
Plastid Phylogenomic Analyses Reveal the Taxonomic Position of Peucedanum franchetii
 , and
, and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Taxon Sampling, DNA Extraction, and Sequencing
2.2. Plastome Assembly and Annotation
2.3. Codon Usage, RNA Editing, and SSRs Analyses
2.4. Comparative Plastome Analyses
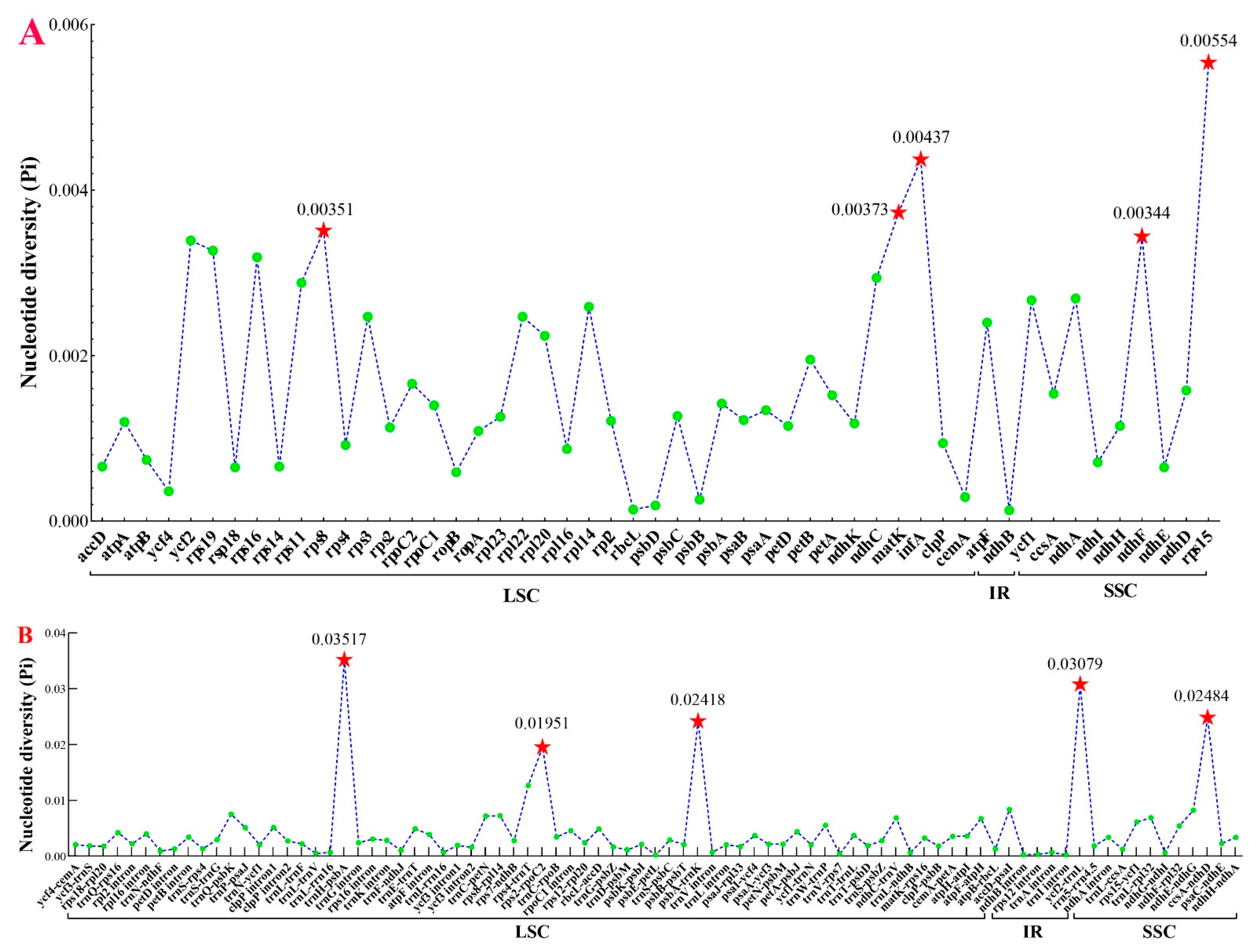
2.5. Identification of Divergence Hotspots
2.6. Phylogenetic Analyses
3. Results
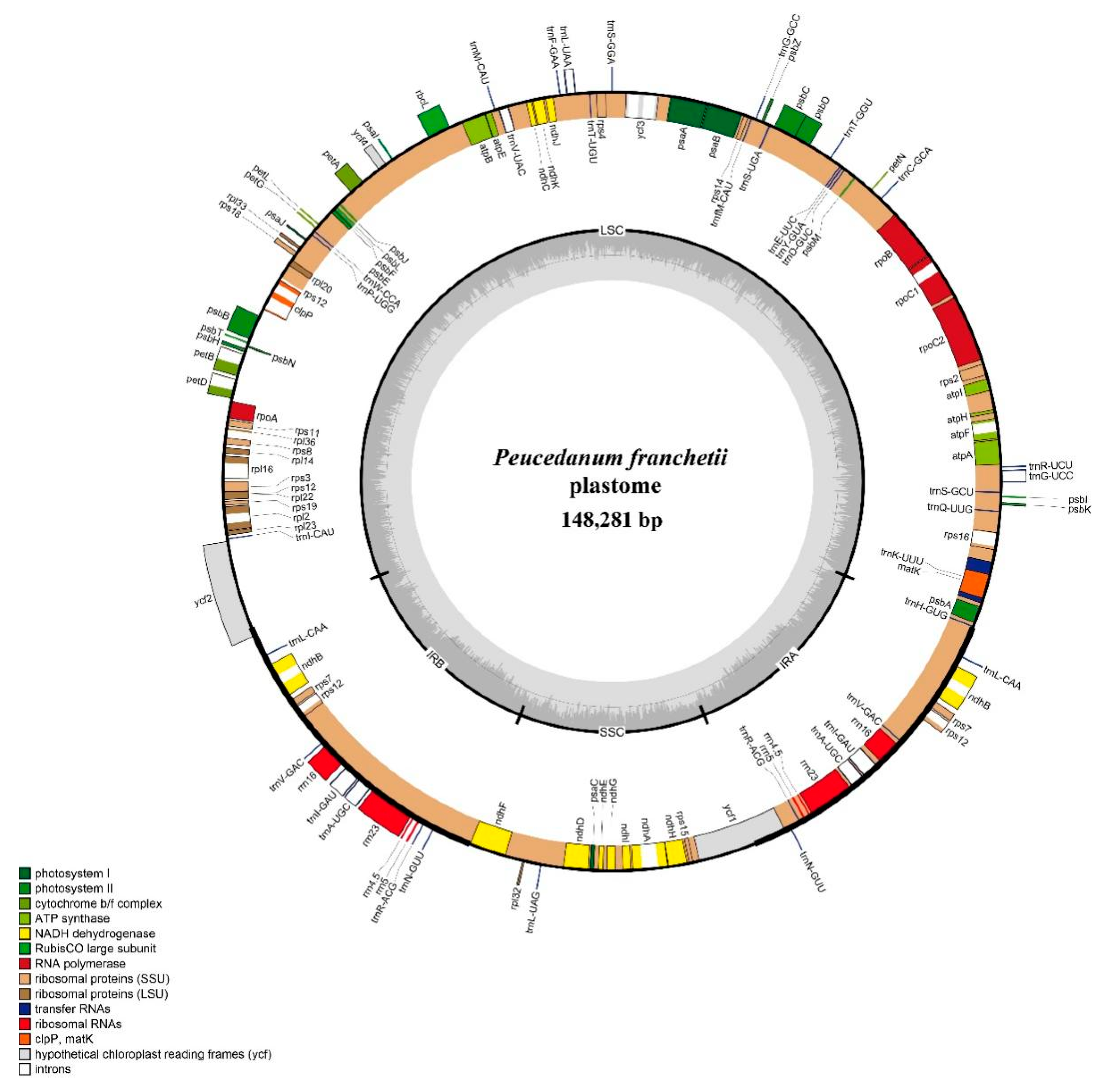
3.1. Characteristics of Plastome
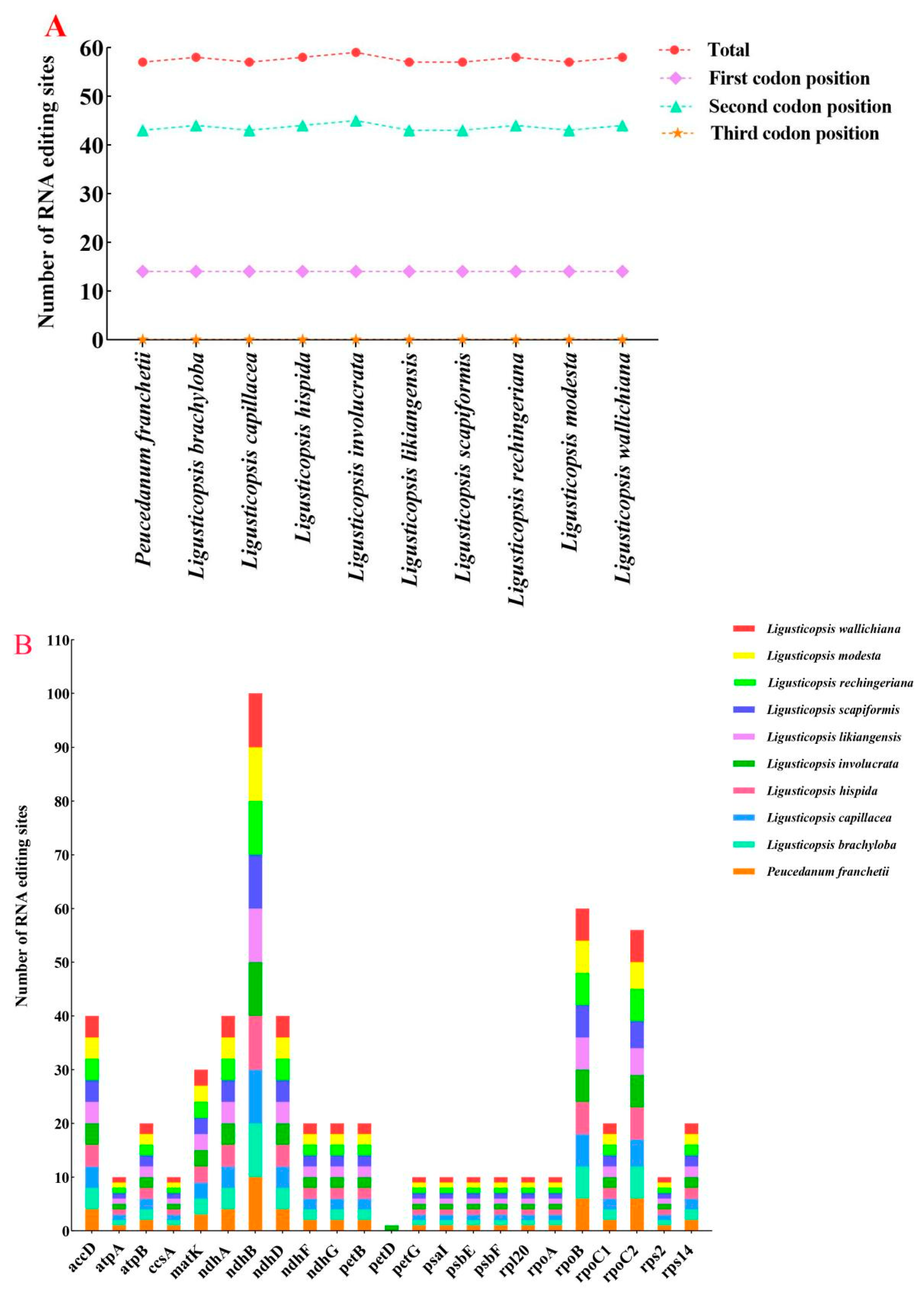
3.2. Codon Usage, RNA Editing in Protein-Coding Genes
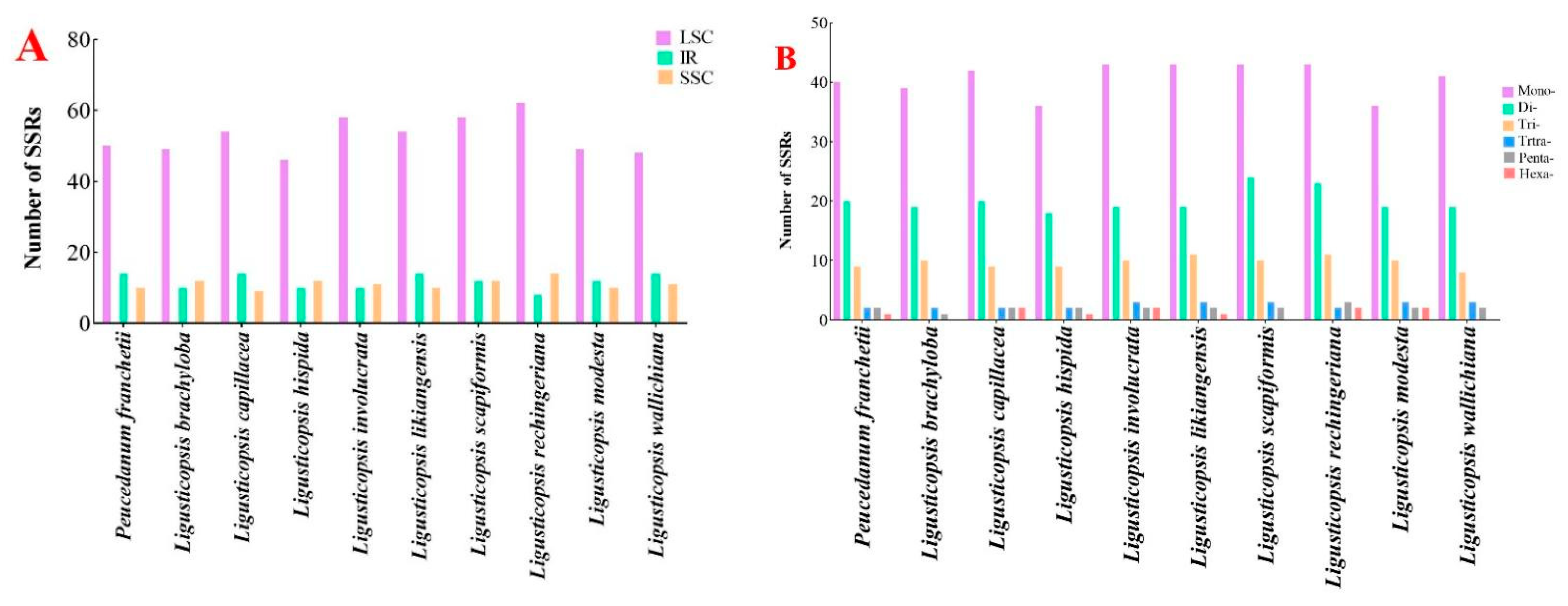
3.3. Simple Sequence Repeats (SSRs)
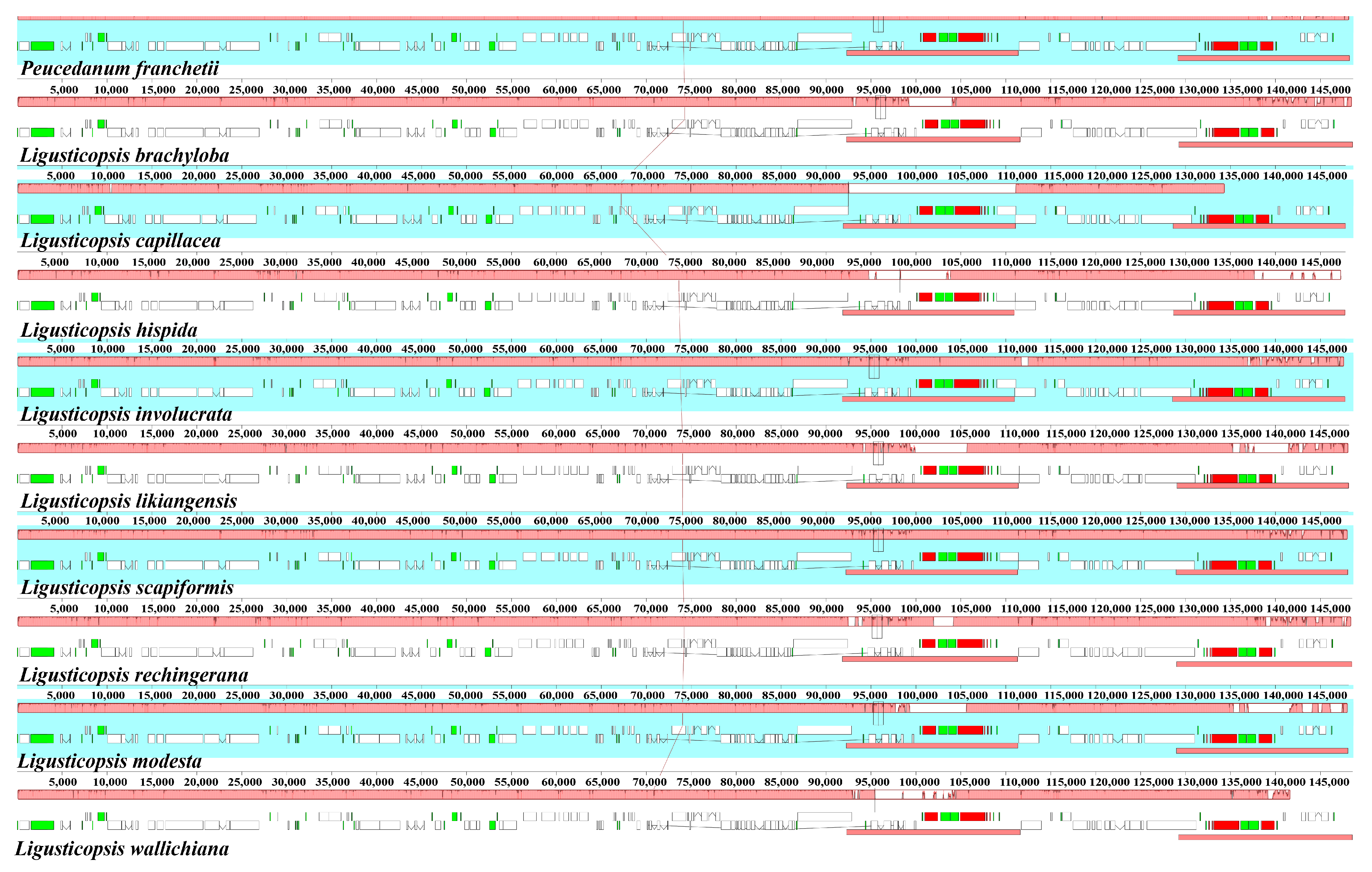
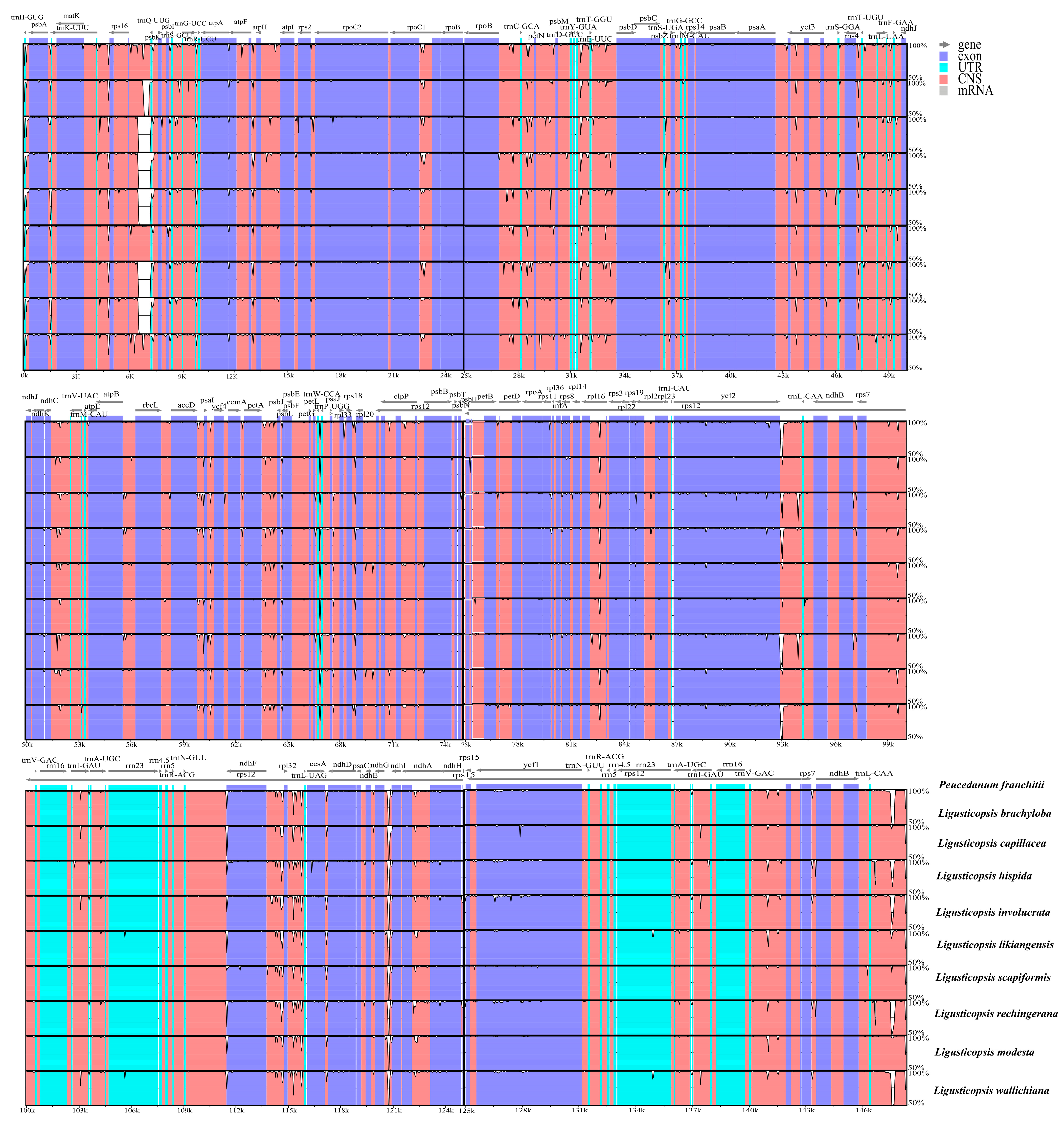
3.4. Plastome Comparison and Hotspots Identification
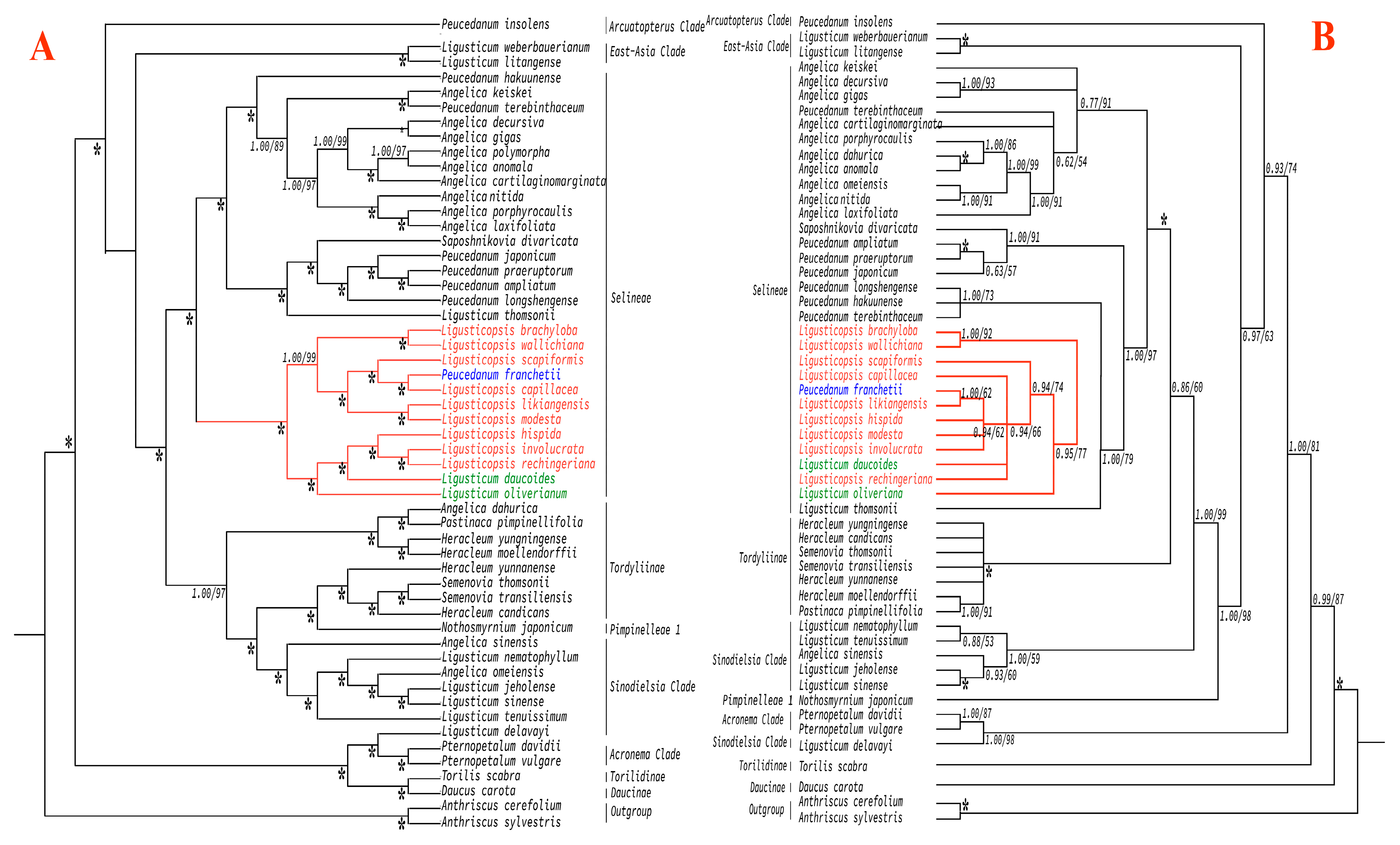
3.5. Phylogenetic Analyses
4. Discussion
4.1. Plastome Characteristics
4.2. Codon Usage Analyses
4.3. RNA Editing Analyses
4.4. Simple Sequence Repeats (SSRs) Analyses
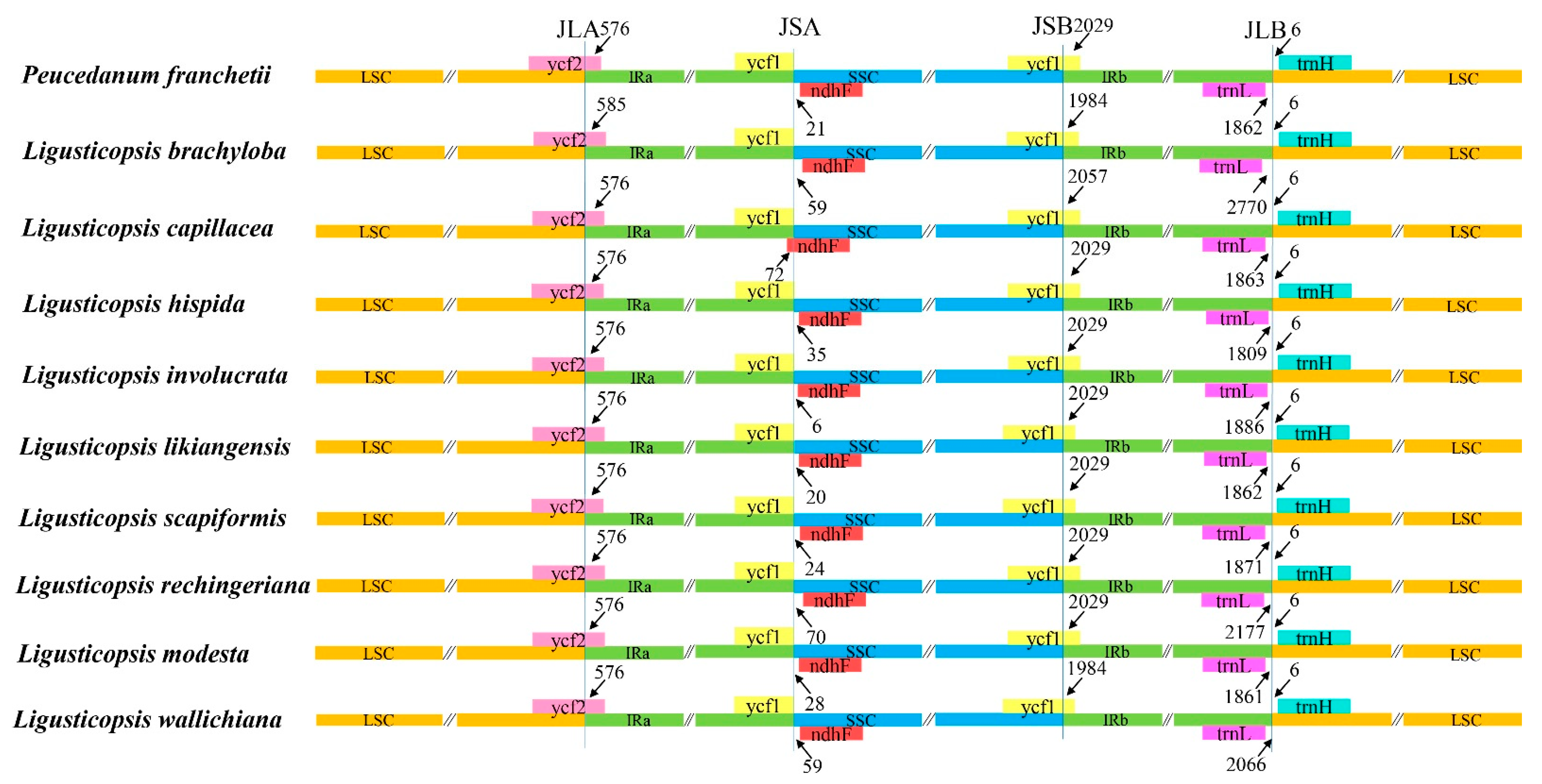
4.5. The IR/SC Boundaries of the Ten Plastomes
4.6. Candidate DNA Barcodes
4.7. Phylogenetic Relationships and Taxonomic Implication of P. franchetii
4.8. Taxonomic Treatment
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Linnaeus, C. Species Plantarum.Laurentii Salvii; Oxford University Press: Stockholm, Sweden, 1753. [Google Scholar]
- Pimenov, M.G.; Leonov, M.V.; Constance, L. The Genera of the Umbelliferae: A Nomenclator[M]; Royal Botanic Gardens: Kew, UK, 1993; ISBN 978-0-947643-58-4. [Google Scholar]
- Zhang, X. Studies on geographic distribution of Peucedanum L. in southwest of China. Chin. Agric. Sci. Bull. 2011, 27, 177–180. [Google Scholar] [CrossRef]
- Spalik, K.; Reduron, J.P.; Downie, S.R. The phylogenetic position of Peucedanum sensu lato and allied genera and their placement in tribe Selineae (Apiaceae, subfamily Apioideae). Plant Syst. Evol. 2004, 243, 189–210. [Google Scholar] [CrossRef]
- Sheh, M.L.; Watson, M.F. Peucedanum Linnaeus. In Flora of China; Science Press & Missouri Botanic Garden Press: Beijing, China; St. Louis, MO, USA, 2005; Volume 14, pp. 182–192. [Google Scholar]
- Calestani, V. Conspectus specierum europaearum generis Peucedani. Bull. Soc. Bot. Ital. Ann. 1905, 1905, 193–201. [Google Scholar]
- Leute, G.H. Die Gattungen Imperatoria L. Und Tommasinia Bertol. (Apiaceae). Ann. Des Nat. Mus.Wien 1965, 69–79. [Google Scholar]
- Pimenov, M.G.; Hedge, I.C.; Lamond, J.M.; Rechinger, K.H. Cervaria. In Umbelliferae; Rechinger, K.H., Iranica, F., Eds.; Springer: Berlin/Heidelberg, Germany, 1987; Volume 162, pp. 83–110. [Google Scholar]
- Frey, R. Taxonomische Revision der Gattung Peucedanum: Sektion Peucedanum und Sektion Palimbioidea (Umbelliferae). Candollea 1989, 44, 257–327. [Google Scholar]
- Tutin, T.G.; Heywood, V.H.; Burges, N.A.; Moore, D.M.; Valentine, D.H.; Walters, S.M.; Webb, D.A. (Eds.) Flora Europaea; Cambridge University Press: Cambridge, UK, 1968; Volume 2. [Google Scholar]
- Reduron, J.-P.; Charpin, A.; Pimenov, M.G. Contribution a la nomenclature generique des Apiaceae (Ombelliferes). J. Bot. Soc. Bot. Fr. 1997, 91–104. [Google Scholar]
- Winter, P.J.D.; Magee, A.R.; Phephu, N.; Tilney, P.M.; Downie, S.R.; van Wyk, B.E. A new generic classification for African peucedanoid species (Apiaceae). Taxon 2008, 57, 347–364. [Google Scholar] [CrossRef]
- Leute, G.H. Untersuchungen über den Verwandtschaftkreis der Gattung Ligusticum L. (Umbelliferae) 1. Ann. Des Nat. Mus. Wien 1969, 73, 55–98. [Google Scholar]
- Doyle, J.; Doyle, J.L. Genomic plant DNA preparation from fresh tissue-CTAB method. Phytochem. Bull. 1987, 19, 11–15. [Google Scholar]
- Davis, J.I.; Mcneal, J.R.; Barrett, C.F.; Chase, M.W.; Cohen, J.I.; Duvall, M.R. Contrasting patterns of support among plastid genes and genomes for major clades of the monocotyledons. Early Events Monocot Evol. 2013, 83, 315–349. [Google Scholar]
- Chen, S.; Zhou, Y.; Chen, Y.; Gu, J. Fastp: An ultra-fast all-in-one FASTQ preprocessor. Bioinformatics 2018, 34, i884–i890. [Google Scholar] [CrossRef] [PubMed]
- White, T.J.; Bruns, S.; Lee, S.; Taylor, J. Amplification and direct sequencing of fungal ribosomal RNA genes for phylogenetics. PCR Protoc. A Guide Methods Appl. 1990, 18, 315–322. [Google Scholar] [CrossRef]
- Burland, T.G. DNASTAR’s Lasergene Sequence Analysis Software. In Bioinformatics Methods and Protocols. Methods in Molecular Biology™; Misener, S., Krawetz, S.A., Eds.; Humana Press: Totowa, NJ, USA, 2000; Volume 132, pp. 71–91. [Google Scholar] [CrossRef]
- Jin, J.J.; Yu, W.B.; Yang, J.B.; Song, Y.; Depamphilis, C.; Yi, T.S.; Li, D.Z. GetOrganelle: A fast and versatile toolkit for accurate de novo assembly of organelle genomes. Genome Biol. 2020, 21, 241. [Google Scholar] [CrossRef] [PubMed]
- Kearse, M.; Moir, R.; Wilson, A.; Stones-Havas, S.; Cheung, M.; Sturrock, S.; Buxton, S.; Cooper, A.; Markowitz, S.; Duran, C.; et al. Geneious basic: An integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics 2012, 28, 1647–1649. [Google Scholar] [CrossRef] [Green Version]
- Qu, X.J.; Moore, M.J.; Li, D.Z.; Yi, T.S. PGA: A software package for rapid, accurate, and flexible batch annotation of plastomes. Plant Methods 2019, 15, 50. [Google Scholar] [CrossRef] [Green Version]
- Tillich, M.; Lehwark, P.; Pellizzer, T.; Ulbricht-Jones, E.S.; Fischer, A.; Bock, R.; Greiner, S. GeSeq–versatile and accurate annotation of organelle genomes. Nucleic Acids Res. 2017, 45, 6–11. [Google Scholar] [CrossRef]
- Schubert, M.; Lindgreen, S.; Orlando, L. AdapterRemoval v2: Rapid adapter trimming, identification, and read merging. BMC Res. Notes 2016, 9, 88. [Google Scholar] [CrossRef] [Green Version]
- Greiner, S.; Lehwark, P.; Bock, R. OrganellarGenomeDRAW (OGDRAW) version 1.3. 1: Expanded toolkit for the graphical visualization of organellar genomes. Nucleic Acids Res. 2019, 47, W59–W64. [Google Scholar] [CrossRef] [Green Version]
- Sharp, P.M.; Li, W.H. An evolutionary perspective on synonymous codon usage in unicellular organisms. J. Mol. Evol. 1986, 24, 28–38. [Google Scholar] [CrossRef]
- Peden, J.F. Analysis of Codon Usage. Ph.D. Thesis, University of Nottingham, Nottingham, UK, 1999. [Google Scholar]
- Chen, C.; Chen, H.; Zhang, Y.; Thomas, H.R.; Frank, M.H.; He, Y.; Xia, R. TBtools: An integrative toolkit developed for interactive analyses of big biological data. Mol. Plant 2020, 13, 1194–1202. [Google Scholar] [CrossRef]
- Mower, J.P. The PREP suite: Predictive RNA editors for plant mitochondrial genes, chloroplast genes, and user-defined alignments. Nucleic Acids Res. 2009, 37, W253–W259. [Google Scholar] [CrossRef] [PubMed]
- Beier, S.; Thiel, T.; Münch, T.; Scholz, U.; Mascher, M. MISA-web: A web server for microsatellite prediction. Bioinformatics 2017, 33, 2583–2585. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Amiryousefi, A.; Hyvönen, J.; Poczai, P. IRscope: An online program to visualize the junction sites of chloroplast genomes. Bioinformatics 2018, 34, 3030–3031. [Google Scholar] [CrossRef] [PubMed]
- Darling, A.C.E.; Mau, B.; Blattner, F.R.; Perna, N.T. Mauve: Multiple alignment of conserved genomic sequence with rearrangements. Genome Res. 2004, 14, 1394–1403. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Frazer, K.A.; Pachter, L.; Poliakov, A.; Rubin, E.M.; Dubchak, I. VISTA: Computational tools for comparative genomics. Nucleic Acids Res. 2004, 32 (Suppl. S2), W273–W279. [Google Scholar] [CrossRef] [Green Version]
- Katoh, K.; Standley, D.M. MAFFT multiple sequence alignment software version 7: Improvements in performance and usability. Mol. Biol. Evol. 2013, 30, 772–780. [Google Scholar] [CrossRef]
- Librado, P.; Rozas, J. DnaSP v5: A software for comprehensive analysis of DNA polymorphism data. Bioinformatics 2009, 25, 1451–1452. [Google Scholar] [CrossRef] [Green Version]
- Wen, J.; Xie, D.F.; Price, M.; Ren, T.; Deng, Y.; Gui, L.; Guo, X.L.; He, X.J. Backbone phylogeny and evolution of Apioideae (Apiaceae): New insights from phylogenomic analyses of plastome data. Mol. Phylogenet. Evol. 2021, 161, 107183. [Google Scholar] [CrossRef]
- Stamatakis, A. RAxML version 8: A tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 2014, 30, 1312–1313. [Google Scholar] [CrossRef] [Green Version]
- Stamatakis, A.; Hoover, P.; Rougemont, J. A rapid bootstrap algorithm for the RAxML. Syst. Biol. 2008, 57, 758–771. [Google Scholar] [CrossRef]
- Ronquist, F.; Teslenko, M.; Van Der Mark, P.; Ayres, D.L.; Darling, A.; Höhna, S.; Larget, B.; Liu, L.; Suchard, M.A.; Huelsenbeck, J.P. MrBayes 3.2: Efficient Bayesian phylogenetic inference and model choice across a large model space. Syst. Biol. 2012, 61, 539–542. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Posada, D.; Crandall, K.A. Modeltest: Testing the model of DNA substitution. Bioinformatics 1998, 14, 817–818. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rambaut, A.; Drummond, A. FigTree, Version 1.4.2. 2015. Available online: http://tree.bio.ed.ac.uk/software/figtree/ (accessed on 4 October 2021).
- Letunic, I.; Bork, P. Interactive Tree of Life (iTOL): An online tool for phylogenetic tree display and annotation. Bioinformatics 2007, 23, 127–128. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kang, L.; Xie, D.F.; Xiao, Q.Y.; Peng, C.; Yu, Y.; He, X.J. Sequencing and analyses on chloroplast genomes of Tetrataenium candicans and two allies give new insights on structural variants, DNA barcoding and phylogeny in Apiaceae subfamily Apioideae. PeerJ 2019, 7, e8063. [Google Scholar] [CrossRef] [Green Version]
- Liu, C.K.; Lei, J.Q.; Jiang, Q.P.; Zhou, S.D.; He, X.J. The complete plastomes of seven Peucedanum plants: Comparative and phylogenetic analyses for the Peucedanum genus. BMC Plant Biol. 2022, 22, 101. [Google Scholar] [CrossRef]
- Ren, T.; Li, Z.X.; Xie, D.F.; Gui, L.J.; Peng, C.; Wen, J.; He, X.J. Plastomes of eight Ligusticum species: Characterization, genome evolution, and phylogenetic relationships. BMC Plant Biol. 2020, 20, 519. [Google Scholar] [CrossRef]
- Li, Z.X.; Guo, X.L.; Price, M.; Zhou, S.D.; He, X.J. Phylogenetic position of Ligusticopsis (Apiaceae, Apioideae): Evidence from molecular data and carpological characters. AoB Plants 2022, 14, plac008. [Google Scholar] [CrossRef]
- Logacheva, M.D.; Krinitsina, A.A.; Belenikin, M.S.; Khafizov, K.; Konorov, E.A.; Kuptsov, S.V.; Speranskaya, A.S. Comparative analysis of inverted repeats of polypod fern (Polypodiales) plastomes reveals two hypervariable regions. BMC Plant Biol. 2017, 17, 61–73. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.; Geng, M.; Xu, Z.; Wang, Q.; Li, L.; Xu, M.; Li, M.M. The complete plastome of Peucedanum praeruptorum (Apiaceae). Mitochondrial DNA B Resour. 2019, 4, 3612–3613. [Google Scholar] [CrossRef] [Green Version]
- Gou, W.; Jia, S.B.; Price, M.; Guo, X.L.; Zhou, S.D.; He, X.J. Complete plastid genome sequencing of eight species from Hansenia, Haplosphaera and Sinodielsia (Apiaceae): Comparative analyses and phylogenetic implications. Plants 2020, 9, 1523. [Google Scholar] [CrossRef]
- Asaf, S.; Khan, A.L.; Khan, M.A.; Imran, Q.M.; Kang, S.-M.; Al-Hosni, K.; Jeong, E.J.; Lee, K.E.; Lee, I.-J. Comparative analysis of complete plastid genomes from wild soybean (Glycine soja) and nine other Glycine species. PLoS ONE 2017, 12, e0182281. [Google Scholar] [CrossRef] [PubMed]
- de Souza, U.J.B.; Nunes, R.; Targueta, C.P.; Diniz-Filho, J.A.F.; de Campos Telles, M.P. The complete chloroplast genome of Stryphnodendron adstringens (Leguminosae-Caesalpinioideae): Comparative analysis with related Mimosoid species. Sci. Rep. 2019, 9, 14206. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lagerkvist, U. “Two out of three”: An alternative method for codon reading. Proc. Natl. Acad. Sci. USA 1978, 75, 1759–1762. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ikemura, T. Codon usage and tRNA content in unicellular and multicellular organisms. Mol. Biol. Evol. 1985, 2, 13–34. [Google Scholar] [CrossRef] [PubMed]
- Plotkin, J.B.; Kudla, G. Synonymous but not the same: The causes and consequences of codon bias. Nat. Rev. Genet. 2011, 12, 32–42. [Google Scholar] [CrossRef] [Green Version]
- Yu, C.H.; Dang, Y.; Zhou, Z.; Wu, C.; Zhao, F.; Sachs, M.S.; Liu, Y. Codon Usage Infuences the Local Rate of Translation Elongation to Regulate Cotranslational Protein Folding. Mol. Cell. 2015, 59, 744–754. [Google Scholar] [CrossRef]
- Mittal, P.; Brindle, J.; Stephen, J.; Plotkin, J.B.; Kudla, G. Codon usage infuences ftness through RNA toxicity. Proc. Natl. Acad. Sci. USA 2018, 115, 8639–8644. [Google Scholar] [CrossRef] [Green Version]
- Shen, X.; Guo, S.; Yin, Y.; Zhang, J.; Yin, X.; Liang, C.; Wang, Z.W.; Huang, B.F.; Liu, Y.H.; Xiao, S.M.; et al. Complete chloroplast genome sequence and phylogenetic analysis of aster tataricus. Molecules 2018, 23, 2426. [Google Scholar] [CrossRef] [Green Version]
- Zhu, B.; Qian, F.; Hou, Y.; Yang, W.; Cai, M.; Wu, X. Complete chloroplast genome features and phylogenetic analysis of Eruca sativa (Brassicaceae). PLoS ONE 2021, 16, 1–19. [Google Scholar] [CrossRef]
- Du, X.; Zeng, T.; Feng, Q.; Hu, L.; Luo, X.; Weng, Q.; He, J.F.; Zhu, B. The complete chloroplast genome sequence of yellow mustard (Sinapisalba L.) and its phylogenetic relationship to other Brassicaceae species. Genes 2020, 731, 144340. [Google Scholar] [CrossRef]
- Tang, D.; Wei, F.; Kashif, M.H.; Munsif, F.; Zhou, R. Identifcation and analysis of RNA editing sites in chloroplast transcripts of kenaf (Hibiscus cannabinus L.). 3 Biotech 2019, 9, 361. [Google Scholar] [CrossRef] [PubMed]
- Redwan, R.M.; Saidin, A.; Kumar, S.V. Complete chloroplast genome sequence of MD-2 pineapple and its comparative analysis among nine other plants from the subclass Commelinidae. BMC Plant Biol. 2015, 15, 196. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yan, C.; Du, J.; Gao, L.; Li, Y.; Hou, X. The complete chloroplast genome sequence of watercress (Nasturtium ofcinale R. Br.): Genome organization, adaptive evolution and phylogenetic relationships in Cardamineae. Gene 2019, 699, 24–36. [Google Scholar] [CrossRef] [PubMed]
- Ren, T.; Yang, Y.C.; Zhou, T.; Liu, Z.L. Comparative plastid genomes of Primula species: Sequence divergence and phylogenetic relationships. Int. J. Mol. Sci. 2018, 19, 1050. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dong, S.; Ying, Z.; Yu, S.; Wang, Q.; Liao, G.; Ge, Y.; Cheng, R. Complete chloroplast genome of Stephania tetrandra (Menispermaceae) from Zhejiang Province: Insights into molecular structures, comparative genome analysis, mutational hotspots and phylogenetic relationships. BMC Genom. 2021, 6, 880. [Google Scholar] [CrossRef]
- Wang, X.; Zhou, T.; Bai, G.; Zhao, Y. Complete chloroplast genome sequence of Fagopyrum dibotrys: Genome features, comparative analysis and phylogenetic relationships. Sci. Rep. 2018, 8, 12379. [Google Scholar] [CrossRef]
- Castandet, B.; Araya, A. RNA editing in plant organelles. Why make it easy? Biochemistry 2011, 76, 924–931. [Google Scholar] [CrossRef]
- Chen, H.; Deng, L.; Jiang, Y.; Lu, P.; Yu, J.N. RNA editing sites exist in protein-coding genes in the chloroplast genome of Cycas taitungensis. J. Integr. Plant Biol. 2011, 53, 961–970. [Google Scholar] [CrossRef]
- Wang, M.; Liu, H.; Ge, L.; Xing, G.W.; Wang, M.; Song, W.N.; Nie, X.J. Identification and analysis of RNA editing sites in the chloroplast transcripts of Aegilops tauschii L. Genes 2016, 8, 13. [Google Scholar] [CrossRef] [Green Version]
- Corneille, S.; Lutz, K.; Maliga, P. Conservation of RNA editing between rice and maize plastids: Are most editing events dispensable? Mol. Gen. Genet. 2000, 264, 419–424. [Google Scholar] [CrossRef]
- Hanson, M.R.; Sutton, C.A.; Lu, B. Plant organelle gene expression: Altered by RNA editing. Trends Plant Sci. 1996, 1, 57–64. [Google Scholar] [CrossRef]
- He, P.; Huang, S.; Xiao, G.; Zhang, Y.; Yu, J. Abundant RNA editing sites of chloroplast protein-coding genes in Ginkgo biloba and an evolutionary pattern analysis. BMC Plant Biol. 2016, 16, 257. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Provan, J.; Powell, W.; Hollingsworth, P.M. Chloroplast microsatellites: New tools for studies in plant ecology and evolution. Trends Ecol. Evol. 2001, 16, 142–147. [Google Scholar] [CrossRef] [PubMed]
- Jakobsson, M.; Säll, T.; Lind-Halldén, C.; Halldén, C. Evolution of chloroplast mononucleotide microsatellites in Arabidopsis thaliana. Theor. Appl. Genet. 2007, 114, 223. [Google Scholar] [CrossRef]
- Powell, W.; Machray, G.C.; Provan, J. Polymorphism revealed by simple sequence repeats. Trends Plant Sci. 1996, 1, 215–222. [Google Scholar] [CrossRef]
- Ranade, S.S.; Lin, Y.-C.; Zuccolo, A.; Van de Peer, Y.; del Rosario García-Gil, M. Comparative in silico analysis of EST-SSRs in angiosperm and gymnosperm tree genera. BMC Plant Biol. 2014, 14, 220. [Google Scholar] [CrossRef]
- Li, J.; Xie, D.F.; Guo, X.L.; Zheng, Z.Y.; He, X.J.; Zhou, S.D. Comparative analysis of the complete plastid genome of five Bupleu rum species and new insights into DNA barcoding and phylogenetic relationship. Plants 2020, 9, 543. [Google Scholar] [CrossRef]
- Tangphatsornruang, S.; Sangsrakru, D.; Chanprasert, J.; Uthaipaisanwong, P.; Yoocha, T.; Jomchai, N.; Tragoonrung, S. The chloroplast genome sequence of mungbean (Vigna radiata) determined by high-throughput pyrosequencing: Structural organization and phylogenetic relationships. DNA Res. 2010, 17, 11–22. [Google Scholar] [CrossRef] [Green Version]
- Yin, D.; Wang, Y.; Zhang, X.; Ma, X.; He, X.; Zhang, J. Development of chloroplast genome resources for peanut (Arachis hypogaea L.) and other species of Arachis. Sci. Rep. 2017, 7, 11649. [Google Scholar] [CrossRef] [Green Version]
- Menezes, A.P.A.; Resende-Moreira, L.C.; Buzatti, R.S.O.; Nazareno, A.G.; Carlsen, M.; Lobo, F.P.; Kalapothakis, E.; Lovato, M.B. Chloroplast genomes of Byrsonima species (Mal pighiaceae): Comparative analysis and screening of high divergence sequences. Sci. Rep. 2018, 8, 2210. [Google Scholar] [CrossRef] [Green Version]
- Mehmood, F.A.; Shahzadi, I.; Waseem, S.; Mirza, B.; Ahmed, I.; Waheed, M.T. Chloroplast genome of Hibiscus rosasinensis (Malvaceae): Comparative analyses and identifcation of mutational hotspots. Genomics 2020, 112, 581–591. [Google Scholar] [CrossRef]
- Song, Y.; Wang, S.; Ding, Y.; Xu, J.; Li, M.F.; Zhu, S.; Chen, N.J. Chloroplast Genomic Resource of Paris for Species Discrimination. Sci. Rep. 2017, 7, 3427. [Google Scholar] [CrossRef] [PubMed]
- Kim, K.J.; Lee, H.L. Complete chloroplast genome sequences from Korean ginseng (Panax schinseng Nees) and comparative analysis of sequence evolution among 17 vascular plants. DNA Res. 2004, 11, 247–261. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Su, N.; Zhang, L.; Tong, R.; Zhang, X.; Wang, J.R.; Chnag, Z.Y.; Zhao, L.; Potter, D. Chloroplast genomes elucidate diversity, phylogeny, and taxonomy of Pulsatilla (Ranunculaceae). Sci. Rep. 2020, 10, 19781. [Google Scholar] [CrossRef] [PubMed]
- Mehmood, F.; Abdullah Ubaid, Z.; Shahzadi, I.; Ahmed, I.; Waheed, M.T.; Poczai, P.; Mirza, B. Plastid genomics of Nicotiana (Solanaceae): Insights into molecular evolution, positive selection and the origin of the maternal genome of Aztec tobacco (Nicotiana rustica). PeerJ 2020, 8, e9552. [Google Scholar] [CrossRef] [PubMed]
- Cao, J.; Jiang, D.; Zhao, Z.; Yuan, S.; Zhang, Y.; Zhang, T.; Zhong Yuan, Q.; Huang, L. Development of Chloroplast Genomic Resources in Chinese Yam (Dioscorea polys tachya). Biomed. Res. Int. 2018, 2018, 6293847. [Google Scholar] [CrossRef]
- Liu, L.; Wang, Y.; He, P.; Li, P.; Lee, J.; Soltis, D.E.; Fu, C.X. Chloroplast genome analyses and genomic resource development for epilithic sister genera Oresitrophe and Mukdenia (Saxifragaceae), using genome skimming data. BMC Genom. 2018, 19, 235. [Google Scholar] [CrossRef] [Green Version]
- Kress, W.J. Plant DNA barcodes: Applications today and in the future. J. Syst. Evol. 2017, 55, 291–307. [Google Scholar] [CrossRef] [Green Version]
- Zhang, D.; Mo, X.; Xiang, J.; Zhou, N. Molecular identification of original plants of Fritillariae cirrhosae bulbus, a Tradtional Chinese Cedicine (TCM) using plant DNA barcoding. Afr. J. Tradit. Complement. Altern. Med. 2016, 13, 74–82. [Google Scholar] [CrossRef] [Green Version]
- Jansen, R.K.; Ruhlman, T.A. Plastid Genomes of Seed Plants. In Genomics of Chloroplasts and Mitochondria; Bock, R., Knoop, V., Eds.; 2012; pp. 103–126. [Google Scholar] [CrossRef]
- Parks, M.; Cronn, R.; Liston, A. Increasing phylogenetic resolution at low taxonomic levels using massively parallel sequencing of chloroplast genomes. BMC Biol. 2009, 7, 84. [Google Scholar] [CrossRef] [Green Version]
- Huang, H.; Shi, C.; Liu, Y.; Mao, S.-Y.; Gao, L.-Z. Thirteen Camellia chloroplast genome sequences determined by high-throughput sequencing: Genome structure and phylogenetic relationships. BMC Evol. Biol. 2014, 14, 151. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, J.; Gao, Y.; Wei, J.; Liu, W.; Downie, S.R. Molecular phylogenetics of Ligusticum (Apiaceae) based on nrDNA ITS sequences: Rampant polyphyly, placement of the Chinese endemic species, and a much-reduced circumscription of the genus. Int. J. Plant Sci. 2020, 181, 306–323. [Google Scholar] [CrossRef]
- Downie, S.R.; Spalik, K.; Katz-Downie, D.S.; Reduron, J.P. Major clades within Apiaceae subfamily Apioideae as inferred by phylogenetic analysis of nrDNA ITS sequences. Plant Divers Evol. 2010, 128, 111–136. [Google Scholar] [CrossRef]
- Pimenov, M.G. Updated checklist of Chinese Umbelliferae: Nomenclature, synonymy, typification, distribution. Turczaninowia 2017, 20, 106–239. [Google Scholar]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Taxon | Length (bp) | Number of Genes (Unique) | |||||||
|---|---|---|---|---|---|---|---|---|---|
| Genome | LSC | SSC | IR | Total | CDS | rRNA | tRNA | GC (%) | |
| P. Franchetii C.Y.Wu & F.T.Pu | 148,281 | 92,298 | 17,691 | 19,146 | 113 | 79 | 4 | 30 | 37.50% |
| L. brachyloba (Franch.) Leute | 148,633 | 92,265 | 17,588 | 19,390 | 113 | 79 | 4 | 30 | 37.40% |
| L. capillacea (H.Wolff) Leute | 147,808 | 91,907 | 17,503 | 19,199 | 113 | 79 | 4 | 30 | 37.50% |
| L. hispida Lavrova & Kljuykov | 147,797 | 91,846 | 17,627 | 19,162 | 113 | 79 | 4 | 30 | 37.40% |
| L. involucrata Lavrova | 147,752 | 91,782 | 17,560 | 19,205 | 113 | 79 | 4 | 30 | 37.40% |
| L. likiangensis (H.Wolff) Lavrova & Kljuykov | 148,196 | 92,305 | 17,575 | 19,158 | 113 | 79 | 4 | 30 | 37.50% |
| L. scapiformis (H.Wolff) Leute | 148,107 | 92,214 | 17,581 | 19,156 | 113 | 79 | 4 | 30 | 37.50% |
| L. rechingeriana Leute | 148,525 | 91,813 | 17,654 | 19,529 | 113 | 79 | 4 | 30 | 37.30% |
| L. modesta (Diels) Leute | 148,133 | 92,247 | 17,568 | 19,159 | 113 | 79 | 4 | 30 | 37.50% |
| L. wallichiana Fedde ex H.Wolff | 148,594 | 92,281 | 17,567 | 19,373 | 113 | 79 | 4 | 30 | 37.40% |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Song, B.; Liu, C.; Xie, D.; Xiao, Y.; Tian, R.; Li, Z.; Zhou, S.; He, X. Plastid Phylogenomic Analyses Reveal the Taxonomic Position of Peucedanum franchetii. Plants 2023, 12, 97. https://doi.org/10.3390/plants12010097
Song B, Liu C, Xie D, Xiao Y, Tian R, Li Z, Zhou S, He X. Plastid Phylogenomic Analyses Reveal the Taxonomic Position of Peucedanum franchetii. Plants. 2023; 12(1):97. https://doi.org/10.3390/plants12010097
Chicago/Turabian StyleSong, Boni, Changkun Liu, Dengfeng Xie, Yulin Xiao, Rongming Tian, Zixuan Li, Songdong Zhou, and Xingjin He. 2023. "Plastid Phylogenomic Analyses Reveal the Taxonomic Position of Peucedanum franchetii" Plants 12, no. 1: 97. https://doi.org/10.3390/plants12010097