
Investigating Variability in Viral Presence and Abundance across Soybean Seed Development Stages Using Transcriptome Analysis

Abstract
:1. Introduction
2. Results
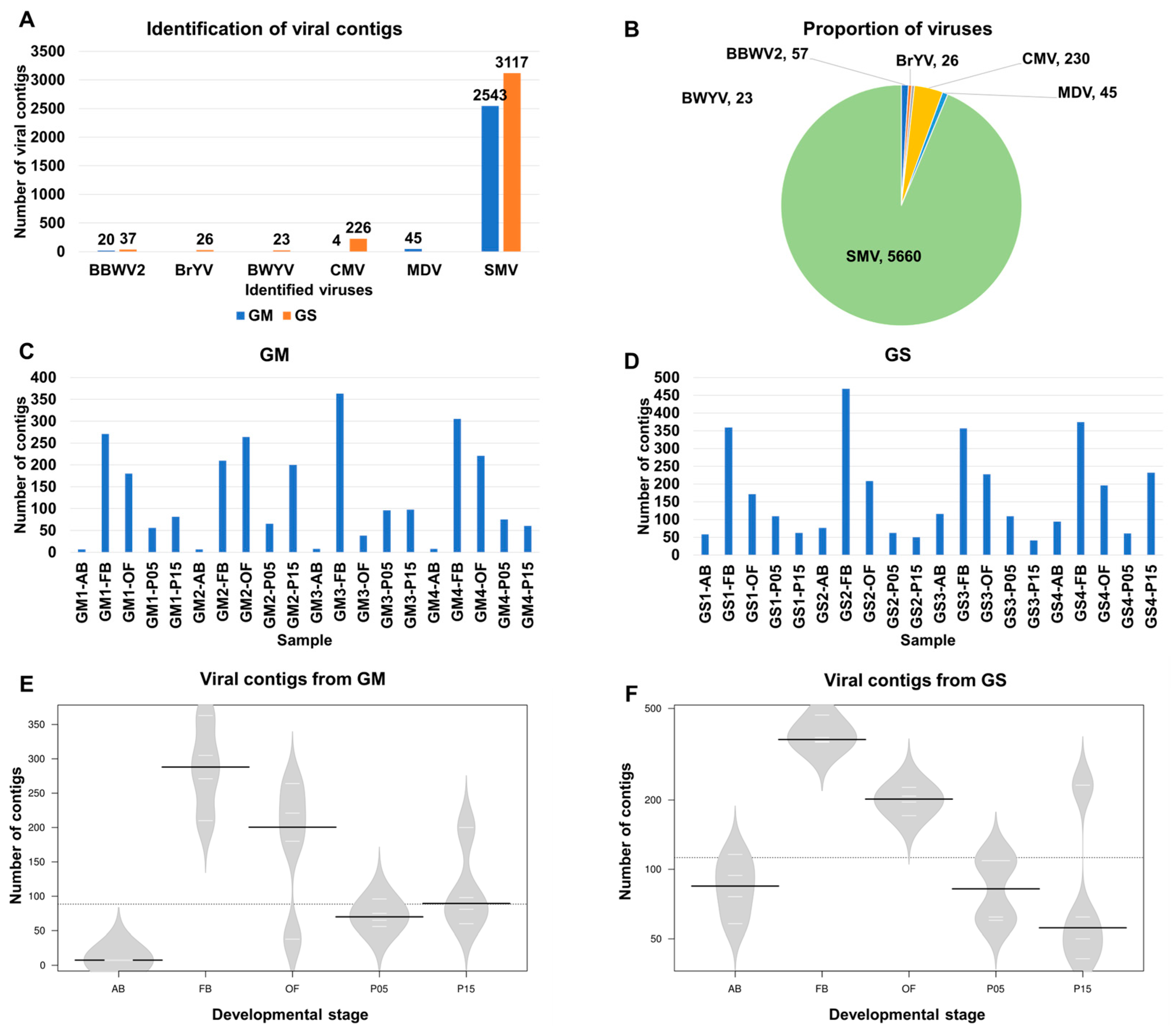
2.1. Identification of Viruses from 40 Soybean Transcriptome Datasets
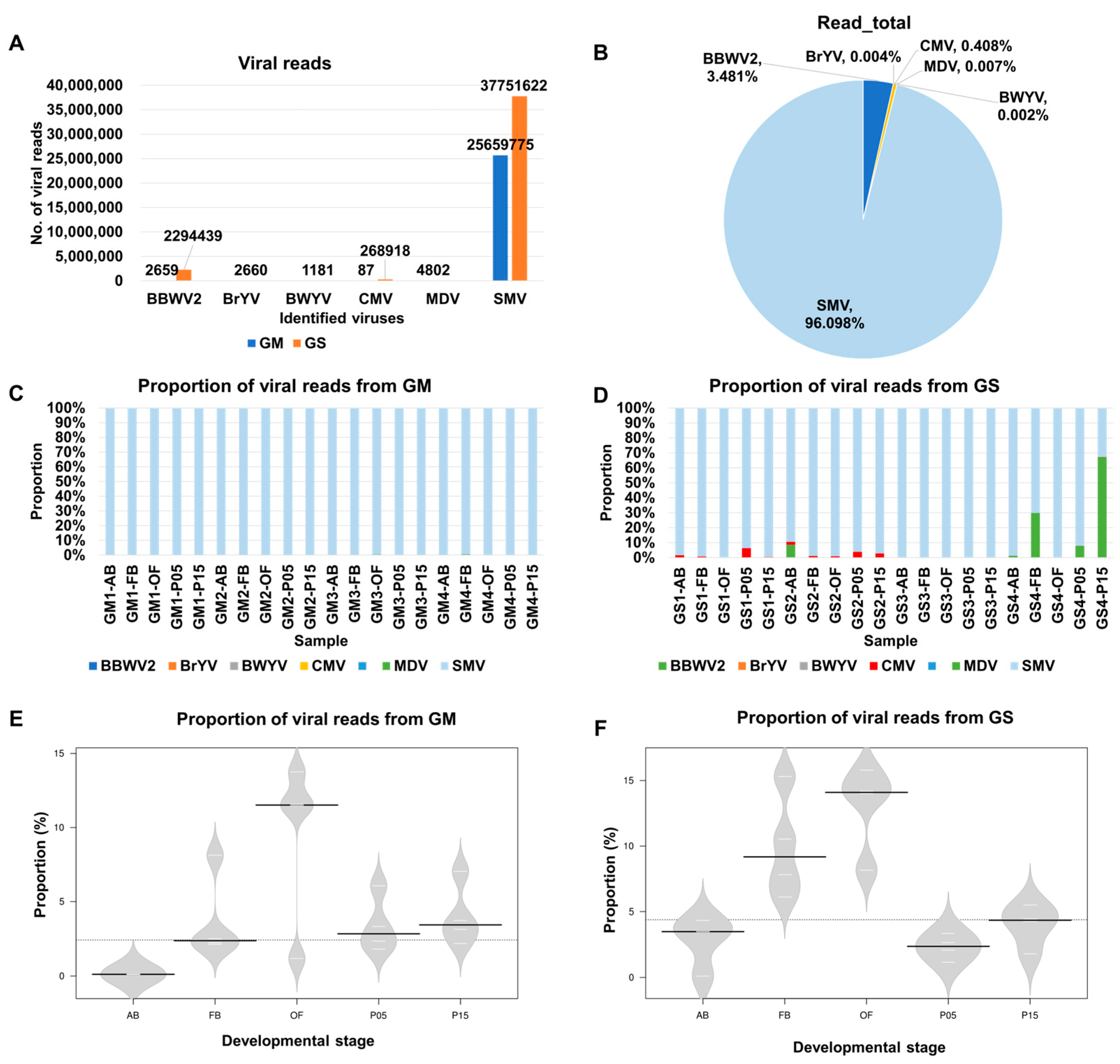
2.2. Analysis of Viral Abundance Using Viral Reads
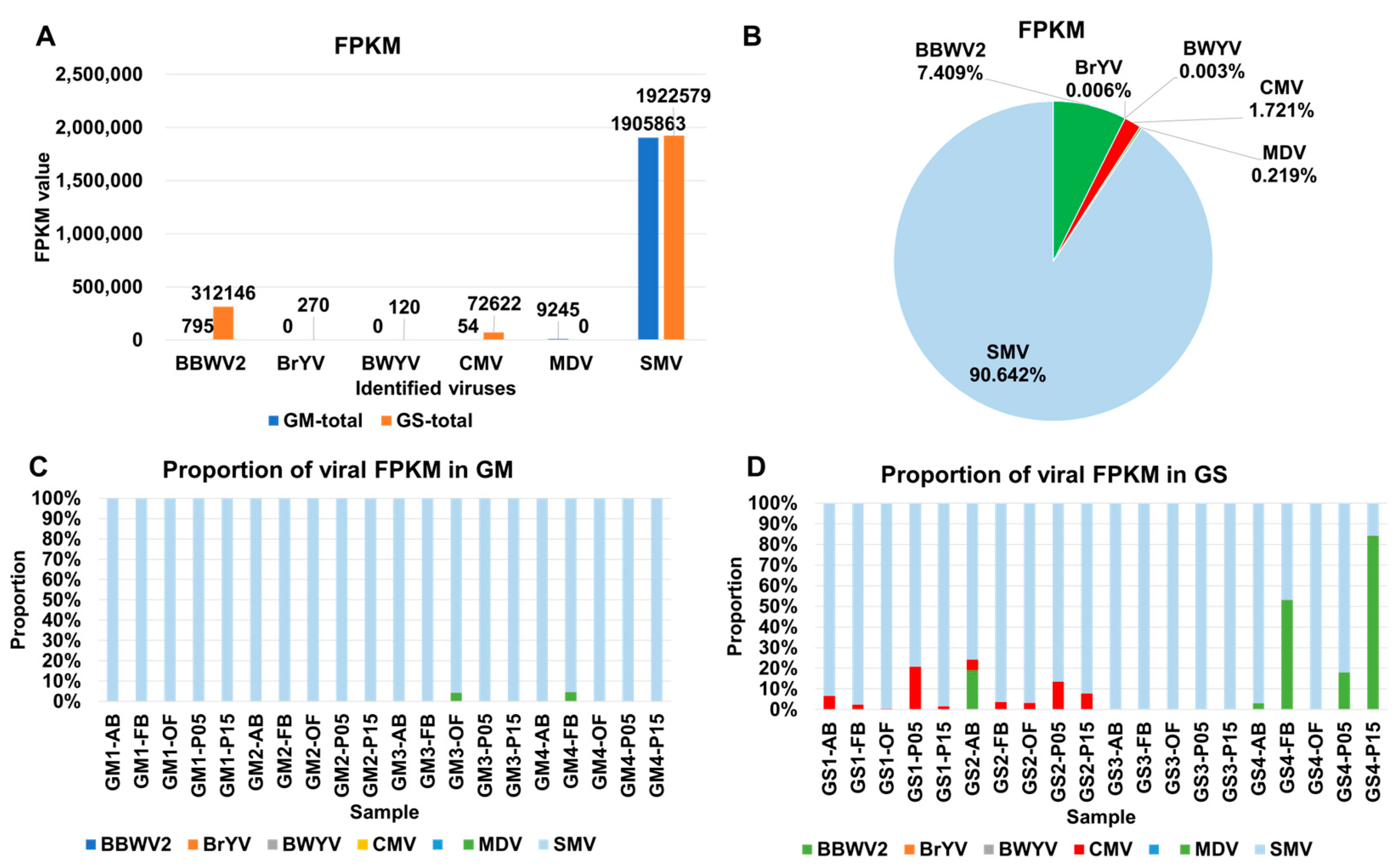
2.3. Analysis of Viral Abundance Using Fragments per Kilobase of Transcript per Million (FPKM) Values
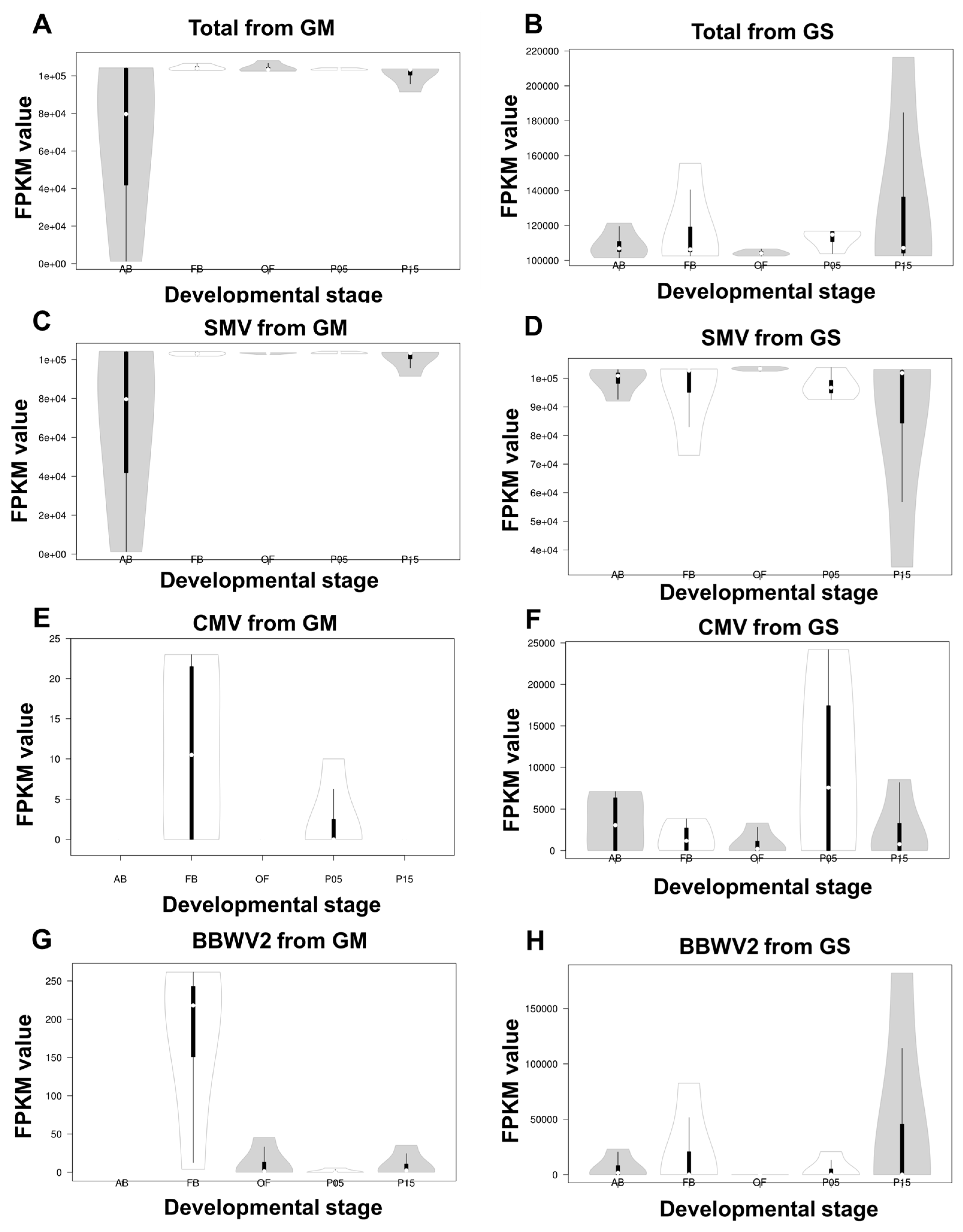
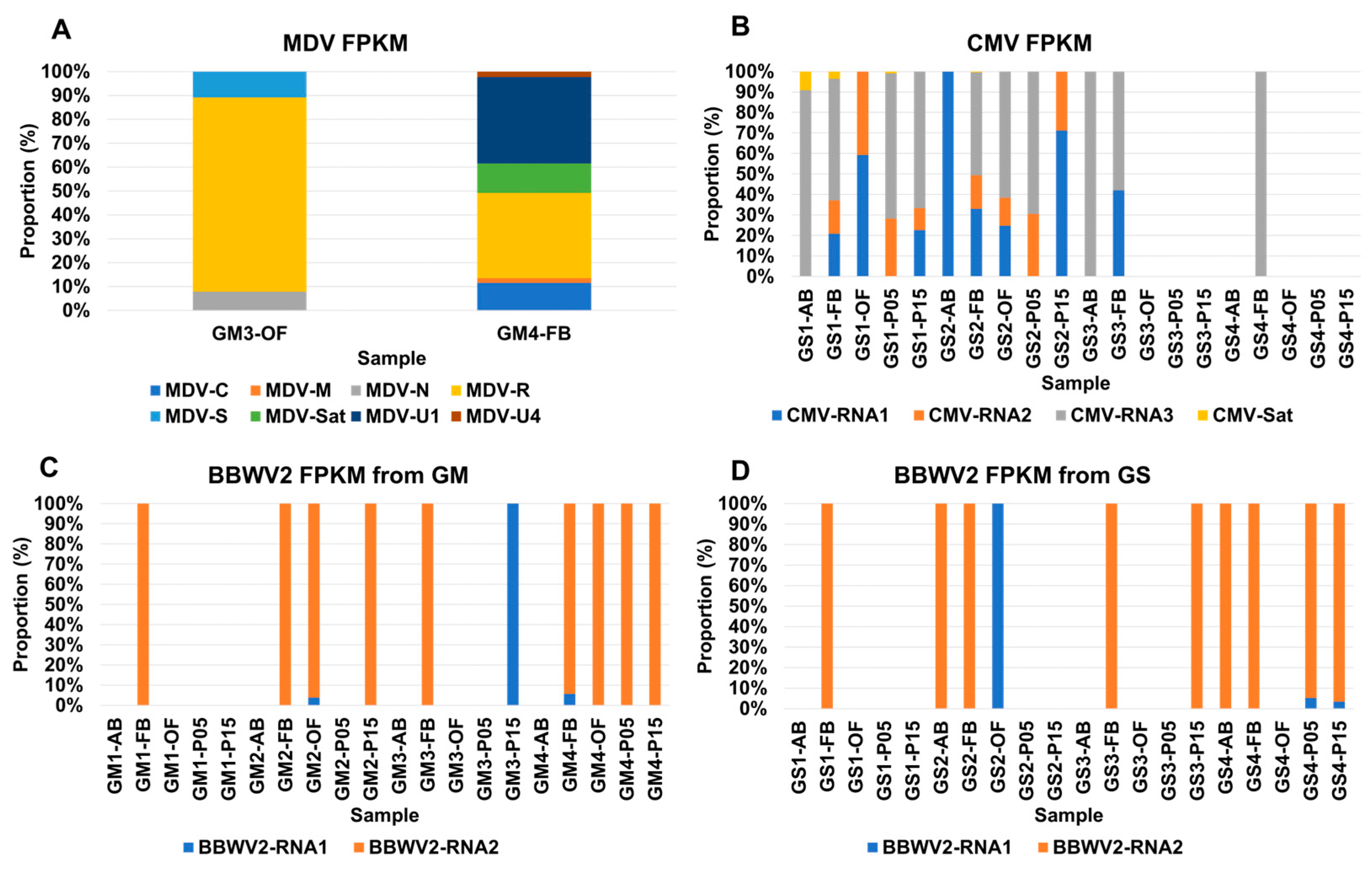
2.4. Comparison of Viral Abundance Based on FPKM Values
2.5. Viral Abundance of Viruses Composed of Multiple Segments
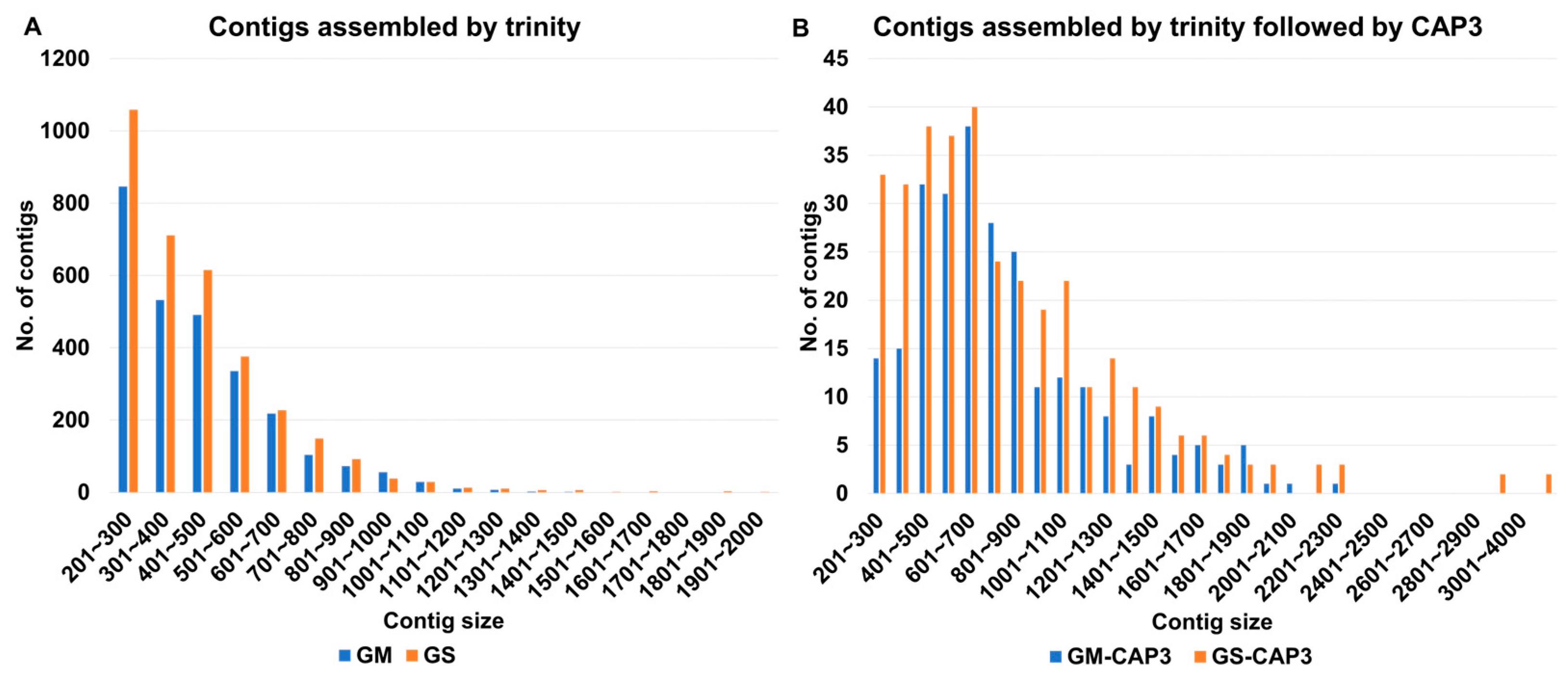
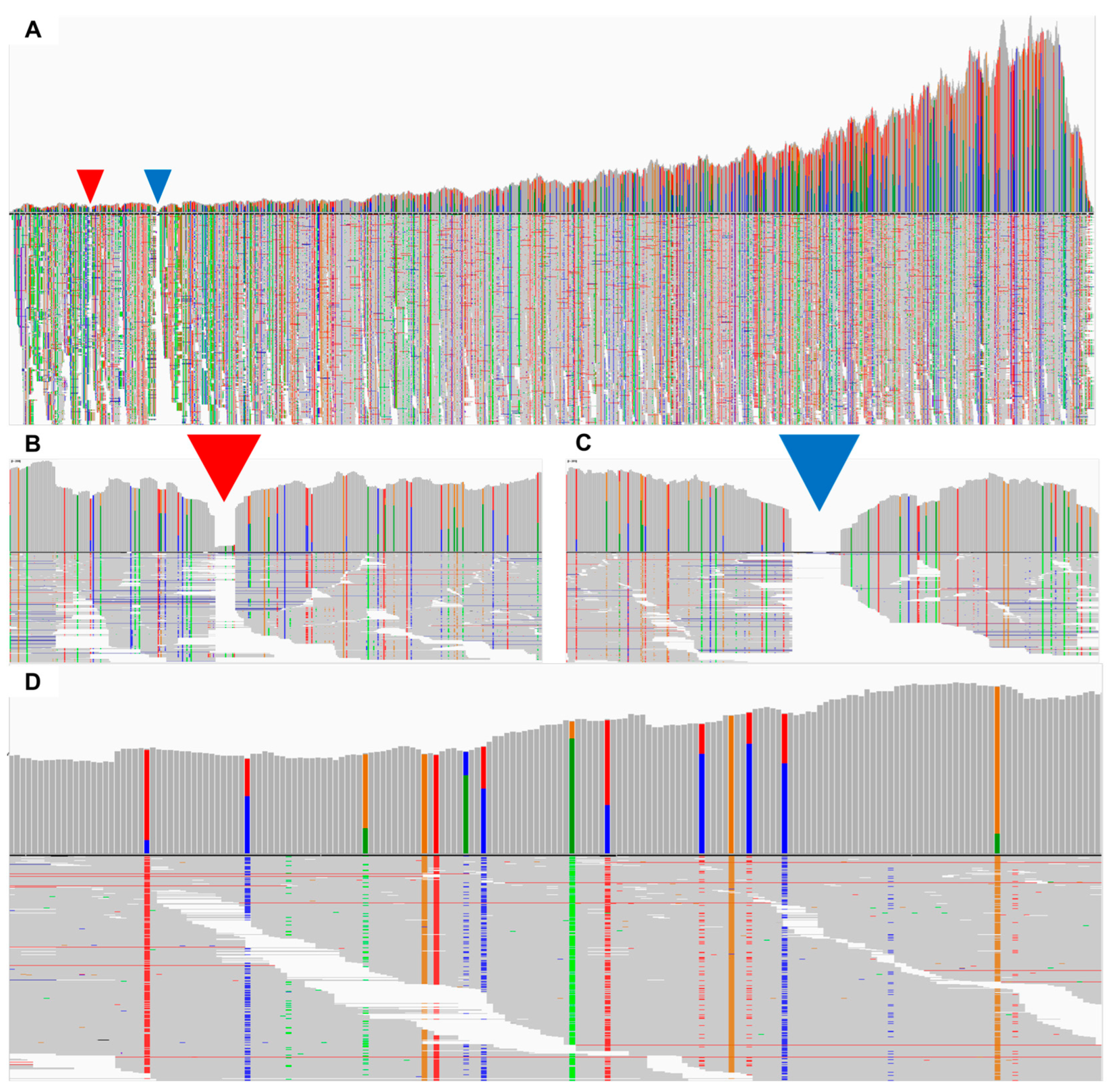
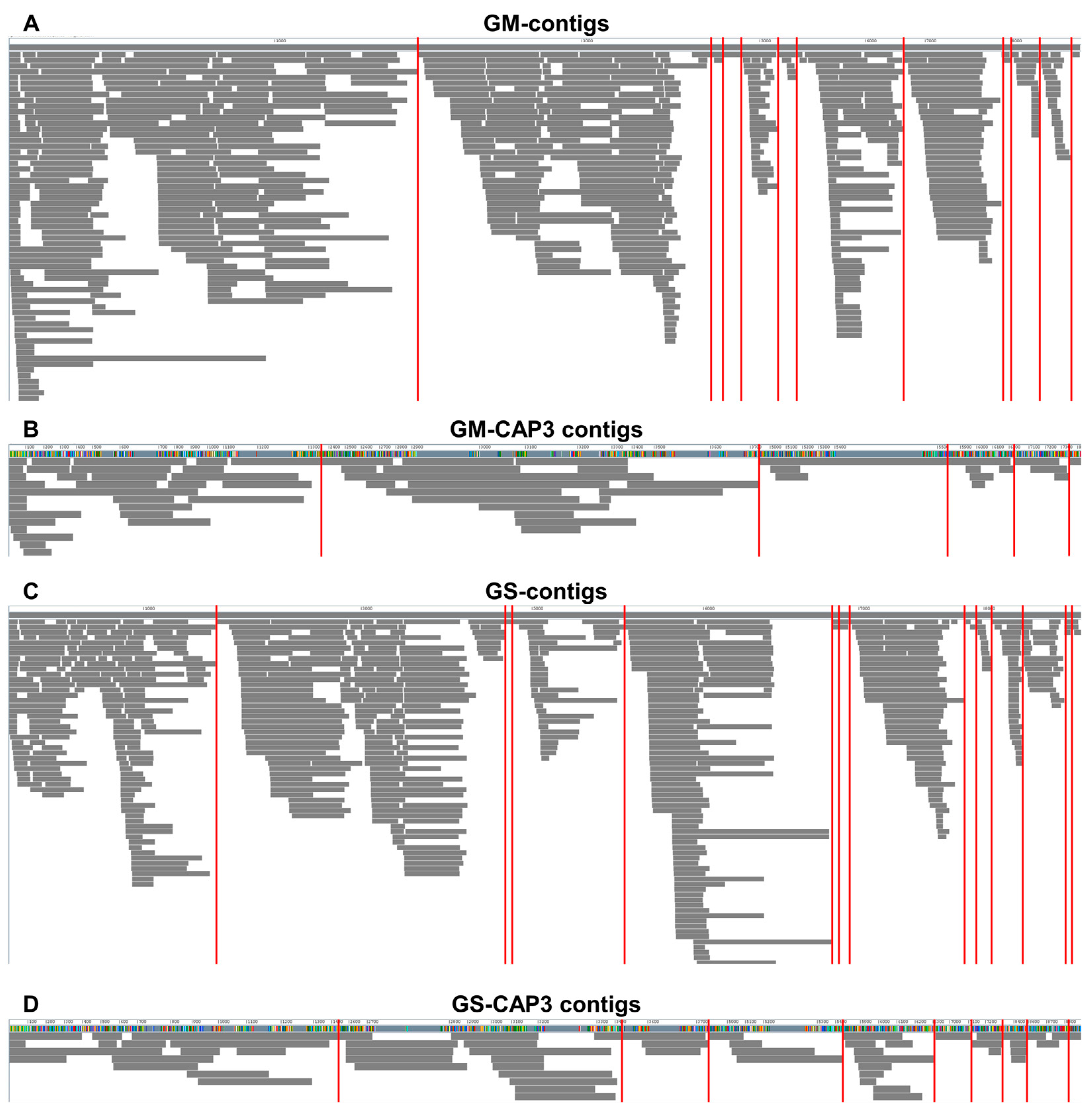
2.6. Assembly and Mapping of SMV-Associated Contigs
3. Discussion
4. Materials and Methods
4.1. Sample Collection and RNA-Sequencing
4.2. Processing of Raw Data and De Novo Assembly of the Transcriptome
4.3. Identification of Viral Contigs through BLASTX Search
4.4. Annotation of the Viral Contigs
4.5. Assembly of Contigs and Mapping of Viral Contigs on the Reference Viral Genome
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Toomer, O.T.; Oviedo, E.O.; Ali, M.; Patino, D.; Joseph, M.; Frinsko, M.; Vu, T.; Maharjan, P.; Fallen, B.; Mian, R. Current agronomic practices, harvest & post-harvest processing of soybeans (Glycine max)—A review. Agronomy 2023, 13, 427. [Google Scholar]
- Elmore, M.G.; Groves, C.L.; Hajimorad, M.; Stewart, T.P.; Gaskill, M.A.; Wise, K.A.; Sikora, E.; Kleczewski, N.M.; Smith, D.L.; Mueller, D.S. Detection and discovery of plant viruses in soybean by metagenomic sequencing. Virol. J. 2022, 19, 149. [Google Scholar] [CrossRef] [PubMed]
- Jo, Y.; Yoon, Y.N.; Jang, Y.-W.; Choi, H.; Lee, Y.-H.; Kim, S.-M.; Choi, S.Y.; Lee, B.C.; Cho, W.K. Soybean Viromes in the Republic of Korea Revealed by RT-PCR and Next-Generation Sequencing. Microorganisms 2020, 8, 1777. [Google Scholar] [CrossRef]
- Hill, J.H.; Whitham, S.A. Control of virus diseases in soybeans. In Advances in Virus Research; Elsevier: Amsterdam, The Netherlands, 2014; Volume 90, pp. 355–390. [Google Scholar]
- Choi, H.; Jo, Y.; Choi, S.Y.; Kim, S.-M.; Choi, Y.M.; Hong, J.-S.; Lee, B.C.; Cho, W.K. Evolution and Phylogeny of Soybean Mosaic Virus Based on 143 Complete Genomes. Int. J. Mol. Sci. 2022, 24, 22. [Google Scholar] [CrossRef] [PubMed]
- Choi, H.; Jo, Y.; Chung, H.; Choi, S.Y.; Kim, S.-M.; Hong, J.-S.; Lee, B.C.; Cho, W.K. Phylogenetic and Phylodynamic Analyses of Soybean Mosaic Virus Using 305 Coat Protein Gene Sequences. Plants 2022, 11, 3256. [Google Scholar] [CrossRef]
- Hajimorad, M.; Domier, L.; Tolin, S.; Whitham, S.; Saghai Maroof, M. Soybean mosaic virus: A successful potyvirus with a wide distribution but restricted natural host range. Mol. Plant Pathol. 2018, 19, 1563–1579. [Google Scholar] [CrossRef]
- Domier, L.L.; Steinlage, T.A.; Hobbs, H.A.; Wang, Y.; Herrera-Rodriguez, G.; Haudenshield, J.S.; McCoppin, N.K.; Hartman, G.L. Similarities in seed and aphid transmission among Soybean mosaic virus isolates. Plant Dis. 2007, 91, 546–550. [Google Scholar] [CrossRef]
- Choi, H.; Jo, Y.; Hong, J.; Chung, H.; Choi, S.; Kim, S.; Lee, J.; Moh, S.; Lee, B.; Cho, W.K. Complete Genome Sequence of a Novel Monopartite Mastrevirus, Soybean Geminivirus B, Isolated from Soybean (Glycine max (L.) Merrill). Plants 2022, 11, 1768. [Google Scholar] [CrossRef]
- Cheng, R.; Mei, R.; Yan, R.; Chen, H.; Miao, D.; Cai, L.; Fan, J.; Li, G.; Xu, R.; Lu, W. A new distinct geminivirus causes soybean stay-green disease. Mol. Plant 2022, 15, 927–930. [Google Scholar] [CrossRef]
- Barba, M.; Czosnek, H.; Hadidi, A. Historical perspective, development and applications of next-generation sequencing in plant virology. Viruses 2014, 6, 106–136. [Google Scholar] [CrossRef]
- Zanardo, L.G.; de Souza, G.B.; Alves, M.S. Transcriptomics of plant–virus interactions: A review. Theor. Exp. Plant Physiol. 2019, 31, 103–125. [Google Scholar] [CrossRef]
- Pagán, I. Transmission through seeds: The unknown life of plant viruses. PLoS Pathog. 2022, 18, e1010707. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Gerstein, M.; Snyder, M. RNA-Seq: A revolutionary tool for transcriptomics. Nat. Rev. Genet. 2009, 10, 57–63. [Google Scholar] [CrossRef] [PubMed]
- Choi, H.; Jo, Y.; Cho, W.K. In Silico Virome Analysis of Chinese Narcissus Transcriptomes Reveals Diverse Virus Species and Genetic Diversity at Different Flower Development Stages. Biology 2023, 12, 1094. [Google Scholar] [CrossRef] [PubMed]
- Jo, Y.; Choi, H.; Kyong Cho, J.; Yoon, J.-Y.; Choi, S.-K.; Kyong Cho, W. In silico approach to reveal viral populations in grapevine cultivar Tannat using transcriptome data. Sci. Rep. 2015, 5, 15841. [Google Scholar] [CrossRef]
- Gao, H.; Wang, Y.; Li, W.; Gu, Y.; Lai, Y.; Bi, Y.; He, C. Transcriptomic comparison reveals genetic variation potentially underlying seed developmental evolution of soybeans. J. Exp. Bot. 2018, 69, 5089–5104. [Google Scholar] [CrossRef]
- Zhao, Y.; Li, M.-C.; Konaté, M.M.; Chen, L.; Das, B.; Karlovich, C.; Williams, P.M.; Evrard, Y.A.; Doroshow, J.H.; McShane, L.M. TPM, FPKM, or normalized counts? A comparative study of quantification measures for the analysis of RNA-seq data from the NCI patient-derived models repository. J. Transl. Med. 2021, 19, 269. [Google Scholar] [CrossRef]
- Villamor, D.; Ho, T.; Al Rwahnih, M.; Martin, R.; Tzanetakis, I. High throughput sequencing for plant virus detection and discovery. Phytopathology 2019, 109, 716–725. [Google Scholar] [CrossRef]
- Vilsker, M.; Moosa, Y.; Nooij, S.; Fonseca, V.; Ghysens, Y.; Dumon, K.; Pauwels, R.; Alcantara, L.C.; Vanden Eynden, E.; Vandamme, A.-M. Genome Detective: An automated system for virus identification from high-throughput sequencing data. Bioinformatics 2019, 35, 871–873. [Google Scholar] [CrossRef]
- Massart, S.; Olmos, A.; Jijakli, H.; Candresse, T. Current impact and future directions of high throughput sequencing in plant virus diagnostics. Virus Res. 2014, 188, 90–96. [Google Scholar] [CrossRef]
- Jo, Y.; Choi, H.; Bae, M.; Kim, S.-M.; Kim, S.-L.; Lee, B.C.; Cho, W.K.; Kim, K.-H. De novo genome assembly and single nucleotide variations for soybean mosaic virus using soybean seed transcriptome data. Plant Pathol. J. 2017, 33, 478. [Google Scholar] [CrossRef]
- Sharma, D.; Priyadarshini, P.; Vrati, S. Unraveling the web of viroinformatics: Computational tools and databases in virus research. J. Virol. 2015, 89, 1489–1501. [Google Scholar] [CrossRef]
- Bowers, G., Jr.; Goodman, R. Soybean mosaic virus: Infection of soybean seed parts and seed transmission. Phytopathology 1979, 69, 569–572. [Google Scholar] [CrossRef]
- Hong, J.; Masuta, C.; Nakano, M.; Abe, J.; Uyeda, I. Adaptation of Cucumber mosaic virus soybean strains (SSVs) to cultivated and wild soybeans. Theor. Appl. Genet. 2003, 107, 49–53. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, N.; Tamada, T. Host range and molecular analysis of Beet leaf yellowing virus, Beet western yellows virus-JP and Brassica yellows virus in Japan. Plant Pathol. 2019, 68, 1045–1058. [Google Scholar] [CrossRef]
- Choi, H.; Jo, Y.; Zhou, Y.; Cho, W. First report of milk vetch dwarf virus infecting lily in Korea. Plant Dis. 2019, 103, 2144. [Google Scholar] [CrossRef]
- Usovsky, M.; Chen, P.; Li, D.; Wang, A.; Shi, A.; Zheng, C.; Shakiba, E.; Lee, D.; Canella Vieira, C.; Lee, Y.C. Decades of genetic research on Soybean mosaic virus resistance in soybean. Viruses 2022, 14, 1122. [Google Scholar] [CrossRef] [PubMed]
- Tisza, M.J.; Pastrana, D.V.; Welch, N.L.; Stewart, B.; Peretti, A.; Starrett, G.J.; Pang, Y.-Y.S.; Krishnamurthy, S.R.; Pesavento, P.A.; McDermott, D.H. Discovery of several thousand highly diverse circular DNA viruses. eLife 2020, 9, e51971. [Google Scholar] [CrossRef]
- Selitsky, S.R.; Marron, D.; Hollern, D.; Mose, L.E.; Hoadley, K.A.; Jones, C.; Parker, J.S.; Dittmer, D.P.; Perou, C.M. Virus expression detection reveals RNA-sequencing contamination in TCGA. BMC Genom. 2020, 21, 79. [Google Scholar] [CrossRef]
- Jo, Y.; Choi, H.; Lee, J.H.; Moh, S.H.; Cho, W.K. Viromes of 15 Pepper (Capsicum annuum L.) Cultivars. Int. J. Mol. Sci. 2022, 23, 10507. [Google Scholar] [CrossRef]
- Holland, J.J.; De La Torre, J.; Steinhauer, D. RNA virus populations as quasispecies. Genet. Divers. RNA Viruses 1992, 176, 1–20. [Google Scholar]
- Li, H.; Durbin, R. Fast and accurate long-read alignment with Burrows–Wheeler transform. Bioinformatics 2010, 26, 589–595. [Google Scholar] [CrossRef] [PubMed]
- Buchfink, B.; Xie, C.; Huson, D.H. Fast and sensitive protein alignment using DIAMOND. Nat. Methods 2015, 12, 59–60. [Google Scholar] [CrossRef] [PubMed]
- Bağcı, C.; Patz, S.; Huson, D.H. DIAMOND+ MEGAN: Fast and easy taxonomic and functional analysis of short and long microbiome sequences. Curr. Protoc. 2021, 1, e59. [Google Scholar] [CrossRef] [PubMed]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cultivar Name | Species | Abbreviations | Flower Color | Seed Coat Color |
|---|---|---|---|---|
| Hefeng 48 | Glycine max | GM1 | purple | yellow |
| Donong 53 | Glycine max | GM2 | white | yellow |
| Nenfeng16 | Glycine max | GM3 | white | yellow |
| Heinong 35 | Glycine max | GM4 | white | yellow |
| 01-737 | Glycine soja | GS1 | purple | black |
| 200201-6 | Glycine soja | GS2 | purple | black |
| 01-590 | Glycine soja | GS3 | purple | brown |
| 01-555 | Glycine soja | GS4 | purple | green |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Choi, H.; Jo, Y.; Chung, H.; Choi, S.Y.; Kim, S.-M.; Hong, J.-S.; Lee, B.C.; Cho, W.K. Investigating Variability in Viral Presence and Abundance across Soybean Seed Development Stages Using Transcriptome Analysis. Plants 2023, 12, 3257. https://doi.org/10.3390/plants12183257
Choi H, Jo Y, Chung H, Choi SY, Kim S-M, Hong J-S, Lee BC, Cho WK. Investigating Variability in Viral Presence and Abundance across Soybean Seed Development Stages Using Transcriptome Analysis. Plants. 2023; 12(18):3257. https://doi.org/10.3390/plants12183257
Chicago/Turabian StyleChoi, Hoseong, Yeonhwa Jo, Hyunjung Chung, Soo Yeon Choi, Sang-Min Kim, Jin-Sung Hong, Bong Choon Lee, and Won Kyong Cho. 2023. "Investigating Variability in Viral Presence and Abundance across Soybean Seed Development Stages Using Transcriptome Analysis" Plants 12, no. 18: 3257. https://doi.org/10.3390/plants12183257