
Integrated Systems Biology Pipeline to Compare Co-Expression Networks in Plants and Elucidate Differential Regulators

{kind=link}
Abstract
:1. Introduction
2. Results
3. Discussion
4. Materials and Methods
- OS: Windows 11 (5.10.102.1-Microsoft-standard-WSL2), and Fedora 36;
- RAM: 16 GB;
- SDD: 256 GB;
- CPU: Intel i7;
- Conda 4.12.0.
4.1. Choosing Plant Materials and Growing Conditions for High-Throughput Sequencing Analysis
4.2. High-Throughput Sequencing Analysis Data
4.3. Co-Expression Network and WGCNA Modules
4.4. Inference of Gene Regulatory Network
| #!/usr/bin/env python3 import pandas as pd from arboreto.algo import grnboost2, genie3 from arboreto.utils import load_tf_names from distributed import LocalCluster, Client tfdf = pd.read_csv(“Auxiliary_File/Arabidopsis_TF and family.csv”) tf_names = list(set(tfdf[‘Protein ID’].values.tolist())) len(tf_names) ex_matrix = pd.read_csv(“1_Expression_data/Expr_Uncut.csv”, sep=‘,’, index_col=0).T ex_matrix.head() local_cluster = LocalCluster(n_workers=10, threads_per_worker=1, memory_limit=8e9) custom_client = Client(local_cluster) network = grnboost2(expression_data=ex_matrix, tf_names=tf_names, verbose=True, client_or_address=custom_client) network.to_csv(‘3_GRN_data/GSE74488_Uncut_arboreto_regnet.tsv’, sep=‘\t’, index=False) network.head() |
4.5. WGCNA Module Enrichment
- a.
- The following command was used to activate the conda environment:$ conda activate POTFUL
- b.
- The POTFUL (v v1.0.1) package was loaded:
from POTFUL import POTFUL
POT = POTFUL()
- c.
- All of the auxiliary files were loaded using the following command:
POT.Load_Auxiliary_Files(WGCNA_COLOR_MAP=“Auxiliary_File/WGCNA_COLOR_MAP.csv”,
TF_Targets=“Auxiliary_File/masterTF-target.txt”,
TF_Family=“Auxiliary_File/Arabidopsis_TF and family.csv”)
- d.
- The pre-analyzed (WGCNA and GRN files) files for both datasets were loaded, the uncut, and 3hpc, using the following command:
# Uncut
POT.Load_Files(Sample_name=“Uncut”,
NODE_File=“2_WGCNA_data/WGCNA_GSE74488_Uncut/Nodes_Uncut.txt”,
EDGE_File=“2_WGCNA_data/WGCNA_GSE74488_Uncut/Edges_Uncut.txt”,
GRN_File=“3_GRN_data/GSE74488_Uncut_arboreto_regnet.tsv”)
# 3hr decapitated root samples
POT.Load_Files(Sample_name=“3hpc”,
NODE_File=“2_WGCNA_data/WGCNA_GSE74488_3hpc/Nodes_3hpc.txt”,
EDGE_File=“2_WGCNA_data/WGCNA_GSE74488_3hpc/Edges_3hpc.txt”,
GRN_File=“3_GRN_data/GSE74488_3hpc_arboreto_regnet.tsv”)
# Uncut
| Samples = POT.Samples for i in range(len(Samples)): print(i, Samples[i]) 0 Uncut 1 3hpc |
- e.
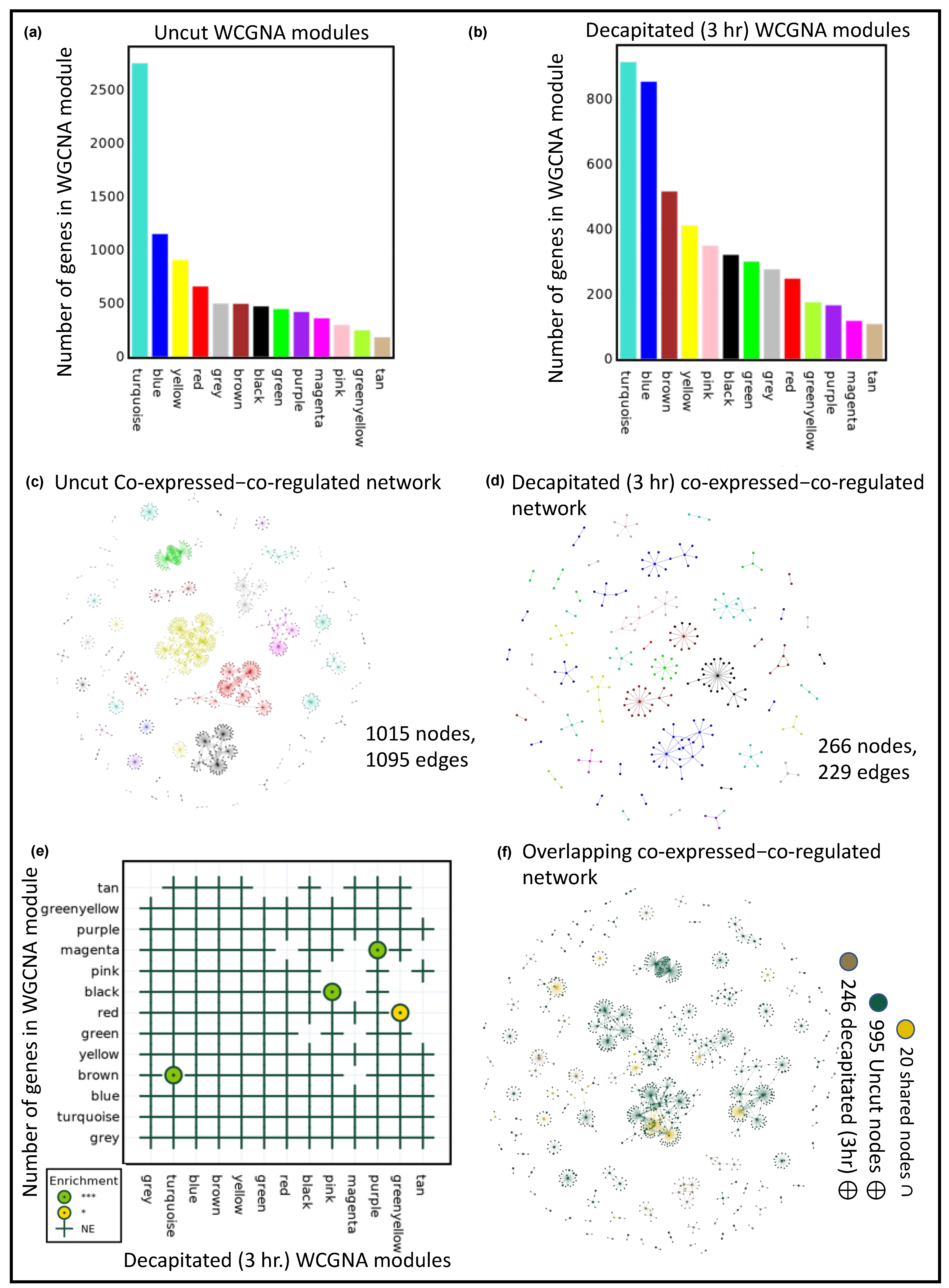
- As part of the enrichment analysis, POTFUL uses Enrichr API (GSEApy); to be able to do so using WIGCNA modules, a GMT (Gene Matrix Transposed file format (*.gmt)) file was created. In the WGCNA module *.gmt file, each row consists of three components, first the name of the WGCNA module (e.g., turquoise, tan, etc.), then the description (e.g., WGCNA3hpc, WGCNAunct, etc.), and finally the list of all of the genes in the module. A *.gmt file was created for both samples for enrichment analysis using the following function for each dataset:
POT.WGCNA_Bucket_GMT()
GMT_base/POTFUL-Uncut.gmt 8921
GMT_base/POTFUL-3hpc.gmt 4756
| for i in range(len(Samples)): print((POT.File[Samples[i]][‘GMT’])) # GMT_base/POTFUL-Uncut.gmt # GMT_base/POTFUL-3hpc.gmt |
- f.
| fig.update_layout(autosize=False, width=350, height=400, xaxis_title=“WGCNA Module”, yaxis_title=“Number of genes”, plot_bgcolor = ‘rgba(0, 0, 0, 0)’, font=dict(family=“Times New Roman”, size=10, color=“black”)) fig.update_xaxes(showline=True, linewidth=2, linecolor=‘black’, mirror=True) fig.update_yaxes(showline=True, linewidth=2, linecolor=‘black’, mirror=True) fig.write_image(“POTFUL_OUT/Uncut.png”, scale=2) fig.write_image(“POTFUL_OUT/Uncut.svg”, scale=2) |
- g.
- Using Fisher’s exact test, the p-value was calculated (hypergeometric test), indicating whether the overlap between the two module gene lists is significant. As the background parameter, the nodes of both co-expression networks that were being compared were used. For assigning significance color codes and significance asterisks, only ‘Adjusted p-value’ is considered by default. An enrichment analysis of modules was performed of one sample concerning another sample using the following command:
POT.WGCNA_Module_Enrichment(Samples[0], Samples[1])
- h.
- Using the following Python command, the enrichment dot plot was generated, and a high-quality image was exported. Every dot in the enrichment dot represents the significance of the enrichment, i.e., green (***), gold (**), and yellow (*). In contrast, the plus (+) symbol represents not significantly enhanced sets.
fig = POT.Plots[“Enrichment_Dotplot”]
fig.update_layout(
autosize=False,
width=490,
height=500,
font=dict(
family=“Arial”,
size=12,
color=“black”))
fig.write_image(POT.OutDir+f”3hpc__UncutEnri_dot.png”, scale=2)
fig.write_image(POT.OutDir+f”3hpc__UncutEnri_dot.svg”)
# Figure 1c
4.6. Co-Expression and GRN Sample Overlap
- a.
- The TF–target pairs that did not belong to the known curated TF–target pairs were filtered out using the following Python command for each sample:
# Uncut
POT.TF_reg(Samples[0], Filter=1)
# 3hpc
POT.TF_reg(Samples[1])
- b.
- Using the following Python command, the remaining GRN-weighted network was matched with the co-expression network to keep only those pairs that are co-expressed and involved in regulation:
# Uncut
POT.merge_reg_coexp(Samples[0])
# 3hpc
POT.merge_reg_coexp(Samples[1])
- c.
- Network centrality analysis was performed on the co-expressed–GRN using the following command (see Appendix C, Problem 4):
# Uncut
POT.network_centrality(Samples[0])
# 3hpc
POT.network_centrality(Samples[1])
- d.
- The GraphML file was generated, and the network visualized using the following command (see Appendix C, Problem 3):
# Uncut
POT.generate_graphml_out(Samples[0])
# 3hpc
POT.generate_graphml_out(Samples[1])
- e.
- f.
- The co-expressed–GRNs of both samples were compared and plotted to check for any overlapping nodes, using the following command:
POT.netowork_overlap(Samples[0], Samples[1])
# There are 20 nodes overlapping between pair of Graphs
(‘AT5G41920’, ‘AT1G58340’, ‘AT1G18330’, ‘AT2G42150’, ‘AT3G03200’, ‘AT3G04030’, ‘AT1G51220’, ‘AT5G62320’, ‘AT2G45660’, ‘AT1G75390’, ‘AT5G42070’, ‘AT4G08940’, ‘AT3G10113’, ‘AT3G01530’, ‘AT1G75820’, ‘AT1G75388’, ‘AT2G18380’, ‘AT4G36900’, ‘AT5G46590’, ‘AT2G45420’)
POT.Plots[‘Overlap_Network_Viz’].show(‘Overlap.html’)
# Figure 1f
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
Appendix A
Appendix B
- The protocol enables the comparison of pairs of co-expression networks in a flexible manner.
- TF–target networks are necessary for the construction of co-expressed–GRNs. POTFUL may not be applicable for non-model plants without a robust list of TFs.
- Different cell types regulate genes differently, and the pattern changes over time. The protocol cannot be applied to samples of different types of cells.
Appendix C
Appendix D
References
- Kumar, N.; Mishra, B.K.; Liu, J.; Mohan, B.; Thingujam, D.; Pajerowska-Mukhtar, K.M.; Mukhtar, M.S. Network Biology Analyses and Dynamic Modeling of Gene Regulatory Networks under Drought Stress Reveal Major Transcriptional Regulators in Arabidopsis. Int. J. Mol. Sci. 2023, 24, 7349. [Google Scholar] [CrossRef] [PubMed]
- Kumar, N.; Mishra, B.; Athar, M.; Mukhtar, S. Inference of Gene Regulatory Network from Single-Cell Transcriptomic Data Using pySCENIC. Methods Mol. Biol. 2021, 2328, 171–182. [Google Scholar] [CrossRef] [PubMed]
- Levine, M.; Davidson, E.H. Gene regulatory networks for development. Proc. Natl. Acad. Sci. USA 2005, 102, 4936–4942. [Google Scholar] [CrossRef] [PubMed]
- Davidson, E.; Levin, M. Gene regulatory networks. Proc. Natl. Acad. Sci. USA 2005, 102, 4935. [Google Scholar] [CrossRef]
- Mishra, B.; Kumar, N.; Mukhtar, M.S. Systems Biology and Machine Learning in Plant-Pathogen Interactions. Mol. Plant Microbe Interact. 2019, 32, 45–55. [Google Scholar] [CrossRef]
- von Dassow, G.; Meir, E.; Munro, E.M.; Odell, G.M. The segment polarity network is a robust developmental module. Nature 2000, 406, 188–192. [Google Scholar] [CrossRef]
- Tyson, J.J.; Chen, K.C.; Novak, B. Sniffers, buzzers, toggles and blinkers: Dynamics of regulatory and signaling pathways in the cell. Curr. Opin. Cell Biol. 2003, 15, 221–231. [Google Scholar] [CrossRef]
- Yin, W.; Mendoza, L.; Monzon-Sandoval, J.; Urrutia, A.O.; Gutierrez, H. Emergence of co-expression in gene regulatory networks. PLoS ONE 2021, 16, e0247671. [Google Scholar] [CrossRef]
- Inoue, K.; Araki, T.; Endo, M. Correction to: Circadian clock during plant development. J. Plant Res. 2018, 131, 571. [Google Scholar] [CrossRef]
- Mishra, B.; Sun, Y.; Howton, T.C.; Kumar, N.; Mukhtar, M.S. Dynamic modeling of transcriptional gene regulatory network uncovers distinct pathways during the onset of Arabidopsis leaf senescence. NPJ Syst. Biol. Appl. 2018, 4, 35. [Google Scholar] [CrossRef]
- Millar, A.J. The Intracellular Dynamics of Circadian Clocks Reach for the Light of Ecology and Evolution. Annu. Rev. Plant Biol. 2016, 67, 595–618. [Google Scholar] [CrossRef] [PubMed]
- Nohales, M.A.; Kay, S.A. Molecular mechanisms at the core of the plant circadian oscillator. Nat. Struct. Mol. Biol. 2016, 23, 1061–1069. [Google Scholar] [CrossRef] [PubMed]
- Huang, R.C. The discoveries of molecular mechanisms for the circadian rhythm: The 2017 Nobel Prize in Physiology or Medicine. Biomed. J. 2018, 41, 5–8. [Google Scholar] [CrossRef]
- Ritonga, F.N.; Chen, S. Physiological and Molecular Mechanism Involved in Cold Stress Tolerance in Plants. Plants 2020, 9, 560. [Google Scholar] [CrossRef] [PubMed]
- Kidokoro, S.; Shinozaki, K.; Yamaguchi-Shinozaki, K. Transcriptional regulatory network of plant cold-stress responses. Trends Plant Sci. 2022, 27, 922–935. [Google Scholar] [CrossRef]
- Hoang, X.L.T.; Nhi, D.N.H.; Thu, N.B.A.; Thao, N.P.; Tran, L.P. Transcription Factors and Their Roles in Signal Transduction in Plants under Abiotic Stresses. Curr. Genom. 2017, 18, 483–497. [Google Scholar] [CrossRef] [PubMed]
- Kumar, N.; Mishra, B.; Mukhtar, M.S. A pipeline of integrating transcriptome and interactome to elucidate central nodes in host-pathogens interactions. STAR Protoc. 2022, 3, 101608. [Google Scholar] [CrossRef]
- Kumar, N.; Mukhtar, M.S. Ranking Plant Network Nodes Based on Their Centrality Measures. Entropy 2023, 25, 676. [Google Scholar] [CrossRef]
- Luscombe, N.M.; Babu, M.M.; Yu, H.; Snyder, M.; Teichmann, S.A.; Gerstein, M. Genomic analysis of regulatory network dynamics reveals large topological changes. Nature 2004, 431, 308–312. [Google Scholar] [CrossRef]
- Kumar, N.; Mukhtar, S. Building Protein-Protein Interaction Graph Database Using Neo4j. Methods Mol. Biol. 2023, 2690, 469–479. [Google Scholar]
- Mishra, B.; Kumar, N.; Mukhtar, M.S. Network biology to uncover functional and structural properties of the plant immune system. Curr. Opin. Plant Biol. 2021, 62, 102057. [Google Scholar] [CrossRef] [PubMed]
- Serin, E.A.; Nijveen, H.; Hilhorst, H.W.; Ligterink, W. Learning from Co-expression Networks: Possibilities and Challenges. Front. Plant Sci. 2016, 7, 444. [Google Scholar] [CrossRef] [PubMed]
- Cortijo, S.; Bhattarai, M.; Locke, J.C.W.; Ahnert, S.E. Co-expression Networks From Gene Expression Variability Between Genetically Identical Seedlings Can Reveal Novel Regulatory Relationships. Front. Plant Sci. 2020, 11, 599464. [Google Scholar] [CrossRef] [PubMed]
- Stuart, J.M.; Segal, E.; Koller, D.; Kim, S.K. A gene-coexpression network for global discovery of conserved genetic modules. Science 2003, 302, 249–255. [Google Scholar] [CrossRef] [PubMed]
- Rao, X.; Dixon, R.A. Co-expression networks for plant biology: Why and how. Acta Biochim. Biophys. Sin. 2019, 51, 981–988. [Google Scholar] [CrossRef]
- Liseron-Monfils, C.; Ware, D. Revealing gene regulation and associations through biological networks. Curr. Plant Biol. 2015, 3, 30–39. [Google Scholar] [CrossRef]
- Li, Y.; Pearl, S.A.; Jackson, S.A. Gene Networks in Plant Biology: Approaches in Reconstruction and Analysis. Trends Plant Sci. 2015, 20, 664–675. [Google Scholar] [CrossRef]
- Kumar, N.; Mishra, B.; Mehmood, A.; Mohammad, A.; Mukhtar, M.S. Integrative Network Biology Framework Elucidates Molecular Mechanisms of SARS-CoV-2 Pathogenesis. iScience 2020, 23, 101526. [Google Scholar] [CrossRef]
- Proost, S.; Mutwil, M. PlaNet: Comparative Co-Expression Network Analyses for Plants. Methods Mol. Biol. 2017, 1533, 213–227. [Google Scholar] [CrossRef]
- Langfelder, P.; Horvath, S. WGCNA: An R package for weighted correlation network analysis. BMC Bioinform. 2008, 9, 559. [Google Scholar] [CrossRef]
- Dai, R.; Xia, Y.; Liu, C.; Chen, C. csuWGCNA: A combination of signed and unsigned WGCNA to capture negative correlations. bioRxiv 2019, 288225. [Google Scholar] [CrossRef]
- Efroni, I.; Mello, A.; Nawy, T.; Ip, P.L.; Rahni, R.; DelRose, N.; Powers, A.; Satija, R.; Birnbaum, K.D. Root Regeneration Triggers an Embryo-like Sequence Guided by Hormonal Interactions. Cell 2016, 165, 1721–1733. [Google Scholar] [CrossRef] [PubMed]
- Gordon, D.E.; Jang, G.M.; Bouhaddou, M.; Xu, J.; Obernier, K.; White, K.M.; O’Meara, M.J.; Rezelj, V.V.; Guo, J.Z.; Swaney, D.L.; et al. A SARS-CoV-2 protein interaction map reveals targets for drug repurposing. Nature 2020, 583, 459–468. [Google Scholar] [CrossRef] [PubMed]
- Mishra, B.; Kumar, N.; Shahid Mukhtar, M. A rice protein interaction network reveals high centrality nodes and candidate pathogen effector targets. Comput. Struct. Biotechnol. J. 2022, 20, 2001–2012. [Google Scholar] [CrossRef] [PubMed]
- Iyer-Pascuzzi, A.S.; Jackson, T.; Cui, H.; Petricka, J.J.; Busch, W.; Tsukagoshi, H.; Benfey, P.N. Cell identity regulators link development and stress responses in the Arabidopsis root. Dev. Cell 2011, 21, 770–782. [Google Scholar] [CrossRef] [PubMed]
- Cejudo, F.J.; Sandalio, L.M.; Van Breusegem, F. Understanding plant responses to stress conditions: Redox-based strategies. J. Exp. Bot. 2021, 72, 5785–5788. [Google Scholar] [CrossRef] [PubMed]
- Nia, A.M.; Chen, T.; Barnette, B.L.; Khanipov, K.; Ullrich, R.L.; Bhavnani, S.K.; Emmett, M.R. Efficient identification of multiple pathways: RNA-Seq analysis of livers from 56Fe ion irradiated mice. BMC Bioinform. 2020, 21, 118. [Google Scholar] [CrossRef]
- Huynh-Thu, V.A.; Irrthum, A.; Wehenkel, L.; Geurts, P. Inferring regulatory networks from expression data using tree-based methods. PLoS ONE 2010, 5, e12776. [Google Scholar] [CrossRef]
- Rocklin, M. Dask: Parallel computation with blocked algorithms and task scheduling. In Proceedings of the 14th Python in Science Conference, Austin, TX, USA, 6–12 July 2015; p. 136. [Google Scholar]
- Friedman, J.H. Stochastic gradient boosting. Comput. Stat. Data Anal. 2002, 38, 367–378. [Google Scholar] [CrossRef]
- Aibar, S.; Gonzalez-Blas, C.B.; Moerman, T.; Huynh-Thu, V.A.; Imrichova, H.; Hulselmans, G.; Rambow, F.; Marine, J.C.; Geurts, P.; Aerts, J.; et al. SCENIC: Single-cell regulatory network inference and clustering. Nat. Methods 2017, 14, 1083–1086. [Google Scholar] [CrossRef]
- Marbach, D.; Costello, J.C.; Kuffner, R.; Vega, N.M.; Prill, R.J.; Camacho, D.M.; Allison, K.R.; Consortium, D.; Kellis, M.; Collins, J.J.; et al. Wisdom of crowds for robust gene network inference. Nat. Methods 2012, 9, 796–804. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Zhou, B.; Pache, L.; Chang, M.; Khodabakhshi, A.H.; Tanaseichuk, O.; Benner, C.; Chanda, S.K. Metascape provides a biologist-oriented resource for the analysis of systems-level datasets. Nat. Commun. 2019, 10, 1523. [Google Scholar] [CrossRef]
- van Dam, S.; Vosa, U.; van der Graaf, A.; Franke, L.; de Magalhaes, J.P. Gene co-expression analysis for functional classification and gene-disease predictions. Brief. Bioinform. 2018, 19, 575–592. [Google Scholar] [CrossRef] [PubMed]
- Amar, D.; Safer, H.; Shamir, R. Dissection of regulatory networks that are altered in disease via differential co-expression. PLoS Comput. Biol. 2013, 9, e1002955. [Google Scholar] [CrossRef]
- Bhar, A.; Haubrock, M.; Mukhopadhyay, A.; Maulik, U.; Bandyopadhyay, S.; Wingender, E. Coexpression and coregulation analysis of time-series gene expression data in estrogen-induced breast cancer cell. Algorithms Mol. Biol. 2013, 8, 9. [Google Scholar] [CrossRef] [PubMed]
- Palaniswamy, S.K.; James, S.; Sun, H.; Lamb, R.S.; Davuluri, R.V.; Grotewold, E. AGRIS and AtRegNet. a platform to link cis-regulatory elements and transcription factors into regulatory networks. Plant Physiol. 2006, 140, 818–829. [Google Scholar] [CrossRef] [PubMed]
- O’Malley, R.C.; Huang, S.C.; Song, L.; Lewsey, M.G.; Bartlett, A.; Nery, J.R.; Galli, M.; Gallavotti, A.; Ecker, J.R. Cistrome and Epicistrome Features Shape the Regulatory DNA Landscape. Cell 2016, 166, 1598. [Google Scholar] [CrossRef]
- Yu, C.P.; Lin, J.J.; Li, W.H. Positional distribution of transcription factor binding sites in Arabidopsis thaliana. Sci. Rep. 2016, 6, 25164. [Google Scholar] [CrossRef]
- Kulkarni, S.R.; Vaneechoutte, D.; Van de Velde, J.; Vandepoele, K. TF2Network: Predicting transcription factor regulators and gene regulatory networks in Arabidopsis using publicly available binding site information. Nucleic Acids Res. 2018, 46, e31. [Google Scholar] [CrossRef]
- Jin, J.; He, K.; Tang, X.; Li, Z.; Lv, L.; Zhao, Y.; Luo, J.; Gao, G. An Arabidopsis Transcriptional Regulatory Map Reveals Distinct Functional and Evolutionary Features of Novel Transcription Factors. Mol. Biol. Evol. 2015, 32, 1767–1773. [Google Scholar] [CrossRef]
- Kilian, J.; Whitehead, D.; Horak, J.; Wanke, D.; Weinl, S.; Batistic, O.; D’Angelo, C.; Bornberg-Bauer, E.; Kudla, J.; Harter, K. The AtGenExpress global stress expression data set: Protocols, evaluation and model data analysis of UV-B light, drought and cold stress responses. Plant J. 2007, 50, 347–363. [Google Scholar] [CrossRef] [PubMed]
- WGCNAfaq. WGCNA Package: Frequently Asked Questions. 2017. Available online: https://horvath.genetics.ucla.edu/html/CoexpressionNetwork/Rpackages/WGCNA/faq.html (accessed on 13 February 2023).
- Hayes, S.M.S.; Sachs, J.R.; Cho, C.R. From complex data to biological insight: ‘DEKER’ feature selection and network inference. J. Pharmacokinet. Pharmacodyn. 2022, 49, 81–99. [Google Scholar] [CrossRef] [PubMed]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kumar, N.; Mukhtar, M.S. Integrated Systems Biology Pipeline to Compare Co-Expression Networks in Plants and Elucidate Differential Regulators. Plants 2023, 12, 3618. https://doi.org/10.3390/plants12203618
Kumar N, Mukhtar MS. Integrated Systems Biology Pipeline to Compare Co-Expression Networks in Plants and Elucidate Differential Regulators. Plants. 2023; 12(20):3618. https://doi.org/10.3390/plants12203618
Chicago/Turabian StyleKumar, Nilesh, and M. Shahid Mukhtar. 2023. "Integrated Systems Biology Pipeline to Compare Co-Expression Networks in Plants and Elucidate Differential Regulators" Plants 12, no. 20: 3618. https://doi.org/10.3390/plants12203618