
Basic Characteristics of Flower Transcriptome Data and Derived Novel EST-SSR Markers of Luculia yunnanensis, an Endangered Species Endemic to Yunnan, Southwestern China

 ,
,
Abstract
:1. Introduction
2. Results and Discussion
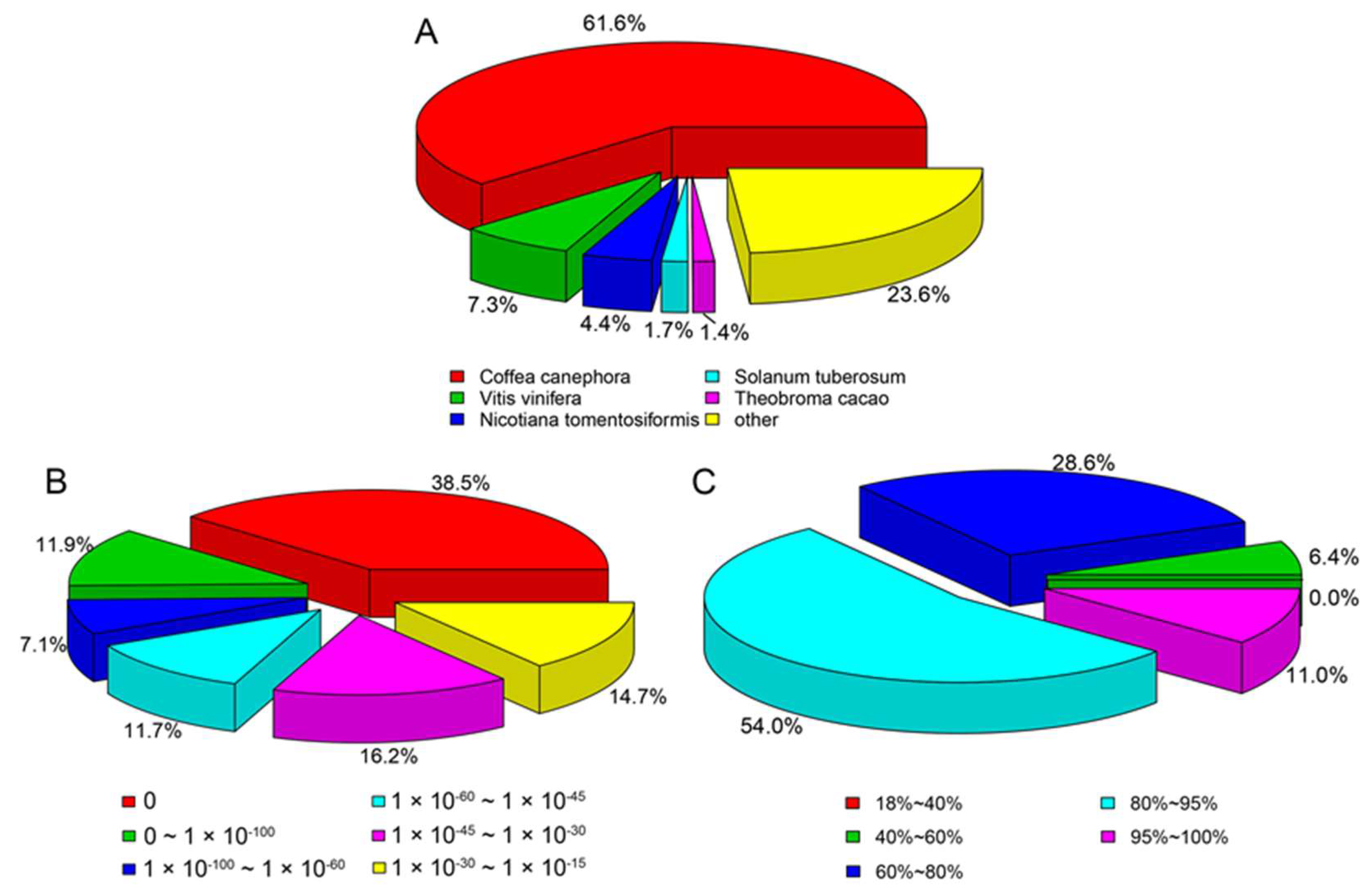
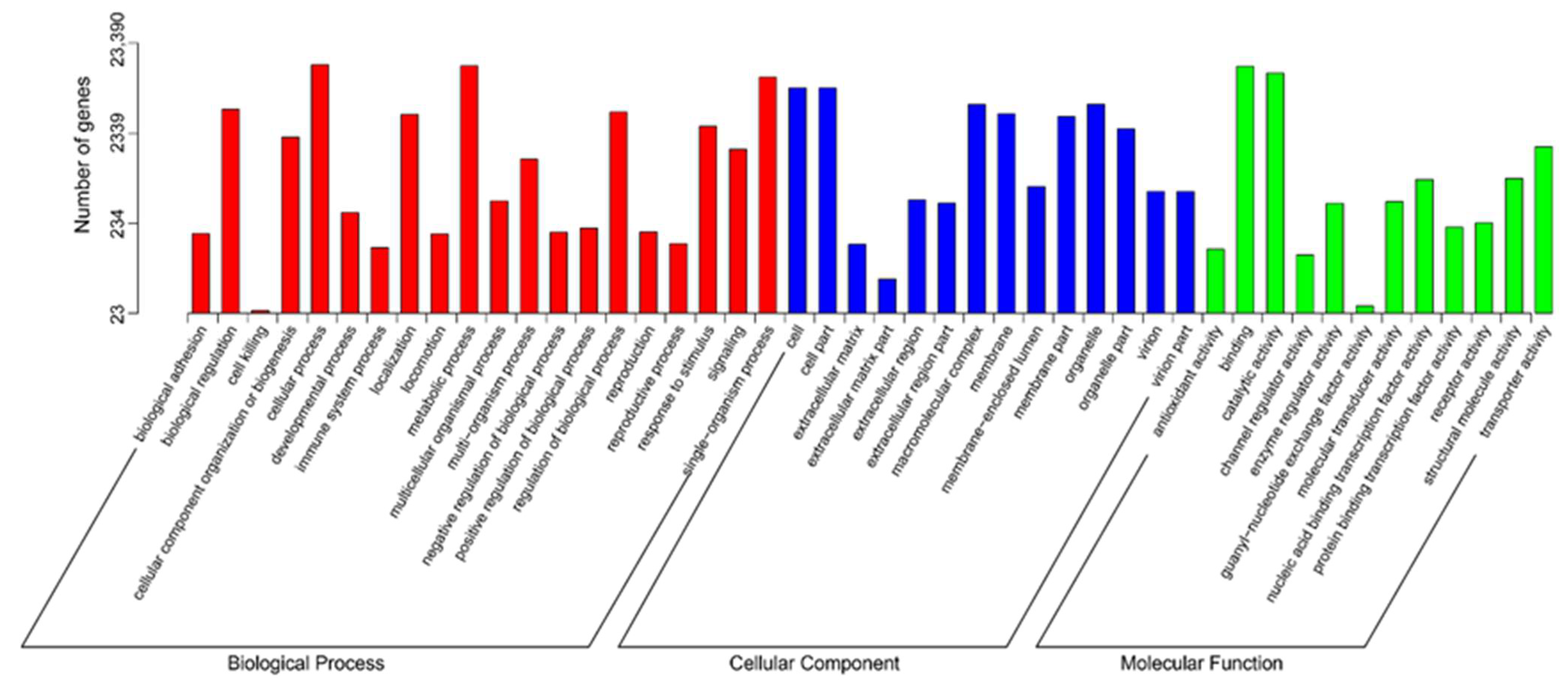
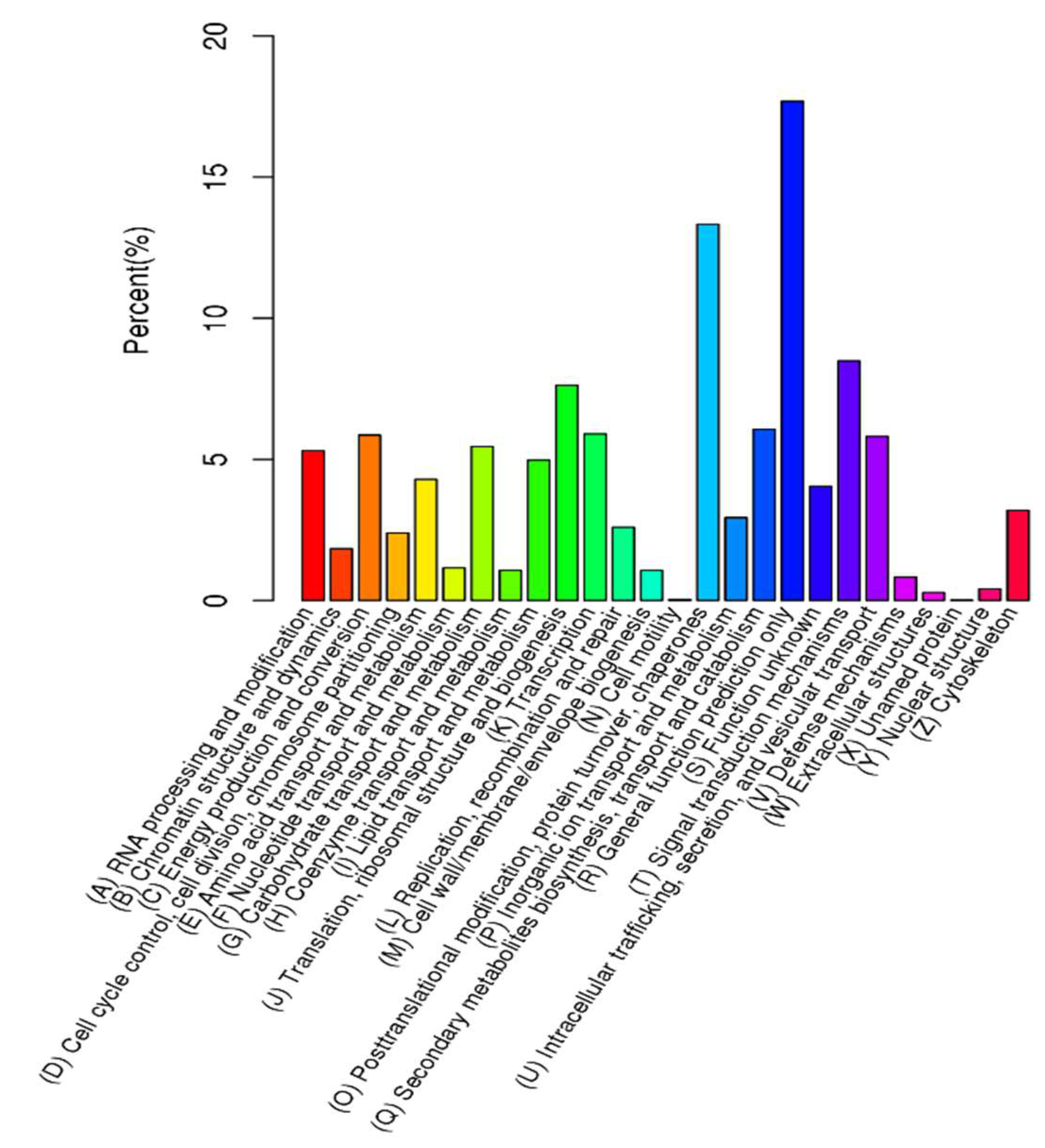
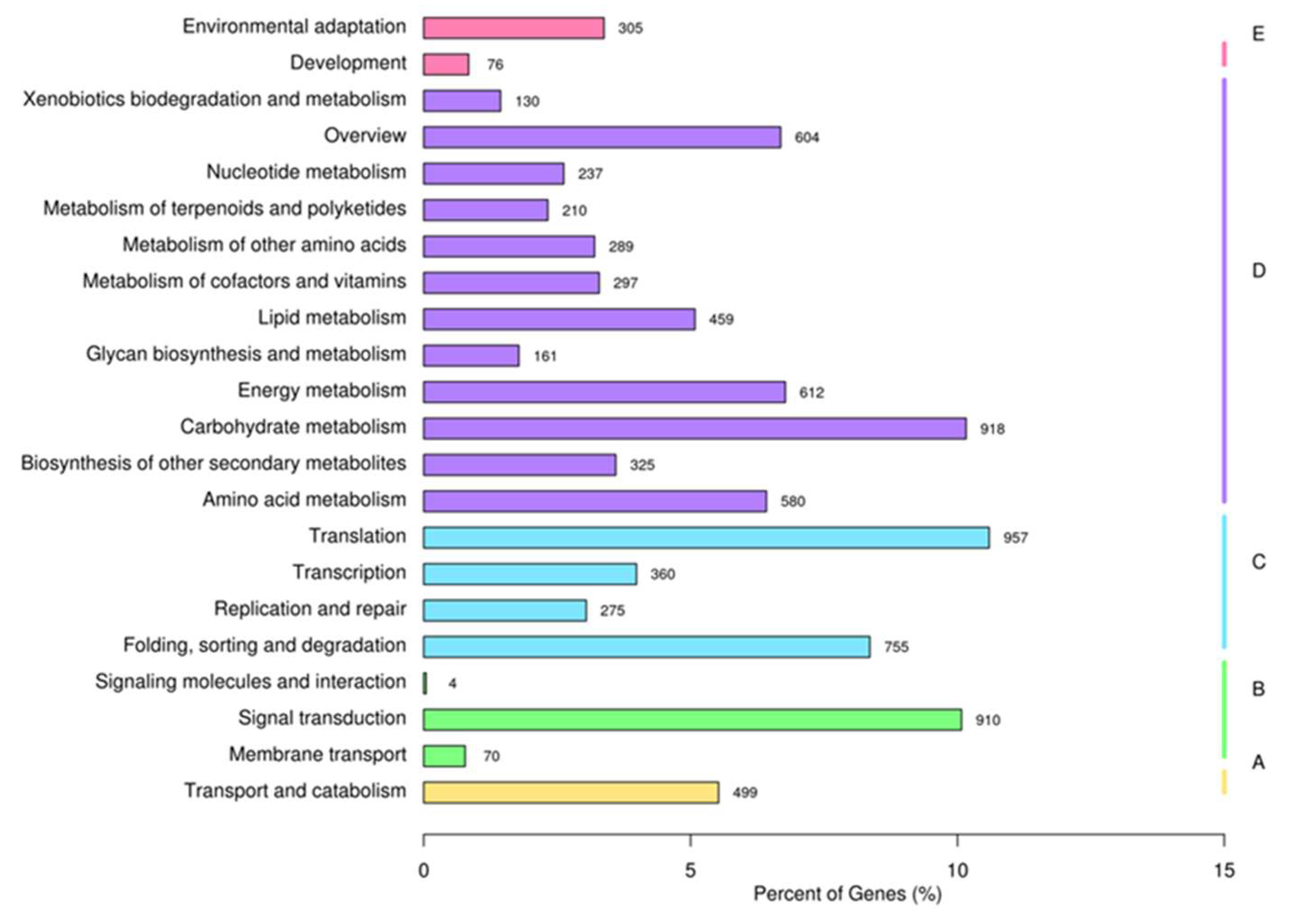
2.1. Functional Annotation and Classification
2.2. Identification of SSRs
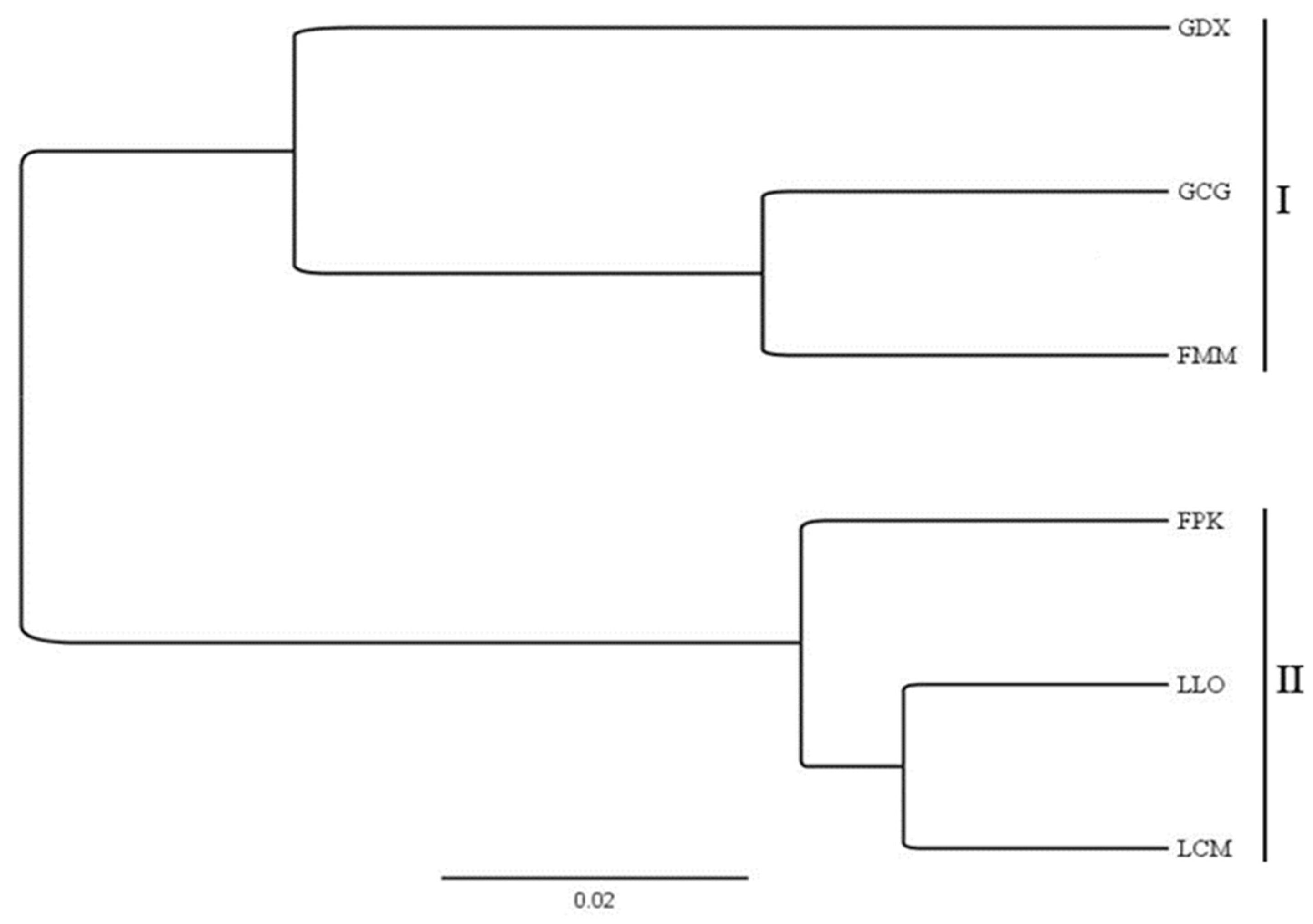
2.3. SSR Primer Screening and Verification
3. Materials and Methods
3.1. Plant Materials
3.2. De Novo Assembly, Functional Annotation, and Classification
3.3. Detection of EST-SSR Markers and Designing of Primers
3.4. DNA Extraction and Quality Check
3.5. EST-SSR Amplification and Data Analysis
4. Conclusions
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Li, Y.Y. Studies on Conservation Genetics of Chinese Endemic Species Luculia yunnanensis Hu. Doctoral Dissertation, Chinese Academy of Forestry, Beijing, China, 2017. [Google Scholar]
- Allendorf, F.; Ryman, N. The role of genetics in population viability analysis. In Population Viability Analysis; Beissinger, S.R., McCullough, D.R., Eds.; The University of Chicago Press: Chicago, IL, USA, 2000; pp. 50–85. [Google Scholar]
- Holsinger, K.E.; Gottlieb, L.D. Conservation of rare and endangered plants: Principles and prospects. In Genetics and Conservation of Rare Plants; Falk, D.A., Holsinger, K.E., Eds.; Oxford University Press: New York, NY, USA, 1991; pp. 195–208. [Google Scholar]
- Lande, R. Risks of population extinction from demographic and environmental stochasticity and random catastrophes. Am. Nat. 1993, 142, 911–927. [Google Scholar] [CrossRef] [PubMed]
- Soule, M.; Simberloff, D. What do genetics and ecology tell us about the design of nature reserves? Biol. Conserv. 1986, 35, 19–40. [Google Scholar] [CrossRef] [Green Version]
- Kaljund, K.; Jaaska, V. No loss of genetic diversity in small and isolated populations of Medicago sativa subsp. falcata. Biochem. Syst. Ecol. 2010, 38, 510–520. [Google Scholar] [CrossRef]
- Varshney, R.K.; Graner, A.; Sorrells, M.E. Genetic microsatellitemarkers in plants: Features and applications. Trends Biotechnol. 2005, 23, 48–55. [Google Scholar] [CrossRef] [PubMed]
- Kalia, R.K.; Rai, M.K.; Kalia, S.; Singh, R.; Dhawan, A.K. Microsatellite markers: An overview of the recent progress in plants. Euphytica 2011, 177, 309–334. [Google Scholar] [CrossRef]
- Liu, G.; Xie, Y.J.; Zhang, D.Q.; Chen, H.P. Analysis of SSR loci and development of SSR primers in Eucalyptus. J. For. Res. 2018, 29, 273–282. [Google Scholar] [CrossRef]
- Liu, Y.; Fang, X.M.; Tang, T.; Wang, Y.D.; Wu, Y.H.; Luo, J.Y.; Wu, H.T.; Wang, Y.Q.; Zhang, J.; Ruan, R.W.; et al. Inflorescence transcriptome sequencing and development of new EST-SSR markers in common buckwheat (Fagopyrum esculentum). Plants 2022, 11, 742. [Google Scholar] [CrossRef]
- Ellis, J.R.; Burke, J.M. EST-SSRs as a resource for population genetic analyses. Heredity 2007, 99, 125–132. [Google Scholar] [CrossRef] [Green Version]
- Yang, X.; Yang, N.; Yuan, Q.; Zhao, G.J.; Li, G.L.; Jia, L.M.; Chen, Z. Development and application of EST-SSR molecular markers in Pinus bungeana. J. Beijing For. Univ. 2021, 43, 1–11. [Google Scholar]
- Xiang, X.Y.; Zhang, Z.X.; Wang, Z.G.; Zhang, X.P.; Wu, G.L. Transcriptome sequencing and development of EST-SSR markers in Pinus dabeshanensis, an endangered conifer endemic to China. Mol. Breed. 2015, 35, 1–10. [Google Scholar] [CrossRef]
- Li, X.Y.; Guo, Q.; Dong, L.; Sun, Y.H.; Niu, D.S.; Liu, J.P.; Wang, H.S.; Li, Y. EST-SSR analysis of genetic diversity of Robinia pseudoacacia clones in Jixian county, Shanxi province of northern China. J. Beijing For. Univ. 2019, 41, 39–48. [Google Scholar]
- Dehkordi, M.K.; Beigzadeh, T.; Sorkheh, K. Novel in silico EST-SSR markers and bioinformatic approaches to detect genetic variation among peach (Prunus persica L.) germplasm. J. For. Res. 2020, 31, 1359–1370. [Google Scholar] [CrossRef]
- Wani, M.S.; Sharma, V.; Gupta, R.C.; Munshi, A.H. Development and characterization of SSR markers in Himalayan species Betula utilis. J. For. Res. 2020, 31, 1453–1460. [Google Scholar] [CrossRef]
- Zhou, X.J.; Dong, Y.; Zhao, J.J.; Huang, L.; Ren, X.P.; Chen, Y.N.; Huang, S.M.; Liao, B.S.; Lei, Y.; Yan, L.Y.; et al. Genomic survey sequencing for development and validation of single-locus SSR markers in peanut (Arachis hypogaea L.). BMC Genom. 2016, 17, 420. [Google Scholar] [CrossRef] [Green Version]
- Ma, H.; Wang, L.; Wan, Y.M.; Li, H.Z.; Li, Z.H.; Liu, X.X.; Liang, N.; Li, W.J. A set of novel microsatellite markers developed for Luculia yunnanensis (Rubiaceae), an endangered plant endemic to Yunnan, China. Int. J. Mol. Sci. 2012, 13, 534–539. [Google Scholar] [CrossRef] [PubMed]
- Ramilowski, J.A.; Sawai, S.; Seki, H.; Mochida, K.; Yoshida, T.; Sakurai, T.; Muranaka, T.; Saito, K.; Daub, C.O. Glycyrrhiza uralensis transcriptome landscape and study of phytochemicals. Plant Cell Physiol. 2013, 54, 697–710. [Google Scholar] [CrossRef] [Green Version]
- Wan, Y.M. Mechanism on photoperiodic control of flowering in Luculia gratissima ‘Xiangfei’. Doctoral Dissertation, Chinese Academy of Forestry, Beijing, China, 2019. [Google Scholar]
- Zhu, Y.H.; Zhang, M.J.; Li, L.; Zhao, L.; Dong, C.M. Transcriptome analysis of Prunella vulgaris and identification of putative genes involved in second metabolism biosynthesis. Zhong Cao Yao 2019, 50, 1220–1226. [Google Scholar]
- Chen, C.L.; Xu, M.L.; Wang, C.P.; Qiao, G.X.; Wang, W.W.; Tan, Z.Y.; Wu, T.T.; Zhang, Z.S. Characterization of the Lycium barbarum fruit transcriptome and development of EST-SSR markers. PLoS ONE 2017, 12, e0187738. [Google Scholar] [CrossRef] [Green Version]
- Xu, Q.W.; Lou, X.Z.; Yang, B.; Lin, E.P.; Tong, Z.K. Transcriptome sequencing and analysis of Rhododendron fortune. J. Zhejiang AF Univ. 2019, 36, 1190–1198. [Google Scholar]
- Liu, Y.L.; Li, S.Q.; Wang, Y.Y.; Liu, P.Y.; Han, W.J. De novo assembly of the seed transcriptome and search for potential EST-SSR markers for an endangered, economically important tree species: Elaeagnus mollis Diels. J. For. Res. 2020, 31, 69–77. [Google Scholar] [CrossRef]
- Zhang, L.; Fan, X.M.; Lin, Q.; Xiang, H.; Wang, W.; Yuan, D.Y. Transcriptome Analysis for developing kernel and expression analysis of starch and sucrose metabolism-related genes in Castanea henryi. J. Plant Genet. Resour. 2015, 16, 603–611. [Google Scholar]
- Cai, N.H.; Deng, L.L.; Xu, Y.L.; Xu, Y.; Zhou, L.; Wang, D.W.; Tian, B.; He, C.Z.; Duan, A.A. Transcriptome analysis for Pinus yunnanensis based on high throughput sequencing. Bull. Bot. Res. 2016, 36, 75–83. [Google Scholar]
- Li, T.Q.; Liu, X.F.; Wan, Y.M.; Li, Z.H.; Qi, G.H.; Li, Y.Y.; Liu, X.X.; He, R.; Ma, Y.; Ma, H. Transcriptome analysis for Rhododendron longipedicellatum (plant species with extremely small populations) based on high throughput sequencing. Bull. Bot. Res. 2017, 37, 825–834. [Google Scholar]
- Liu, X.F.; Li, T.Q.; Li, Z.H.; Wan, Y.M.; Liu, X.X.; Zhang, X.; An, J.; Ma, H. Transcriptome analysis for Phyllanthus emblica distributed in dry-hot valleys in Yunnan, China. Fore. Res. 2018, 31, 1–8. [Google Scholar]
- Xia, L.F.; Zhu, X.Z.; Liang, M.Z.; Ma, W.; Song, W.X.; Tian, Y.P.; Zhou, M.; Chen, L.B. Transcriptome analysis of flower from tea plant [Camellia sinensis (L.) O. Kuntz] via RNA-Seq technology. Southwest China J. Agric. Sci. 2016, 29, 2058–2062. [Google Scholar]
- Biswas, M.K.; Chai, L.J.; Mayer, C.; Xu, Q.; Guo, W.W.; Deng, X.X. Exploiting BAC−end sequences for the mining, characterization and utility of new short sequences repeat (SSR) markers in Citrus. Mol. Biol. Rep. 2012, 39, 5373–5386. [Google Scholar] [CrossRef] [PubMed]
- Zhong, H.Q.; Lin, R.Y.; Lin, B.; Wu, J.S.; Fan, R.H.; Ye, X.X. Analysis on SSR information in transcriptome and development of EST-SSR markers for hybrid cymbidium. Chin. J. Cell. Biol. 2020, 42, 286–295. [Google Scholar]
- Zhao, T.; Chang, S.X.; Leng, Q.Y.; Xu, S.S.; Yin, J.M.; Niu, J.H. Development of SSR molecular markers based on transcriptome sequencing of Bougainvillea. Mol. Plant Breed. 2019, 17, 4331–4341. [Google Scholar]
- Wang, H.; Xie, Y.; Sun, Z.C.; Gao, Y.J.; Zhang, D.H.; Song, Y.X.; Gao, Y.X. Analysis on SSR locis in mulberry fruit transcriptome. Mol. Plant Breed. 2020, 18, 208–215. [Google Scholar]
- Wu, J.; Cai, C.; Cheng, F.; Cui, H.; Zhou, H. Characterisation and development of EST-SSR markers in tree peony using transcriptome sequences. Mol. Breed. 2014, 34, 1853–1866. [Google Scholar] [CrossRef]
- Yi, M.; Zhang, L.; Lei, L.; Cheng, Z.S.; Sun, S.W.; Lai, M. Analysis of SSR information in transcriptome and development of EST-SSR molecular markers in Pinus elliottii Engelm. J. Nanjing. For. Univ. 2020, 44, 75–83. [Google Scholar]
- Feng, S.; He, R.; Lu, J.; Jiang, M.; Shen, X.; Yan, J.; Wang, Z.; Wang, H. Development of SSR markers and assessment of genetic diversity in medicinal Chrysanthemum morifolium cultivars. Front. Genet. 2016, 7, 113. [Google Scholar] [CrossRef] [PubMed]
- Kumpatal, S.P.; Mukhopadhyay, S. Mining and survey of simple sequence repeats in expressed sequence tags of dicotyledonous species. Genome 2005, 48, 985–998. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Li, B.; Guo, S.X. SSR information in transcriptome of Dendrobium nobile. China J. Chin. Mater. Med. 2017, 42, 63–69. [Google Scholar]
- Zou, M.L.; Xia, Z.Q.; Chen, J.R.; Wang, W.Q. Analysis of SSR information and annotation in EST resource of Caster Bean (Ricinus communis). Chin. J. Trop. Crop. 2012, 33, 2138–2143. [Google Scholar]
- Liu, Y.L.; Zhang, P.F.; Song, M.L.; Hou, J.L.; Mei, Q.; Wang, W.Q.; Liu, C.S. Transcriptome analysis and development of SSR molecular markers in Glycyrrhiza uralensis Fisch. PLoS ONE 2015, 10, e0143017. [Google Scholar] [CrossRef] [PubMed]
- Zhou, W.; Wang, H.; Li, D.Z.; Yang, J.B.; Zhou, W. Isolation and characterization of 13 microsatellite loci from Luculia pinceana (Rubiaceae), a typical distylous species. Hortscience 2010, 45, 840–841. [Google Scholar] [CrossRef]
- Temnykh, S.; De Clerck, G.; Lukashova, A.; Lipovich, L.; Cartinhour, S.; Mc Couch, S. Computational and experimental analysis of microsatellites in rice (Oryza sativa L.): Frequency, length variation, transposon associations, and genetic marker potential. Genome Res. 2001, 11, 1441–1452. [Google Scholar] [CrossRef] [Green Version]
- Dreisigacker, S.; Zhang, P.; Warburton, M.L.; Van Ginkel, M.; Hoisington, D.; Bohn, M.; Melchinger, A.E. SSR and pedigree analyses of genetic diversity among CIMMYT wheat lines targeted to different megaenvironments. Crop. Sci. 2004, 44, 381–388. [Google Scholar] [CrossRef]
- Liu, S.S.; Qiao, Z.Q.; Zeng, H.J.; Li, Y.X.; Cai, N.; Liu, X.M.; Wang, X.M. Analysis on SSR loci in transcriptome and development of EST-SSR molecular markers in Lonicera macranthoides. Mol. Plant Breed. 2021, 19, 3015–3021. [Google Scholar]
- Botstein, D.; White, R.L.; Skolnick, M.; Davis, R.W. Construction of a genetic linkage map in man using restriction fragment length polymorphisms. Am. J. Hum. Genet. 1980, 32, 314–331. [Google Scholar] [PubMed]
- Li, D.Z.; Yang, X.Y.; Hugh, W.P. Problems and challenges in germplasm preservation. Bull. Chin. Acad. Sci. 2010, 25, 533–540. [Google Scholar]
- Ding, H.X. Development and Application of SSR Markers in Chrysanthemum Based on Full-Length Transcriptome Sequence. Master Dissertation, Henan University, Kaifeng, China, 2020. [Google Scholar]
- Pertea, G.; Huang, X.Q.; Liang, F.; Antonescu, V.; Sultana, R.; Karamycheva, S.; Lee, Y.D.; White, J.; Cheung, F.; Parvizi, B.; et al. TIGR Gene Indices clustering tools (TGICL): A software system for fast clustering of large EST datasets. Bioinformatics 2003, 19, 651–652. [Google Scholar] [CrossRef] [Green Version]
- Grabherr, G.M.; Haas, B.J.; Yassour, M.; Levin, J.Z.; Thompson, D.A.; Amit, I.; Adiconis, X.; Fan, L.; Raychowdhury, R.; Zeng, Q.D.; et al. Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nat. Biotechnol. 2011, 29, 644–652. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Altschul, S.F.; Gish, W.; Miller, W.; Myers, E.W.; Lipman, D.J. Basic local alignment search tool. J. Mol. Biol. 1990, 215, 403–410. [Google Scholar] [CrossRef]
- Moriya, Y.; Itoh, M.; Okuda, S.; Yoshizawa, A.C.; Kanehisa, M. KAAS: An automatic genome annotation and pathway reconstruction server. Nucleic. Acids. Res. 2007, 35, W182–W185. [Google Scholar] [CrossRef] [Green Version]
- Götz, S.; García-Gómez, J.M.; Terol, J.; Williams, T.D.; Nagaraj, S.H.; Nueda, M.J.; Robles, M.; Talón, M.; Dopazo, J.; Conesa, A. High-throughput functional annotation and data mining with the Blast2GO suite. Nucleic. Acids. Res. 2008, 36, 3420–3435. [Google Scholar] [CrossRef]
- Beier, S.; Thiel, T.; Munch, T.; Scholz, U.; Mascher, M. MISA-web: A web server for microsatellite prediction. Bioinformatics 2017, 33, 2583–2585. [Google Scholar] [CrossRef] [Green Version]
- Rozen, S.; Skaletsky, H. Primer3 on the WWW for general users and for biologist programmers. Methods Mol. Biol. 2000, 132, 365–386. [Google Scholar]
- Yeh, F.C.; Yang, R.C.; Boyle, T.; Ye, Z.H.; Mao, J.X. POPGENE, the User-Friendly Shareware for Population Genetic Analysis; Molecular Biology and Biotechnology Centre, University of Alberta: Edmonton, AB, Canada, 1997; Volume 10, pp. 295–301. [Google Scholar]
- Kalinowski, S.T.; Taper, M.L.; Marshall, T.C. Revising how the computer program CERVUS accommodates genotyping error increases success in paternity assignment. Mol. Ecol. 2007, 16, 1099–1106. [Google Scholar] [CrossRef]
- Populations 1.2.30: Population Genetic Software (Individuals or Populations Distances, Phylogenetic Trees). Available online: http://bioinformatics.org/~tryphon/populations/ (accessed on 13 April 2022).
- Letunic, I.; Bork, P. Interactive tree of life (iTOL) v3: An online tool for the display and annotation of phylogenetic and other trees. Nucleic Acids Res. 2016, 44, W242–W245. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Transcript Length Interval | 200–500 bp | 500–1 kbp | 1 k–2 kbp | >2 kbp | Total |
|---|---|---|---|---|---|
| Number of transcripts | 71,181 (50.83%) | 25,812 (18.43%) | 23,641 (16.88%) | 19,408 (13.86%) | 140,042 |
| Number of unigenes | 62,121 (63.14%) | 17,747 (18.04%) | 10,870 (11.05%) | 7651 (7.78%) | 98,389 |
| Annotation | Number of Annotated Unigenes | Percentage of Annotated Unigenes (%) | Percentage of Total Unigenes (%) |
|---|---|---|---|
| Annotated in Nr | 31,859 | 32.38 | 22.75 |
| Annotated in Nt | 13,853 | 14.07 | 9.89 |
| Annotated in KO | 9722 | 9.88 | 6.94 |
| Annotated in Swiss-Prot | 21,416 | 21.76 | 15.29 |
| Annotated in Pfam | 22,684 | 23.05 | 16.20 |
| Annotated in GO | 23,390 | 23.77 | 16.70 |
| Annotated in KOG | 10,947 | 11.12 | 7.82 |
| Annotated in all databases | 4273 | 4.34 | 3.05 |
| Annotated in at least one database | 36,497 | 37.09 | 26.06 |
| Total annotated unigenes | 98,389 | 100 | 70.26 |
| Repeat Types | No. | Frequency (%) | Maximum Repeat Motif (Number and %) |
|---|---|---|---|
| Mononucleotide repeat (p1) | 8889 | 60.29 | A/T (8816, 99.18%) |
| Dinucleotide repeat (p2) | 3340 | 22.65 | AG/CT (2174, 65.09%) |
| Trinucleotide repeat (p3) | 2309 | 15.66 | AAG/CTT (428, 18.54%) |
| Tetranucleotide repeat (p4) | 158 | 1.07 | AAAG/CTTT (22, 13.92%) |
| Pentanucleotide repeat (p5) | 23 | 0.16 | CCTTC/GAAGG (2, 8.70%) GAGAC/GTCTC (2, 8.70%) |
| Hexanucleotide repeat (p6) | 25 | 0.16 | CCTTC/GAAGG (2, 8.70%) GAGAC/GTCTC (2, 8.70%) |
| Repeat Types | Repeat Times | ||||||
|---|---|---|---|---|---|---|---|
| 5 | 6 | 7 | 8 | 9 | 10 | >10 | |
| Dinucleotide repeat | — | 965 | 688 | 558 | 591 | 417 | 121 |
| Trinucleotide repeat | 1303 | 604 | 374 | 23 | 5 | — | |
| Tetranucleotide repeat | 132 | 25 | — | — | — | 1 | — |
| Pentanucleotide repeat | 14 | 6 | 2 | — | 1 | — | — |
| Hexanucleotide repeat | 7 | 9 | 5 | 4 | — | — | — |
| Primer ID | Primer Sequence (5′-3′) | Repeat Motif | Ta (°C) | Size (bp) | Na | Ne | I | PIC |
|---|---|---|---|---|---|---|---|---|
| N3 | CAAATTCGCGCACCAAAACG | (TGGCGT)5 | 52 | 261–279 | 4 | 2.538 | 1.061 | 0.542 |
| GCTAGAGAGAAAAGGGGCCG | ||||||||
| N6 | ACTGCGTACCTCTCCCTCTT | (TAATT)5 | 58 | 212–228 | 5 | 1.933 | 0.890 | 0.426 |
| TCTCTCTCTCTCGGACGGAC | ||||||||
| N9 | GACCCCAAGTTGGCTGATCA | (TGTTAC)8 | 60 | 127–187 | 6 | 2.212 | 0.923 | 0.446 |
| AGGGCACTTCTGTCATTTCGA | ||||||||
| N10 | CTGGTGCACGAGGATTGAGT | (TCAATT)7 | 60 | 194–224 | 2 | 1.427 | 0.476 | 0.255 |
| GAAGAGTGCCATGGAAACTGC | ||||||||
| Z13 | CCTCCCATAGCAGCAGCAAT | (ACC)5 | 54 | 117–132 | 5 | 1.699 | 0.833 | 0.382 |
| AGTAGTATTAATAATGGCTGGAGGT | ||||||||
| N22 | CGCTTCTGTGTTCGAAACCA | (ACAAC)7 | 60 | 165–180 | 4 | 2.575 | 1.091 | 0.549 |
| CAAAGCTTCCCGTCAACAGC | ||||||||
| N24 | CCCACCGAGCAATACCCAAA | (GAAA)5 | 56 | 268–280 | 4 | 1.344 | 0.555 | 0.245 |
| ACCTTCTCTGTACTCTGCCT | ||||||||
| Z31 | GCAATCCTACTCGTGCTGGT | (GGC)6 | 54 | 228–240 | 5 | 2.583 | 1.239 | 0.580 |
| AGCCAAGACTCGGCAGAAAA | ||||||||
| Z32 | TGCACTCCATAAAAGAAGAAAACACA | (TATT)5 | 53 | 114–122 | 3 | 1.405 | 0.544 | 0.264 |
| TGCAGTAACTTCGTGCCCTT | ||||||||
| Z33 | CCCAACCCACCACACAAGT | (TCT)6 | 54 | 255–270 | 5 | 1.749 | 0.887 | 0.404 |
| AGAGAGGAGGATCGAGGACG | ||||||||
| Z36 | TCGGGTCCTAGGGCTTTCTT | (CTTT)5 | 54 | 211–219 | 3 | 1.766 | 0.768 | 0.390 |
| GGCCCTCCTTGAGCATTGAT | ||||||||
| Z38 | ACCCAAGGAACTCTGTCTCT | (AAT)6 | 53 | 109–118 | 4 | 1.522 | 0.684 | 0.321 |
| ACACTTTCGTCGTCCTTAGGT | ||||||||
| N41 | GCCAGAAGGATAGCTTTCGC | (TCCT)5 | 53 | 197–221 | 6 | 1.298 | 0.547 | 0.223 |
| GGTTTGTGGTGGTTTTTGGGA | ||||||||
| Z48 | AGGAAGGGCTTGTTTTTAAGGT | (AG)8 | 52 | 215–225 | 6 | 1.724 | 0.914 | 0.400 |
| GAGCCAATGACGATCCAGCT | ||||||||
| Z50 | TCTGCTGCATCCAATGTACTGT | (GT)8 | 54 | 144–154 | 5 | 2.690 | 1.205 | 0.576 |
| CCTGCCATAGGTGCCCATTT | ||||||||
| Z54 | AGTAAGTGGGTGGAGGTGGT | (TTGA)5 | 59 | 212–216 | 2 | 1.910 | 0.670 | 0.363 |
| AGGGGCTGATTCTCTAGCGA | ||||||||
| X70 | AGCTGGAAACTAAAGGTGGAGG | (ATC)5 | 58.5 | 244–250 | 3 | 2.355 | 0.944 | 0.493 |
| CTCAGTCTGTCAGGCCTGTG |
| Code | GDX | GCG | FMM | FPK | LLO | LCM |
|---|---|---|---|---|---|---|
| Location | Dulongjiang township, Gongshan County | Cikai town, Gongshan County | Maji township, Fugong County | Pihe township, Fugong County | Luobenzhuo township, Lushui County | Chenggan township, Lushui County |
| Altitude (m) | 1324–1788 | 1325–1778 | 1354–1455 | 1738–1752 | 2029–2153 | 1664–1736 |
| Longitude and latitude (°) | 98.3188 −98.3466 /27.6915 −27.9160 | 98.6088 −98.8334 /27.5254 −27.7472 | 98.8388 −98.8453 /27.3632 −27.3651 | 98.9102 −98.9106 /26.4799 −27.4801 | 98.8116 −98.8172 /26.4780 −26.4812 | 98.8253 −98.8312 /26.3540 −26.3563 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, Y.; Liu, X.; Li, Y.; Liu, X.; Ma, H.; Qu, S.; Li, Z. Basic Characteristics of Flower Transcriptome Data and Derived Novel EST-SSR Markers of Luculia yunnanensis, an Endangered Species Endemic to Yunnan, Southwestern China. Plants 2022, 11, 1204. https://doi.org/10.3390/plants11091204
Zhang Y, Liu X, Li Y, Liu X, Ma H, Qu S, Li Z. Basic Characteristics of Flower Transcriptome Data and Derived Novel EST-SSR Markers of Luculia yunnanensis, an Endangered Species Endemic to Yunnan, Southwestern China. Plants. 2022; 11(9):1204. https://doi.org/10.3390/plants11091204
Chicago/Turabian StyleZhang, Yao, Xi Liu, Yuying Li, Xiongfang Liu, Hong Ma, Suping Qu, and Zhenghong Li. 2022. "Basic Characteristics of Flower Transcriptome Data and Derived Novel EST-SSR Markers of Luculia yunnanensis, an Endangered Species Endemic to Yunnan, Southwestern China" Plants 11, no. 9: 1204. https://doi.org/10.3390/plants11091204