front. Plant Sci. 2015, 6, 128. [Google Scholar] [CrossRef] [Green Version]
- Charmont, S.; Jamet, E.; Pont-Lezica, R.; Canut, H. Proteomic analysis of secreted proteins from Arabidopsis thaliana seedlings: Improved recovery following removal of phenolic compounds. Phytochemistry 2005, 66, 453–461. [Google Scholar] [CrossRef] [Green Version]
- Minic, Z.; Boudart, G.; Albenne, C.; Canut, H.; Jamet, E.; Pont-Lezica, R.F. Cell Wall Proteome. In Plant Proteomics; Šamaj, J., Thelen, J.J., Eds.; Springer: Berlin/Heidelberg, Germany, 2007; pp. 169–185. [Google Scholar] [CrossRef]
- Kwon, H.-K.; Yokoyama, R.; Nishitani, K. A Proteomic Approach to Apoplastic Proteins Involved in Cell Wall Regeneration in Protoplasts of Arabidopsis Suspension-Cultured Cells. Plant Cell Physiol. 2005, 46, 843–857. [Google Scholar] [CrossRef] [Green Version]
- Robertson, D.; Mitchell, G.P.; Gilroy, J.S.; Gerrish, C.; Bolwell, G.P.; Slabas, A.R. Differential Extraction and Protein Sequencing Reveals Major Differences in Patterns of Primary Cell Wall Proteins from Plants. J. Biol. Chem. 1997, 272, 15841–15848. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tran, H.T.; Plaxton, W.C. Proteomic analysis of alterations in the secretome of Arabidopsis thaliana suspension cells subjected to nutritional phosphate deficiency. Proteomics 2008, 8, 4317–4326. [Google Scholar] [CrossRef] [PubMed]
- Borderies, G.; Jamet, E.; Lafitte, C.; Rossignol, M.; Jauneau, A.; Boudart, G.; Monsarrat, B.; Esquerré-Tugayé, M.-T.; Boudet, A.; Pont-Lezica, R. Proteomics of loosely bound cell wall proteins of Arabidopsis thaliana cell suspension cultures: A critical analysis. Electrophoresis 2003, 24, 3421–3432. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Irshad, M.; Canut, H.; Borderies, G.; Pont-Lezica, R.; Jamet, E. A new picture of cell wall protein dynamics in elongating cells of Arabidopsis thaliana: Confirmed actors and newcomers. BMC Plant Biol. 2008, 8, 94. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oh, I.S.; Park, A.R.; Bae, M.S.; Kwon, S.J.; Kim, Y.S.; Lee, J.E.; Kang, N.Y.; Lee, S.; Cheong, H.; Park, O.K. Secretome Analysis Reveals an Arabidopsis Lipase Involved in Defense against Alternaria brassicicola. Plant Cell Online 2005, 17, 2832–2847. [Google Scholar] [CrossRef] [Green Version]
- Bayer, E.M.; Bottrill, A.R.; Walshaw, J.; Vigouroux, M.; Naldrett, M.J.; Thomas, C.L.; Maule, A.J. Arabidopsis cell wall proteome defined using multidimensional protein identification technology. Proteomics 2006, 6, 301–311. [Google Scholar] [CrossRef]
- Feiz, L.; Irshad, M.; Pont-Lezica, R.F.; Canut, H.; Jamet, E. Evaluation of cell wall preparations for proteomics: A new procedure for purifying cell walls from Arabidopsis hypocotyls. Plant Methods 2006, 2, 10. [Google Scholar] [CrossRef] [Green Version]
- Borner, G.H.H.; Lilley, K.S.; Stevens, T.; Dupree, P. Identification of Glycosylphosphatidylinositol-Anchored Proteins in Arabidopsis. A Proteomic and Genomic Analysis. Plant Physiol. 2003, 132, 568–577. [Google Scholar] [CrossRef] [Green Version]
- Casasoli, M.; Spadoni, S.; Lilley, K.S.; Cervone, F.; de Lorenzo, G.; Mattei, B. Identification by 2-D DIGE of apoplastic proteins regulated by oligogalacturonides in Arabidopsis thaliana. Proteomics 2008, 8, 1042–1054. [Google Scholar] [CrossRef]
- Chivasa, S.; Ndimba, B.K.; Simon, W.J.; Robertson, D.; Yu, X.-L.; Knox, P.; Bolwell, P.; Slabas, A.R. Proteomic analysis of the Arabidopsis thaliana cell wall. Electrophoresis 2002, 23, 1754–1765. [Google Scholar] [CrossRef]
- Schultz, C.J.; Ferguson, K.L.; Lahnstein, J.; Bacic, A. Post-translational Modifications of Arabinogalactan-peptides of Arabidopsis thaliana: Endoplasmic Reticulum and Glycosylphosphatidylinositol-Anchor Signal Cleavage Sites and Hydroxylation of Proline. J. Biol. Chem. 2004, 279, 45503–45511. [Google Scholar] [CrossRef] [Green Version]
- Krogh, A.; Larsson, B.; von Heijne, G.; Sonnhammer, E.L. Predicting transmembrane protein topology with a hidden markov model: Application to complete genomes. J. Mol. Biol. 2001, 305, 567–580. [Google Scholar] [CrossRef] [Green Version]
- Eisenhaber, B.; Bork, P.; Eisenhaber, F. Prediction of Potential GPI-modification Sites in Proprotein Sequences. J. Mol. Biol. 1999, 292, 741–758. [Google Scholar] [CrossRef]
- Uhlén, M.; Fagerberg, L.; Hallström, B.M.; Lindskog, C.; Oksvold, P.; Mardinoglu, A.; Sivertsson, Å.; Kampf, C.; Sjöstedt, E.; Asplund, A.; et al. Proteomics. Tissue-Based Map of the Human Proteome. Science 2015, 347, 1260419. [Google Scholar] [CrossRef]
- Brandão, M.M.; Dantas, L.L.; Silva-Filho, M.C. AtPIN: Arabidopsis thaliana Protein Interaction Network. BMC Bioinform. 2009, 10, 454. [Google Scholar] [CrossRef] [Green Version]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
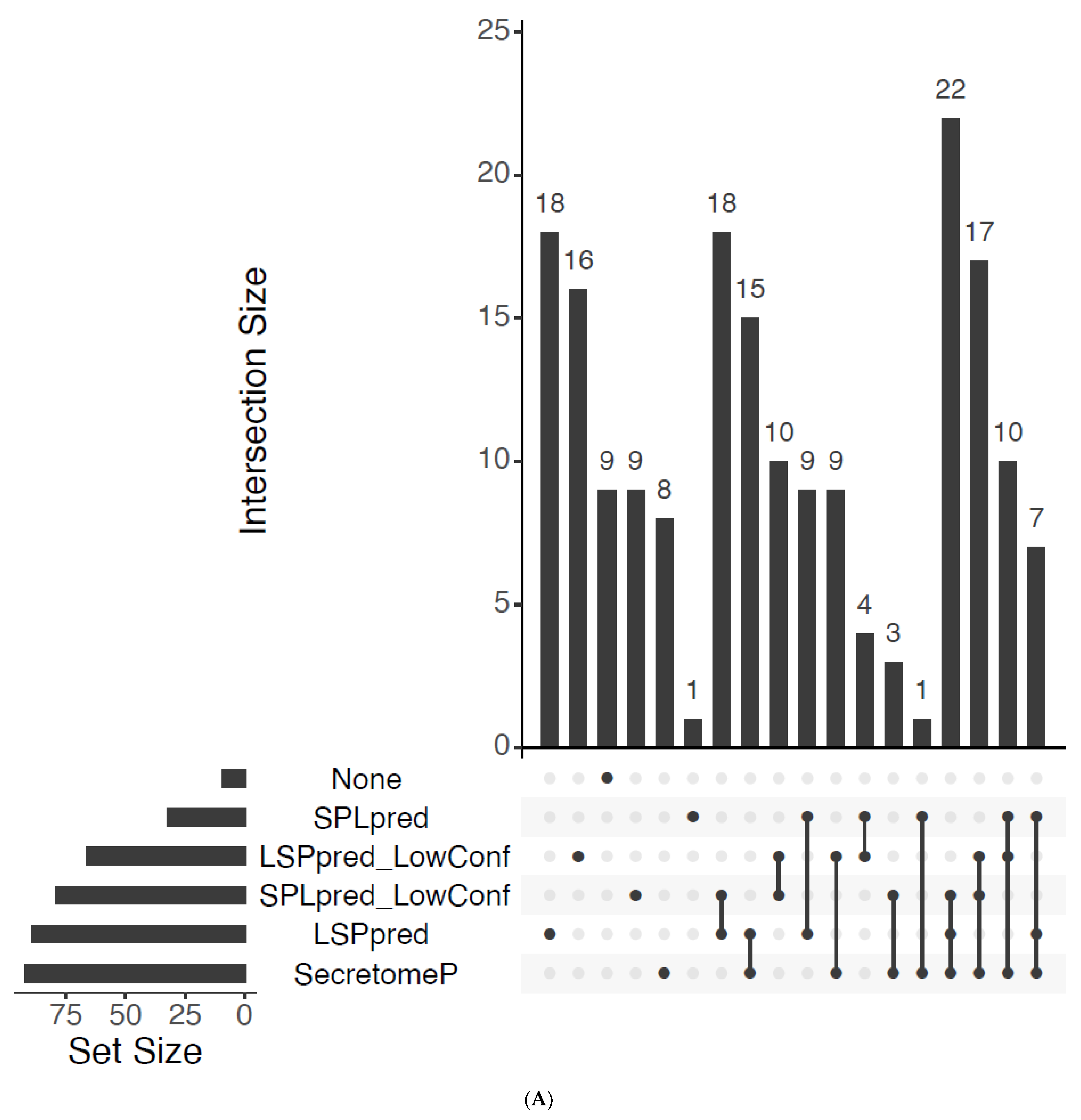{kind=link}
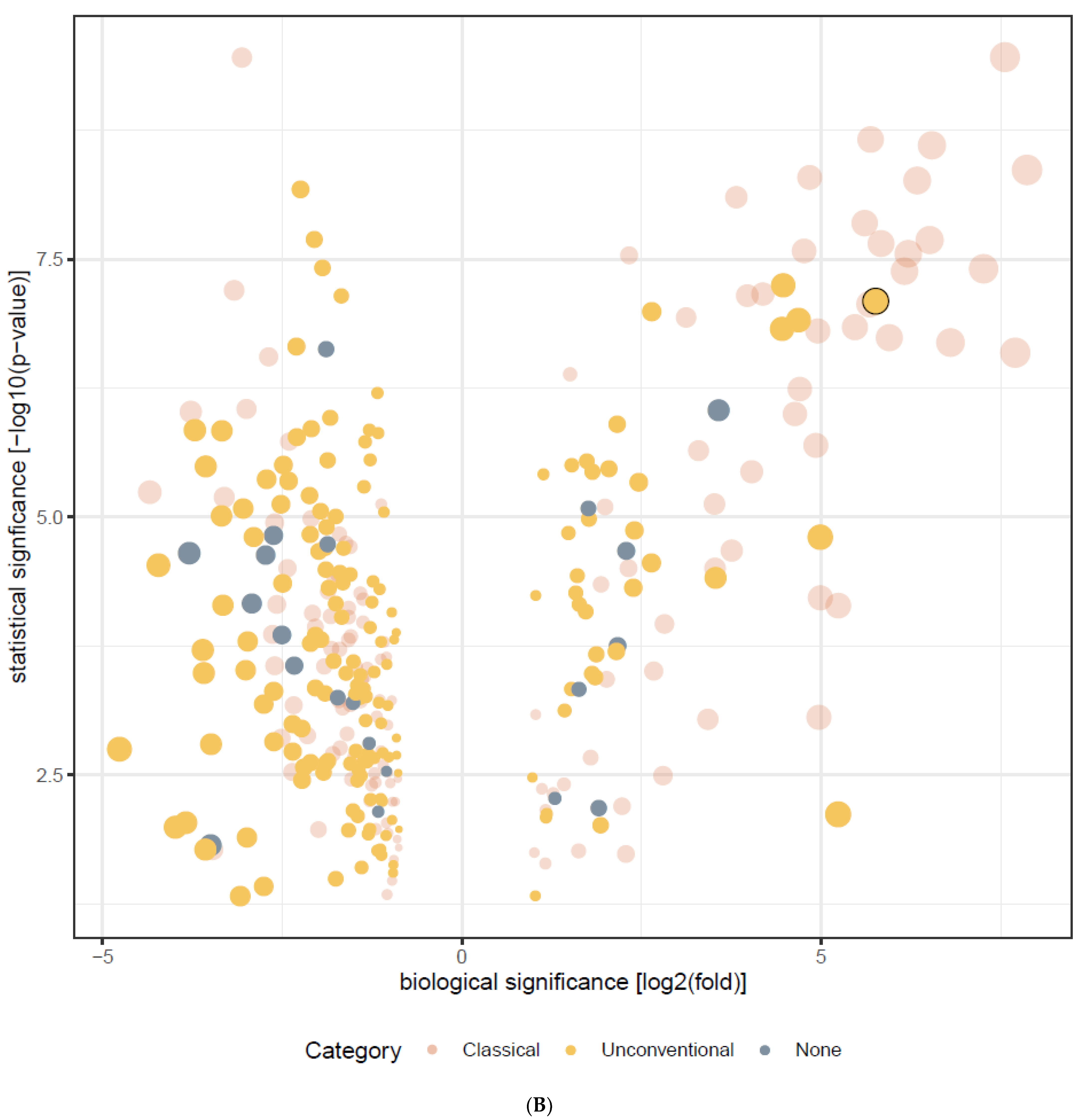{kind=link}
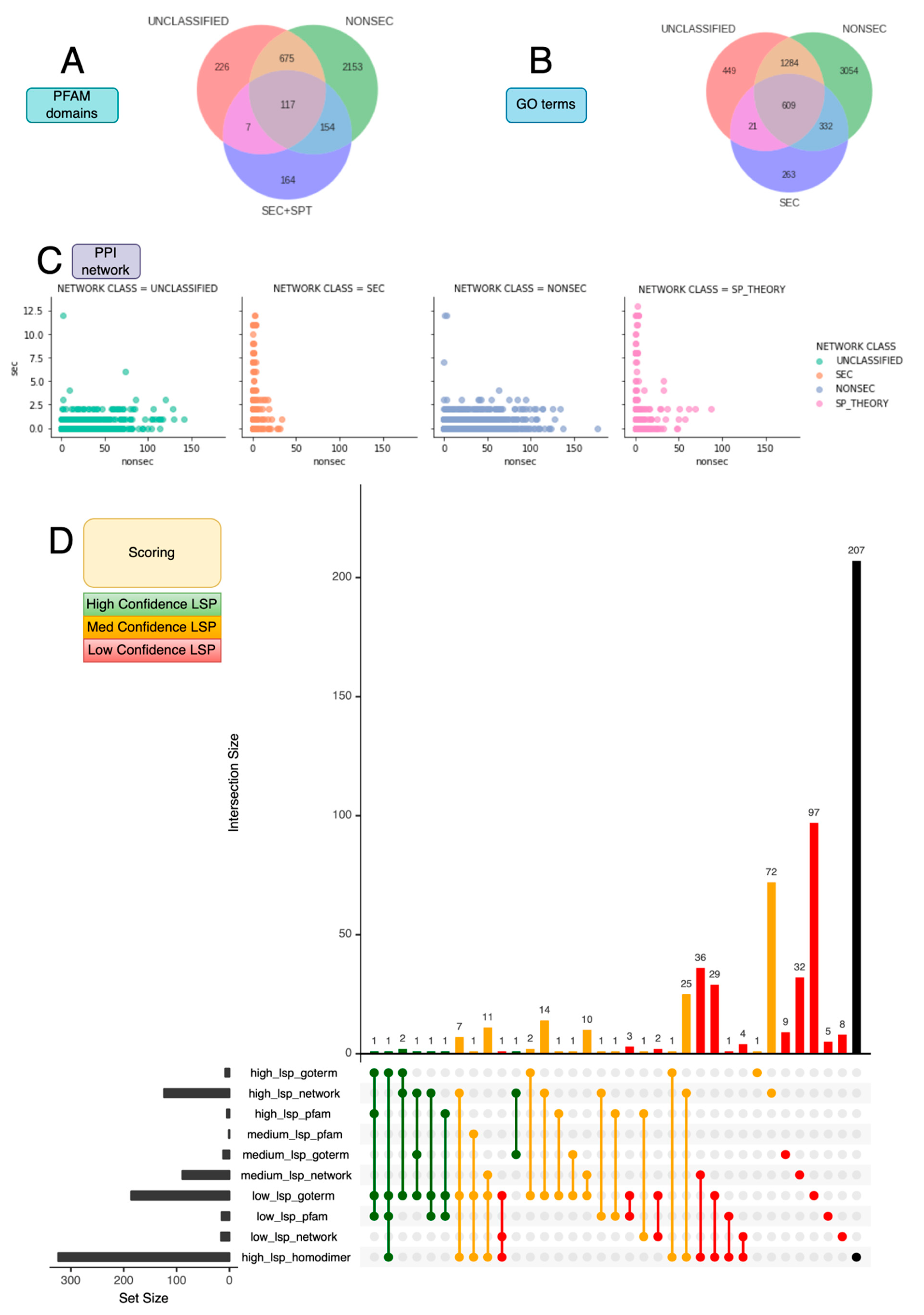
| Confidence (Overall Score) | Network | PFAM Domain | GO Term |
|---|---|---|---|
| Tier 1 (1 point) | All UNCLASSIFIED proteins with SPT interactions matching the profile of SPT with >1 SEC and 0 NONSEC (16 proteins) | PFAMs predominantly in SEC, possibly in SPT, must be in UNCLASSIFIED (15 proteins) | GO predominantly in SEC, possibly in SPT, must be in UNCLASSIFIED (186 proteins) |
| Tier 2 (2 points) | All UNCLASSIFIED proteins with SEC interactions matching the profile, >1 SEC > 33 NONSEC (89 proteins) | PFAMs exclusive to SEC, SPT and UNCLASSIFIED (1 protein) | GO exclusive to SEC, SPT and UNCLASSIFIED (12 proteins) |
| Tier 3 (3 points) | All UNCLASSIFIED proteins with SEC interactions matching the profile, >1 SEC ≤ 33 NONSEC (124 proteins) | PFAMs exclusive to SEC and UNCLASSIFIED (5 proteins) | GO exclusive to SEC and UNCLASSIFIED (8 proteins) |
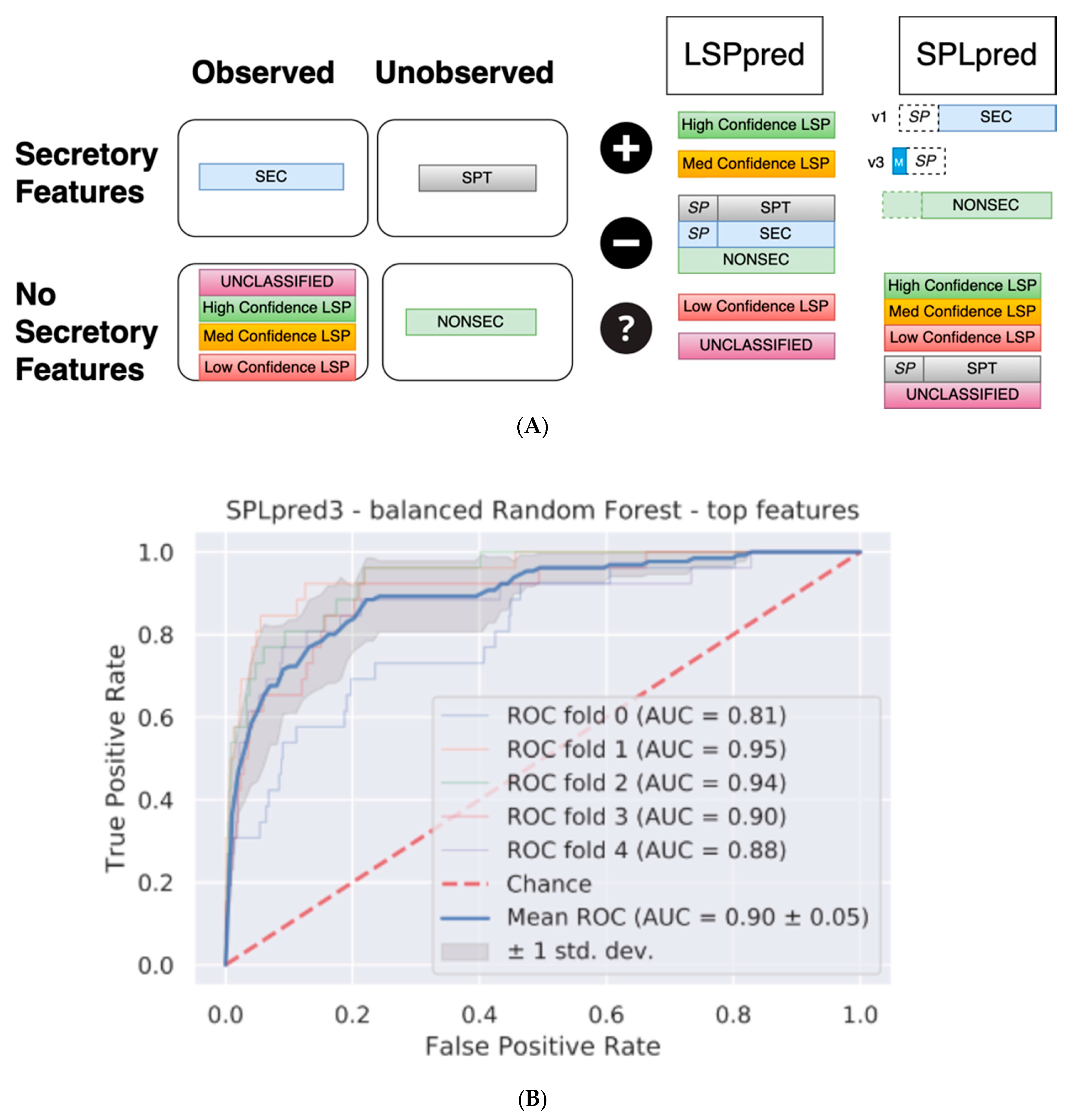
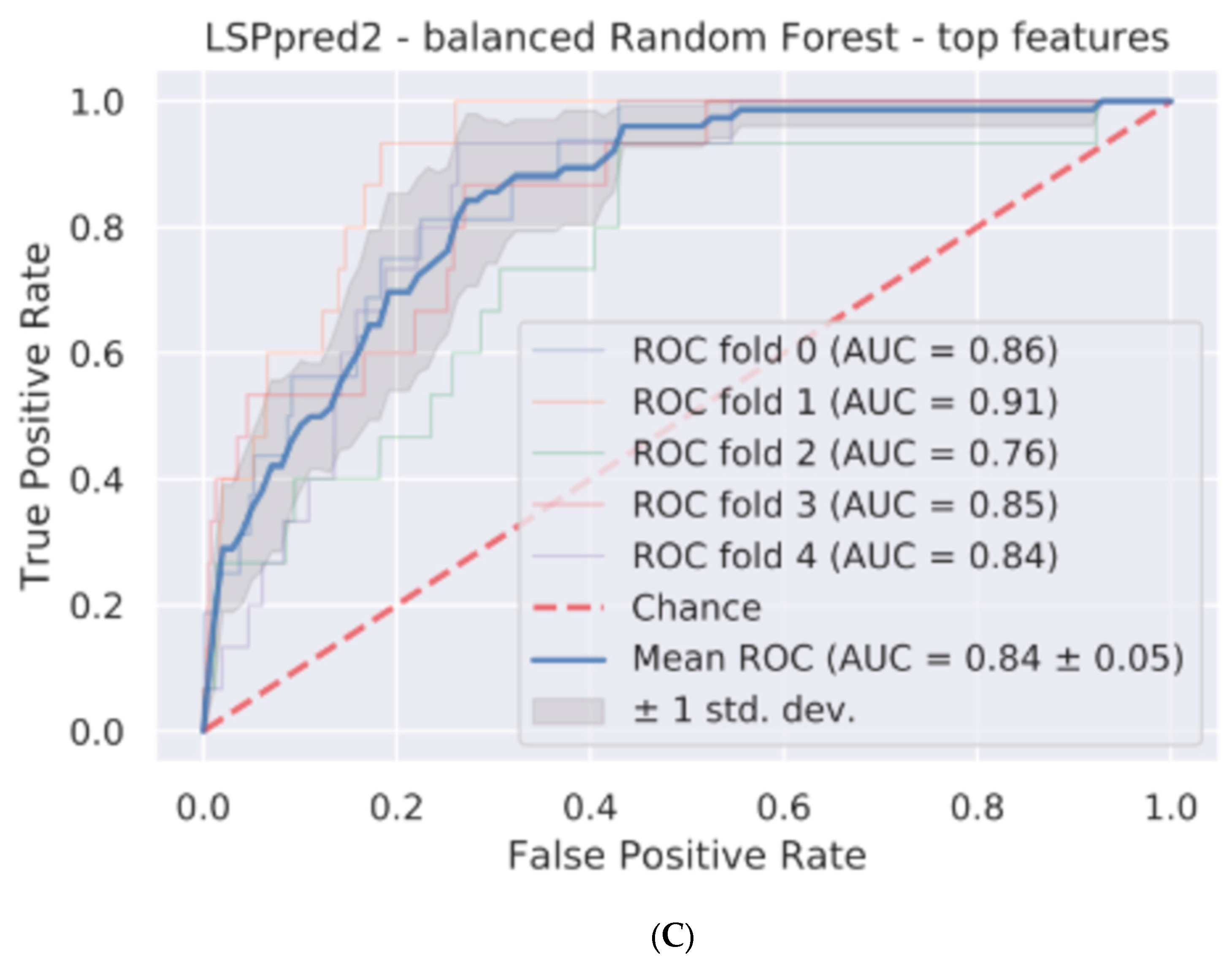
| Tool | Version | AUROC | Mean Threshold for 0.05 FPR | TPR for Threshold | Balanced Accuracy (Controlled Threshold) | Balance Accuracy (0.5) |
|---|---|---|---|---|---|---|
| LSPpred | 1 | 0.82 ± 0.05 | 0.65 | 0.3675 | 0.6 | 0.71 |
| LSPpred | 2 | 0.84 ± 0.05 | 0.68 | 0.355 | 0.63 | 0.65 |
| SPLpred | 1 | 0.99 ± 0.01 | 0.61 | 0.946 | 0.95 | 0.94 |
| SPLpred | 2 | 0.88 ± 0.06 | 0.70 | 0.62 | 0.72 | 0.81 |
| SPLpred | 3 | 0.90 ± 0.05 | 0.70 | 0.62 | 0.76 | 0.82 |
| SPLpred | 4 | 0.89 ± 0.04 | 0.67 | 0.58 | 0.83 | 0.84 |
| Tool | HYGR | SAMS2 | MDH | Helja |
|---|---|---|---|---|
| SecretomeP (Mammalian) | No 0.513 | No 0.421 | No 0.450 | No 0.447 |
| SecretomeP (Gram-) | No 0.078 | No 0.173 | No 0.108 | Yes 0.55 |
| SecretomeP (Gram+) | No 0.087 | No 0.153 | No 0.086 | No 0.402 |
| DeepLoc | Peroxisome, Soluble 0.386 Extracellular 0.007 | Cytoplasm, Soluble 0.730 Extracellular 0.0018 | Cytoplasm, Soluble 0.554 Extracellular 0.0042 | Cytoplasm, Soluble 0.742 Extracellular 0.0878 |
| SRTpred | Non-Secretory Protein −0.362 | Non-Secretory Protein −1.5007 | Non-Secretory Protein −0.9236 | Secretory Protein 0.05923 |
| ApoplastP | Non-apoplastic 0.93 | Non-apoplastic 0.8 | Non-apoplastic 0.82 | Apoplastic 0.81 |
| PeNGaRoo (Gram+) | No 0.417 | Yes 0.575 | Yes 0.626 | Yes 0.582 |
| OutCyte | Intracellular 0.9343 | Intracellular 0.5631 | UPS 0.5345 | Intracellular 0.551 |
| ExoPred | NA | NA | NA | Y |
| EffectorP 3.0 | N 0.885 | N 0.501 | N 0.632 | Y 0.912 |
| SPLpred (high) SPLpred (low) | No No 0.474 | Yes Yes 0.716 | No Yes 0.666 | Yes Yes 0.941 |
| LSPpred (high) LSPpred (low) | Yes Yes 0.714 | Yes Yes 0.839 | Yes Yes 0.789 | Yes Yes 0.690 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lonsdale, A.; Ceballos-Laita, L.; Takahashi, D.; Uemura, M.; Abadía, J.; Davis, M.J.; Bacic, A.; Doblin, M.S. LSPpred Suite: Tools for Leaderless Secretory Protein Prediction in Plants. Plants 2023, 12, 1428. https://doi.org/10.3390/plants12071428
Lonsdale A, Ceballos-Laita L, Takahashi D, Uemura M, Abadía J, Davis MJ, Bacic A, Doblin MS. LSPpred Suite: Tools for Leaderless Secretory Protein Prediction in Plants. Plants. 2023; 12(7):1428. https://doi.org/10.3390/plants12071428
Chicago/Turabian StyleLonsdale, Andrew, Laura Ceballos-Laita, Daisuke Takahashi, Matsuo Uemura, Javier Abadía, Melissa J. Davis, Antony Bacic, and Monika S. Doblin. 2023. "LSPpred Suite: Tools for Leaderless Secretory Protein Prediction in Plants" Plants 12, no. 7: 1428. https://doi.org/10.3390/plants12071428