
Gene Coexpression Analysis Identifies Genes Associated with Chlorophyll Content and Relative Water Content in Pearl Millet
 ,
, 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
2.1. Transcript Quantification
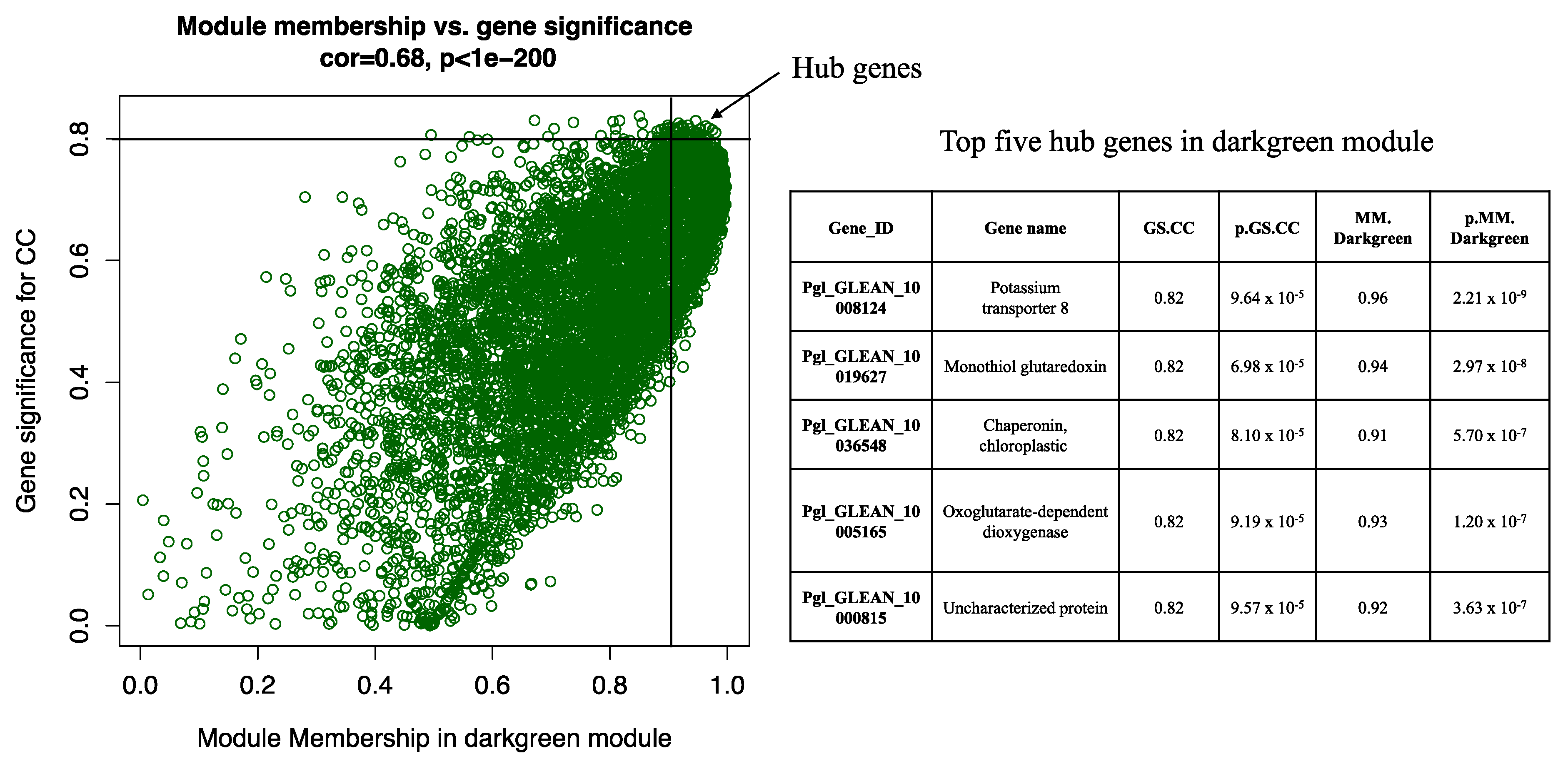
2.2. Weighted Gene Coexpression Network Analysis (WGCNA)
2.3. Gene Clustering Analysis
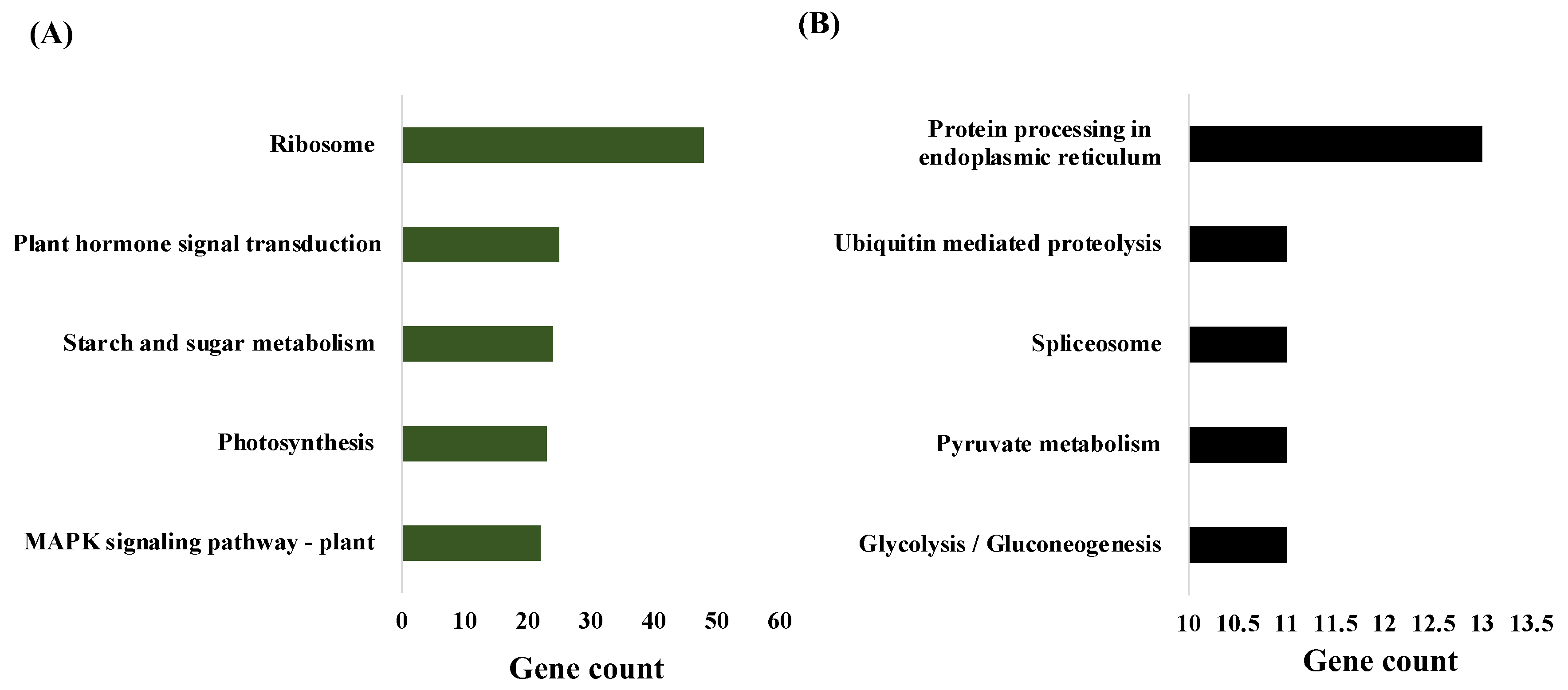
2.4. Metabolic Pathway Analysis
3. Discussion
4. Materials and Methods
4.1. RNA Sequencing Data Preprocessing
4.2. Transcript Quantification
4.3. Weighted Gene Coexpression Network Analysis (WGCNA)
4.4. Gene Clustering Analysis
4.5. Metabolic Pathway Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Xu, C.; Kohler, T.A.; Lenton, T.M.; Svenning, J.C.; Scheffer, M. Future of the Human Climate Niche. Available online: https://www.pnas.org (accessed on 15 October 2022).
- Satyavathi, C.T.; Ambawat, S.; Khandelwal, V.; Srivastava, R.K. Pearl Millet: A Climate-Resilient Nutricereal for Mitigating Hidden Hunger and Provide Nutritional Security. Front. Plant Sci. 2021, 12, 659938. [Google Scholar] [CrossRef]
- Gupta, S.; Rai, K.; Singh, P.; Ameta, V.; Jayalekha, A.; Mahala, R.; Pareek, S.; Swami, M.; Verma, Y. Seed set variability under high temperatures during flowering period in pearl millet (Pennisetum glaucum L. (R.) Br.). Field Crops Res. 2015, 171, 41–53. [Google Scholar] [CrossRef] [Green Version]
- Shrestha, N.; Hu, H.; Shrestha, K.; Doust, A.N. Pearl millet response to drought: A review. Front. Plant Sci. 2023, 14, 1059574. [Google Scholar] [CrossRef]
- Bani Hani, N.; Aukour, F.J.; Al-Qinna, M.I. Investigating the Pearl Millet (Pennisetum glaucum) as a Climate-Smart Drought-Tolerant Crop under Jordanian Arid Environments. Sustainability 2022, 14, 12249. [Google Scholar] [CrossRef]
- Srivastava, R.K.; Yadav, O.P.; Kaliamoorthy, S.; Gupta, S.K.; Serba, D.D.; Choudhary, S.; Govindaraj, M.; Kholová, J.; Murugesan, T.; Satyavathi, C.T.; et al. Breeding Drought-Tolerant Pearl Millet Using Conventional and Genomic Approaches: Achievements and Prospects. Front. Plant Sci. 2022, 13, 781524. [Google Scholar] [CrossRef]
- Varshney, R.K.; Shi, C.; Thudi, M.; Mariac, C.; Wallace, J.; Qi, P.; Zhang, H.; Zhao, Y.; Wang, X.; Rathore, A.; et al. Pearl millet genome sequence provides a resource to improve agronomic traits in arid environments. Nat. Biotechnol. 2017, 35, 969–976. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fritsche-Neto, R.; Borém, A. Plant Physiology for Abiotic Stress Tolerance; Springer: Berlin/Heidelberg, Germany, 2012. [Google Scholar]
- Shinde, H.; Tanaka, K.; Dudhate, A.; Tsugama, D.; Mine, Y.; Kamiya, T.; Gupta, S.K.; Liu, S.; Takano, T. Comparative de novo transcriptomic profiling of the salinity stress responsiveness in contrasting pearl millet lines. Environ. Exp. Bot. 2018, 155, 619–627. [Google Scholar] [CrossRef]
- Dudhate, A.; Shinde, H.; Tsugama, D.; Liu, S.; Takano, T. Transcriptomic analysis reveals the differentially expressed genes and pathways involved in drought tolerance in pearl millet [Pennisetum glaucum (L.) R. Br]. PLoS ONE 2018, 13, e0195908. [Google Scholar] [CrossRef] [Green Version]
- Shinde, H.; Dudhate, A.; Kadam, U.S.; Hong, J.C. RNA methylation in plants: An overview. Front. Plant Sci. 2023, 14, 1132959. [Google Scholar] [CrossRef]
- Dudhate, A.; Shinde, H.; Yu, P.; Tsugama, D.; Gupta, S.K.; Liu, S.; Takano, T. Comprehensive analysis of NAC transcription factor family uncovers drought and salinity stress response in pearl millet (Pennisetum glaucum). BMC Genom. 2021, 22, 70. [Google Scholar] [CrossRef] [PubMed]
- Yu, P.; Shinde, H.; Dudhate, A.; Tsugama, D.; Gupta, S.K.; Liu, S.; Takano, T. Genome-wide investigation of SQUAMOSA promoter binding protein-like transcription factor family in pearl millet (Pennisetum glaucum (L) R. Br.). Plant Gene 2021, 27, 100313. [Google Scholar] [CrossRef]
- Reddy, P.S.; Dhaware, M.G.; Sivasakthi, K.; Divya, K.; Nagaraju, M.; Cindhuri, K.S.; Kishor, P.B.K.; Bhatnagar-Mathur, P.; Vadez, V.; Sharma, K.K. Pearl Millet Aquaporin Gene PgPIP2;6 Improves Abiotic Stress Tolerance in Transgenic Tobacco. Front. Plant Sci. 2022, 13, 820996. [Google Scholar] [CrossRef]
- Shinde, H.; Dudhate, A.; Tsugama, D.; Gupta, S.K.; Liu, S.; Takano, T. Pearl millet stress-responsive NAC transcription factor PgNAC21 enhances salinity stress tolerance in Arabidopsis. Plant Physiol. Biochem. 2019, 135, 546–553. [Google Scholar] [CrossRef]
- Shinde, H.; Dudhate, A.; Anand, L.; Tsugama, D.; Gupta, S.K.; Liu, S.; Takano, T. Small RNA sequencing reveals the role of pearl millet miRNAs and their targets in salinity stress responses. S. Afr. J. Bot. 2020, 132, 395–402. [Google Scholar] [CrossRef]
- Langfelder, P.; Horvath, S. WGCNA: An R package for weighted correlation network analysis. BMC Bioinform. 2008, 9, 559. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jin, Z.; Liu, S.; Zhu, P.; Tang, M.; Wang, Y.; Tian, Y.; Li, D.; Zhu, X.; Yan, D.; Zhu, Z. Cross-species gene expression analysis reveals gene modules implicated in human osteosarcoma. Front. Genet. 2019, 10, 697. [Google Scholar] [CrossRef] [Green Version]
- Zhu, M.; Xie, H.; Wei, X.; Dossa, K.; Yu, Y.; Hui, S.; Tang, G.; Zeng, X.; Yu, Y.; Hu, P.; et al. WGCNA analysis of salt-responsive core transcriptome identifies novel hub genes in rice. Genes 2019, 10, 719. [Google Scholar] [CrossRef] [Green Version]
- Abu-Jamous, B.; Kelly, S. Clust: Automatic extraction of optimal co-expressed gene clusters from gene expression data. Genome Biol. 2018, 19, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Satyavathi, C.T.; Tomar, R.S.; Ambawat, S.; Kheni, J.; Padhiyar, S.M.; Desai, H.; Bhatt, S.B.; Shitap, M.S.; Meena, R.C.; Singhal, T.; et al. Stage specific comparative transcriptomic analysis to reveal gene networks regulating iron and zinc content in pearl millet [Pennisetum glaucum (L.) R. Br.]. Sci. Rep. 2022, 12, 276. [Google Scholar] [CrossRef]
- Sun, M.; Huang, D.; Zhang, A.; Khan, I.; Yan, H.; Wang, X.; Zhang, X.; Zhang, J.; Huang, L. Transcriptome analysis of heat stress and drought stress in pearl millet based on Pacbio full-length transcriptome sequencing. BMC Plant Biol. 2020, 20, 323. [Google Scholar] [CrossRef] [PubMed]
- Jaiswal, S.; Antala, T.J.; Mandavia, M.K.; Chopra, M.; Jasrotia, R.S.; Tomar, R.S.; Kheni, J.; Angadi, U.B.; Iquebal, M.A.; Golakia, B.A.; et al. Transcriptomic signature of drought response in pearl millet (Pennisetum glaucum (L.) and development of web-genomic resources. Sci. Rep. 2018, 8, 1–16. [Google Scholar]
- Kanfany, G.; Serba, D.D.; Rhodes, D.; Amand, P.S.; Bernardo, A.; Gangashetty, P.; Kane, N.A.; Bai, G. Genomic diversity in pearl millet inbred lines derived from landraces and improved varieties. BMC Genom. 2020, 21, 469. [Google Scholar] [CrossRef]
- Srivastava, R.K.; Singh, R.B.; Pujarula, V.L.; Bollam, S.; Pusuluri, M.; Chellapilla, T.S.; Yadav, R.S.; Gupta, R. Genome-Wide Association Studies and Genomic Selection in Pearl Millet: Advances and Prospects. Front. Genet. 2020, 10, 1389. [Google Scholar] [CrossRef] [Green Version]
- Singh, M.; Nara, U. Genetic insights in pearl millet breeding in the genomic era: Challenges and prospects. Plant Biotechnol. Rep. 2022, 17, 15–37. [Google Scholar] [CrossRef]
- Ramakrishnan, J. Ribosome Structure and the Mechanism of Translation. Cell 2002, 108, 557–572. [Google Scholar] [CrossRef] [Green Version]
- Vind, A.C.; Genzor, A.V.; Bekker-Jensen, S. Ribosomal stress-surveillance: Three pathways is a magic number. Nucleic Acids Res. 2020, 48, 10648–10661. [Google Scholar] [CrossRef]
- Wang, J.; Wang, X.; Ma, C.; Li, P. A Review on the Mechanism of Ribosome Stress Response in Plants. Chin. Bull. Bot. 2022, 57, 80–89. [Google Scholar]
- Dias-Fields, L.; Adamala, K.P. Engineering Ribosomes to Alleviate Abiotic Stress in Plants: A Perspective. Plants 2022, 11, 2097. [Google Scholar] [CrossRef]
- Verma, V.; Ravindran, P.; Kumar, P.P. Plant hormone-mediated regulation of stress responses. BMC Plant Biol. 2016, 16, 86. [Google Scholar] [CrossRef] [Green Version]
- Couchoud, M.; Der, C.; Girodet, S.; Vernoud, V.; Prudent, M.; Leborgne-Castel, N. Drought stress stimulates endocytosis and modifies membrane lipid order of rhizodermal cells of Medicago truncatula in a genotype-dependent manner. BMC Plant Biol. 2019, 19, 221. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hamamoto, S.; Horie, T.; Hauser, F.; Deinlein, U.; Schroeder, J.I.; Uozumi, N. HKT transporters mediate salt stress resistance in plants: From structure and function to the field. Curr. Opin. Biotechnol. 2015, 32, 113–120. [Google Scholar] [CrossRef] [Green Version]
- Assaha, D.V.M.; Ueda, A.; Saneoka, H.; Al-Yahyai, R.; Yaish, M.W. The role of Na+ and K+ transporters in salt stress adaptation in glycophytes. Front. Physiol. 2017, 8, 509. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Patro, R.; Duggal, G.; Love, M.I.; Irizarry, R.A.; Kingsford, C. Salmon provides fast and bias-aware quantification of transcript expression. Nat. Methods 2017, 14, 417–419. [Google Scholar] [CrossRef] [Green Version]
- Moriya, Y.; Itoh, M.; Okuda, S.; Yoshizawa, A.C.; Kanehisa, M. KAAS: An automatic genome annotation and pathway reconstruction server. Nucleic Acids Res. 2007, 35 (Suppl. S2), W182–W185. [Google Scholar] [CrossRef] [Green Version]





Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shinde, H.; Dudhate, A.; Sathe, A.; Paserkar, N.; Wagh, S.G.; Kadam, U.S. Gene Coexpression Analysis Identifies Genes Associated with Chlorophyll Content and Relative Water Content in Pearl Millet. Plants 2023, 12, 1412. https://doi.org/10.3390/plants12061412
Shinde H, Dudhate A, Sathe A, Paserkar N, Wagh SG, Kadam US. Gene Coexpression Analysis Identifies Genes Associated with Chlorophyll Content and Relative Water Content in Pearl Millet. Plants. 2023; 12(6):1412. https://doi.org/10.3390/plants12061412
Chicago/Turabian StyleShinde, Harshraj, Ambika Dudhate, Atul Sathe, Neha Paserkar, Sopan Ganpatrao Wagh, and Ulhas Sopanrao Kadam. 2023. "Gene Coexpression Analysis Identifies Genes Associated with Chlorophyll Content and Relative Water Content in Pearl Millet" Plants 12, no. 6: 1412. https://doi.org/10.3390/plants12061412