
Insights into the Genomic Architecture of Seed and Pod Quality Traits in the U.S. Peanut Mini-Core Diversity Panel


Abstract
:1. Introduction
2. Materials and Methods
2.1. Plant Materials and Phenotyping for Seed Quality and Germination
2.2. Assessment for Seed Dormancy and Germination
2.3. Evaluation for Seed Grading
2.4. SNP Genotyping Using SNP Array
2.5. Data Analysis
2.6. Candidate Gene Search
3. Results
3.1. Correlation and Distribution of Dormancy and Seed Quality Traits
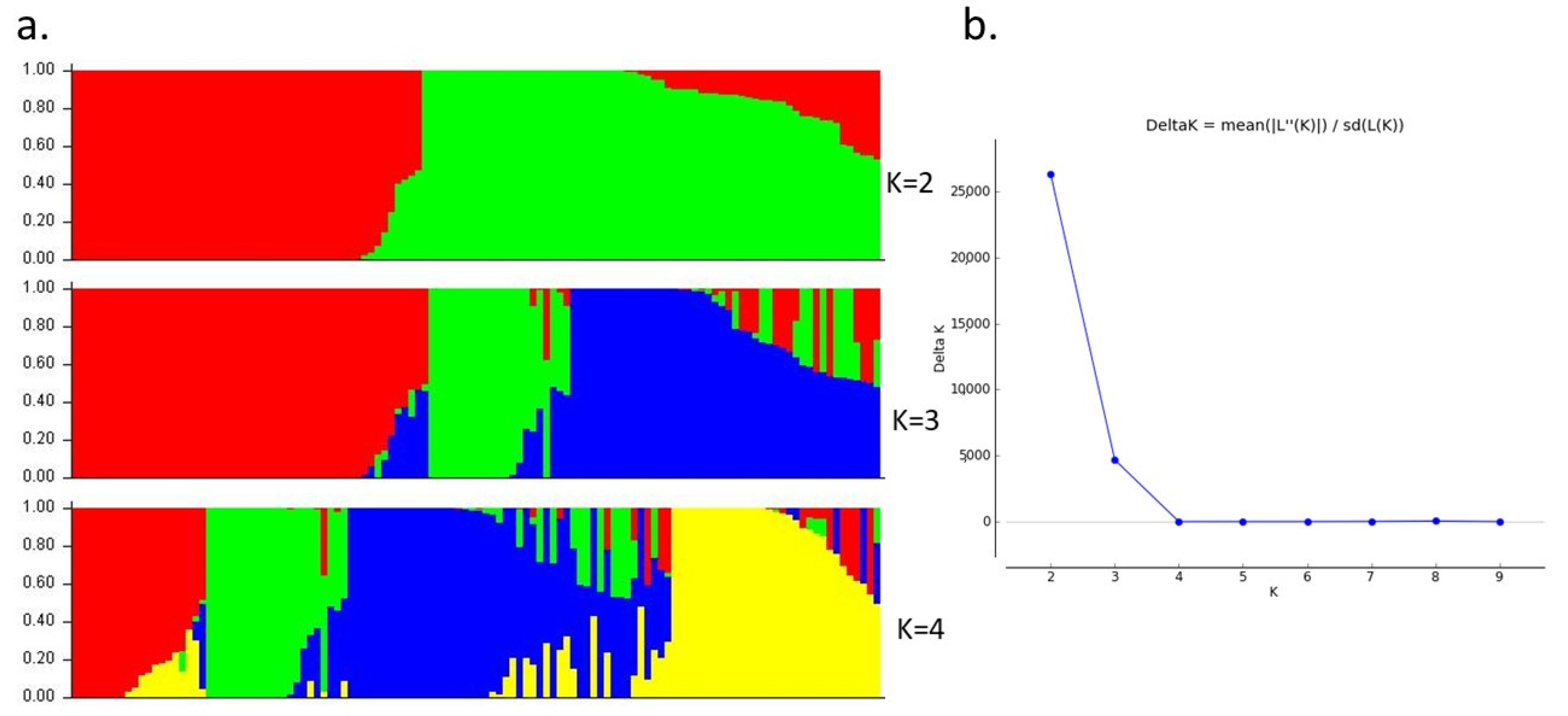
3.2. Distribution of SNP Markers, Linkage Disequilibrium, and Population Structure
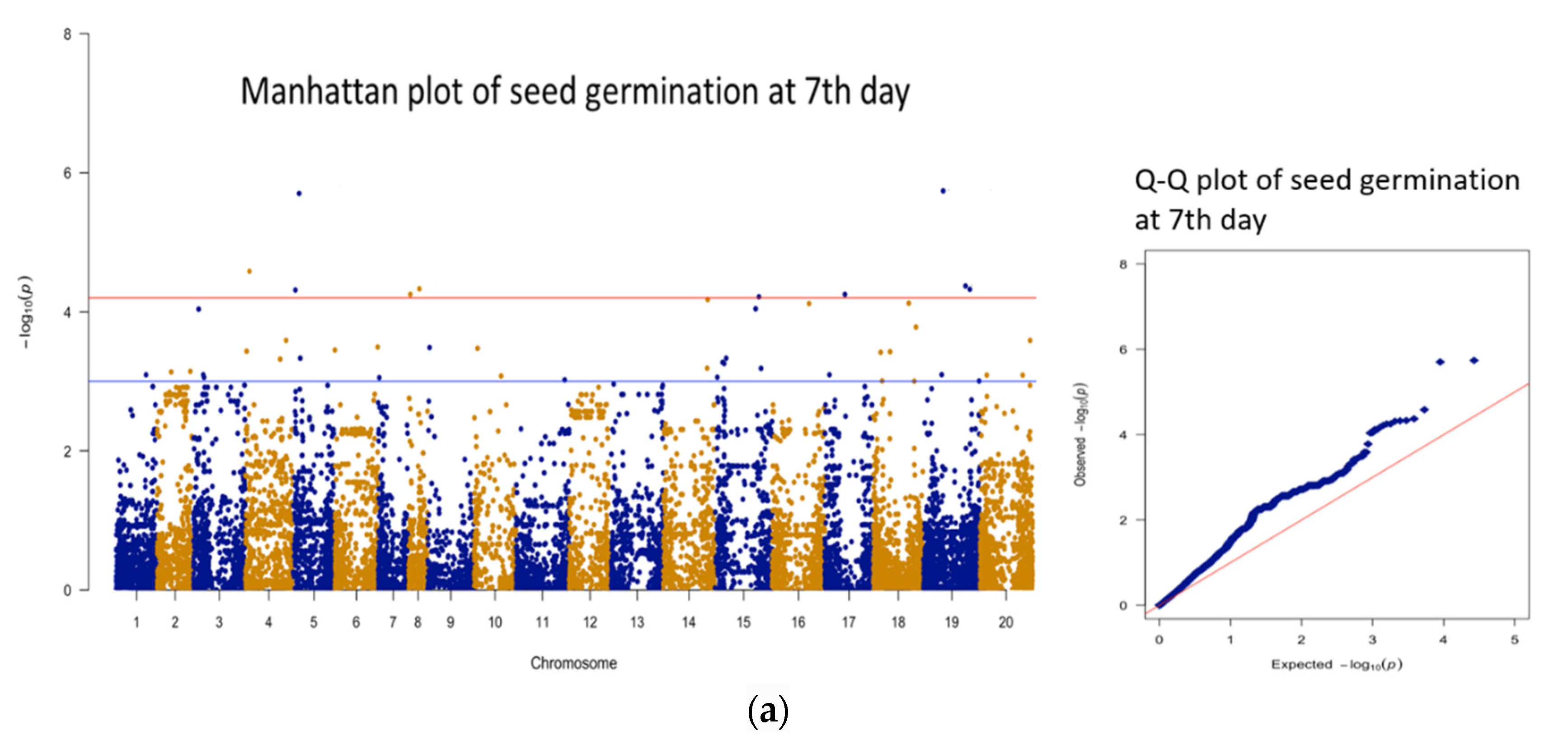
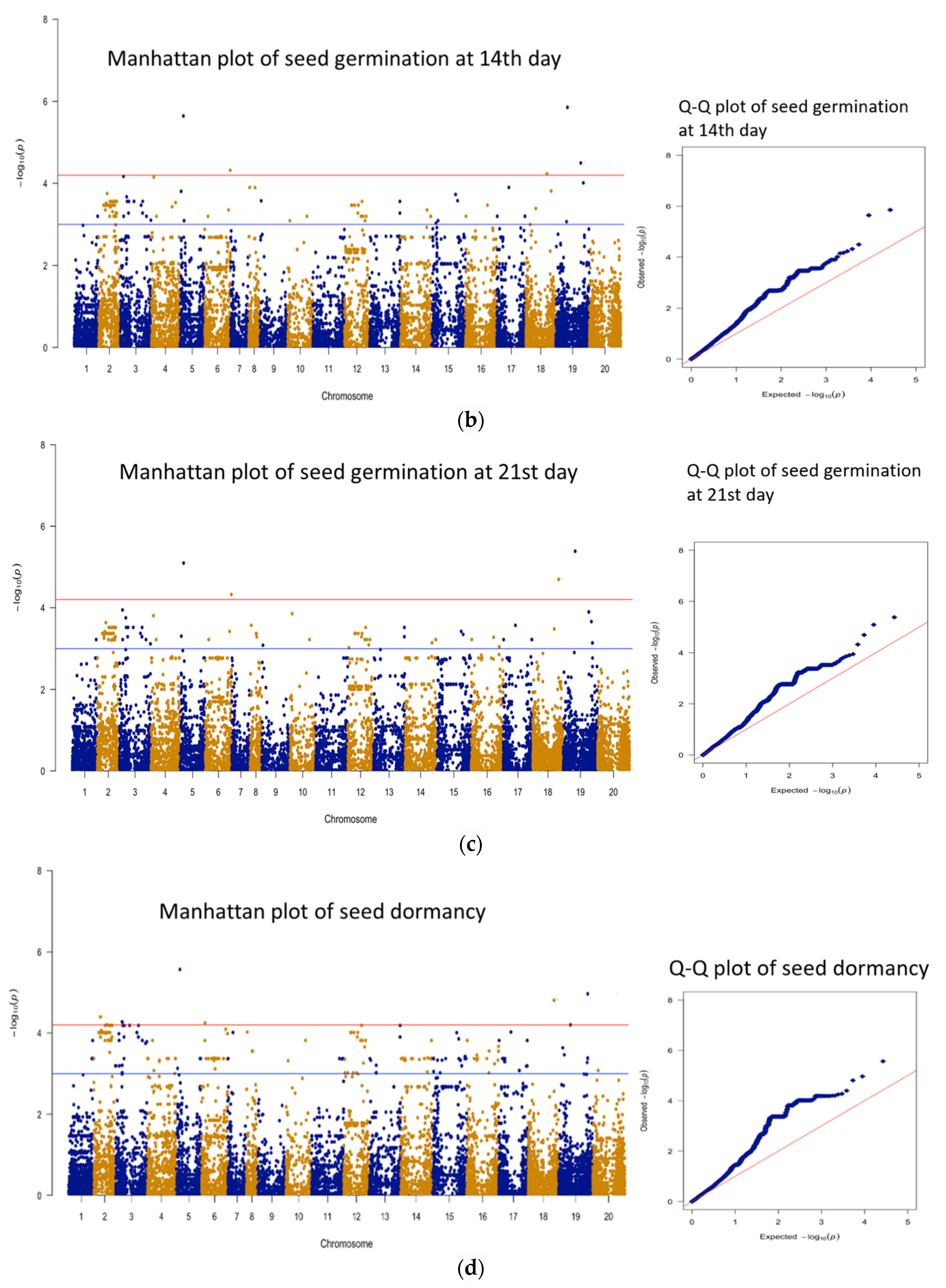
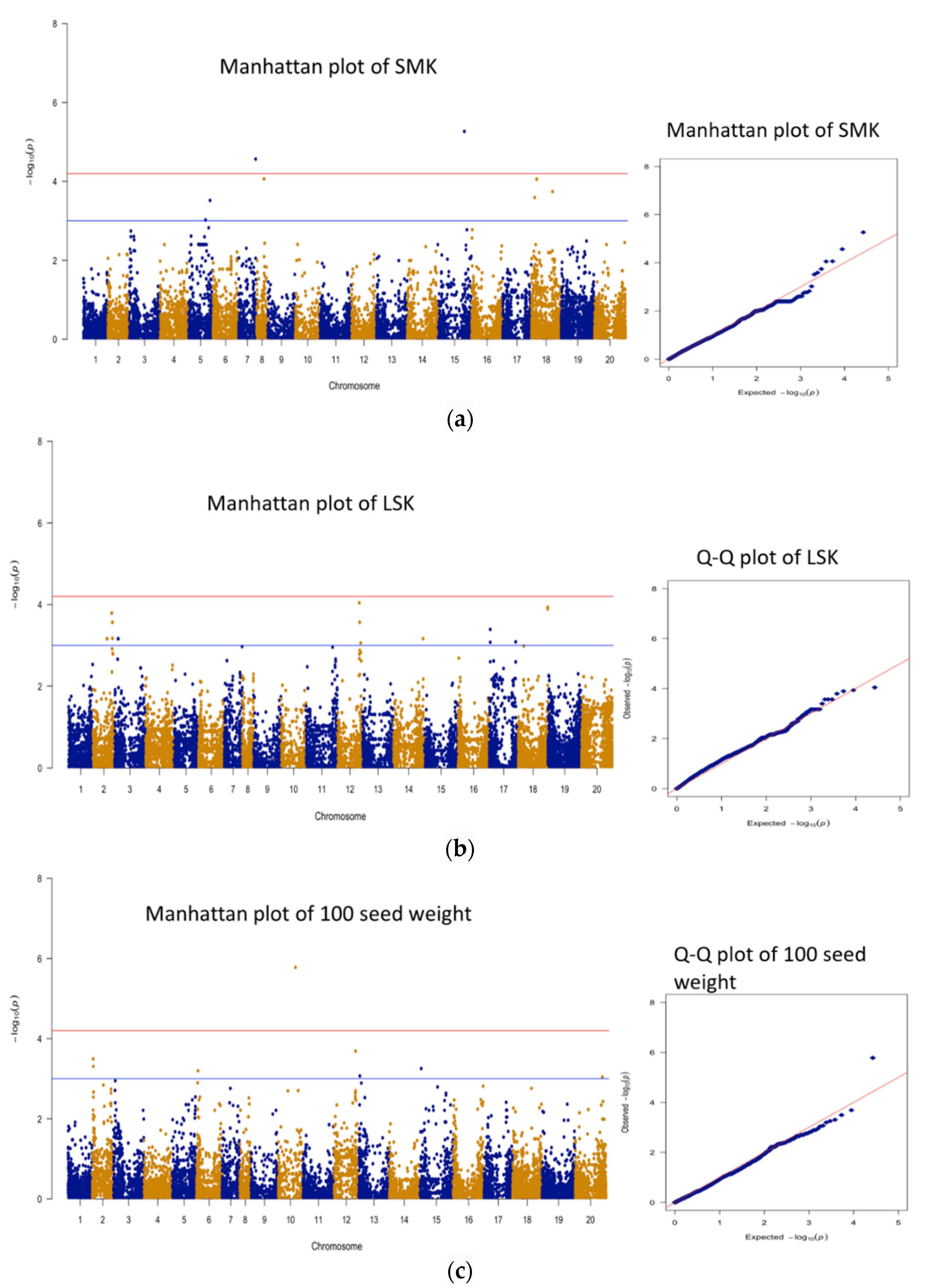
3.3. Genomic Regions Associated with Quantitative Traits and Perusal for Candidate Genes
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
References
- Yin, D.; Ji, C.; Ma, X.; Li, H.; Zhang, W.; Li, S.; Liu, F.; Zhao, K.; Li, F.; Li, K.; et al. Genome of an allotetraploid wild peanut Arachis monticola: A de novo assembly. Gigascience 2018, 7, giy066. [Google Scholar] [CrossRef]
- Seijo, G.; Lavia, G.I.; Fernandez, A.; Krapovickas, A.; Ducasse, D.A.; Bertioli, D.J.; Moscone, E.A. Genomic relationships between the cultivated peanut (Arachis hypogaea, Leguminosae) and its close relatives revealed by double GISH. Am. J. Bot. 2007, 94, 1963–1971. [Google Scholar] [CrossRef] [PubMed]
- Bertioli, D.J.; Jenkins, J.; Clevenger, J.; Dudchenko, O.; Gao, D.; Seijo, G.; Leal-Bertioli, S.C.M.; Ren, L.; Farmer, A.D.; Pandey, M.K.; et al. The genome sequence of segmental allotetraploid peanut Arachis hypogaea. Nat. Genet. 2019, 51, 877–884. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Savage, G.P.; Keenan, J.I. The composition and nutritive value of groundnut kernels. In The Groundnut Crop; Springer: Berlin/Heidelberg, Germany, 1994; pp. 173–213. [Google Scholar]
- Shirasawa, K.; Koilkonda, P.; Aoki, K.; Hirakawa, H.; Tabata, S.; Watanabe, M.; Hasegawa, M.; Kiyoshima, H.; Suzuki, S.; Kuwata, C.; et al. In silico polymorphism analysis for the development of simple sequence repeat and transposon markers and construction of linkage map in cultivated peanut. BMC Plant Biol. 2012, 12, 80. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, W.; Jiao, Y.; Cheng, L.; Huang, L.; Liao, B.; Tang, M.; Ren, X.; Zhou, X.; Chen, Y.; Jiang, H. Quantitative trait locus analysis for pod- and kernel-related traits in the cultivated peanut (Arachis hypogaea L.). BMC Genet. 2016, 17, 25. [Google Scholar] [CrossRef] [Green Version]
- Zhang, S.; Hu, X.; Miao, H.; Chu, Y.; Cui, F.; Yang, W.; Wang, C.; Shen, Y.; Xu, T.; Zhao, L.; et al. QTL identification for seed weight and size based on a high-density SLAF-seq genetic map in peanut (Arachis hypogaea L.). BMC Plant Biol. 2019, 19, 537. [Google Scholar] [CrossRef]
- Li, C.; Ni, P.; Francki, M.; Hunter, A.; Zhang, Y.; Schibeci, D.; Li, H.; Tarr, A.; Wang, J.; Cakir, M.; et al. Genes controlling seed dormancy and pre-harvest sprouting in a rice-wheat-barley comparison. Funct. Integr. Genom. 2004, 4, 84–93. [Google Scholar] [CrossRef]
- Fang, J.; Chu, C. Abscisic acid and the pre-harvest sprouting in cereals. Plant Signal. Behav. 2008, 3, 1046–1048. [Google Scholar] [CrossRef] [Green Version]
- Vishwakarma, M.K.; Pandey, M.K.; Shasidhar, Y.; Manohar, S.S.; Nagesh, P.; Janila, P.; Varshney, R.K. Identification of two major quantitative trait locus for fresh seed dormancy using the diversity arrays technology and diversity arrays technology-seq based genetic map in Spanish-type peanuts. Plant Breed. 2016, 135, 367–375. [Google Scholar] [CrossRef] [Green Version]
- Wang, M.; Chen, C.; Pinnow, D.; Barkley, N.; Pittman, R.; Lamb, M.; Pederson, G. Seed dormancy variability in the US peanut mini-core collection. Res. J. Seed. Sci. 2012, 5, 84–95. [Google Scholar] [CrossRef] [Green Version]
- Hu, B.; Wan, X.R.; Liu, X.H.; Guo, D.L.; Li, L. Abscisic acid (ABA)-mediated inhibition of seed germination involves a positive feedback regulation of ABA biosynthesis in Arachis hypogaea L. Afr. J. Biotechnol. 2010, 9, 1578–1586. [Google Scholar]
- Issa, F.; Danièl, F.; Jean-François, R.; Hodo-Abolo, T.; Ndoye, S.M.; Tahir, D.A.; Ousmane, N. Inheritance of fresh seed dormancy in Spanish-type peanut (Arachis hypogaea L.): Bias introduced by inadvertent selfed flowers as revealed by microsatellite markers control. Afr. J. Biotechnol. 2010, 9, 1905–1910. [Google Scholar]
- Kumar, R.; Janila, P.; Vishwakarma, M.K.; Khan, A.W.; Manohar, S.S.; Gangurde, S.S.; Variath, M.T.; Shasidhar, Y.; Pandey, M.K.; Varshney, R.K. Whole genome re-sequencing-based QTL-seq identified candidate genes and molecular markers for fresh seed dormancy in groundnut. Plant Biotechnol. J. 2019, 18, 992–1003. [Google Scholar] [CrossRef] [PubMed]
- Krapovickas, A.; Gregory, W. Taxonomía del Género. Arachis (Leguminosae); Instituto de Botánica del Nordeste: Corrientes, Argentina, 1994; pp. 1–186. [Google Scholar]
- Holbrook, C.C.; Dong, W. Development and Evaluation of a Mini-core Collection for the U.S. Peanut Germplasm Collection. Crop Sci. 2005, 45, 1540–1544. [Google Scholar] [CrossRef] [Green Version]
- Clevenger, J.; Chu, Y.; Chavarro, C.; Agarwal, G.; Bertioli, D.J.; Leal-Bertioli, S.C.; Pandey, M.K.; Vaughn, J.; Abernathy, B.; Barkley, N.A. Genome-wide SNP genotyping resolves signatures of selection and tetrasomic recombination in peanut. Mol. Plant 2017, 10, 309–322. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Clevenger, J.P.; Ozias-Akins, P. SWEEP: A tool for filtering high-quality SNPs in polyploid crops. G3 Genes Genomes Genet. 2015, 5, 1797–1803. [Google Scholar] [CrossRef] [Green Version]
- Lipka, A.E.; Tian, F.; Wang, Q.; Peiffer, J.; Li, M.; Bradbury, P.J.; Gore, M.A.; Buckler, E.S.; Zhang, Z. GAPIT: Genome association and prediction integrated tool. Bioinformatics 2012, 28, 2397–2399. [Google Scholar] [CrossRef] [Green Version]
- Pritchard, J.K.; Stephens, M.; Donnelly, P. Inference of population structure using multilocus genotype data. Genetics 2000, 155, 945–959. [Google Scholar] [CrossRef]
- VanRaden, P.M. Efficient methods to compute genomic predictions. J. Dairy Sci. 2008, 91, 4414–4423. [Google Scholar] [CrossRef] [Green Version]
- Earl, D.A.; Vonholdt, B.M. Structure Harvester: A website and program for visualizing STRUCTURE output and implementing the Evanno method. Conserv. Genet. Resour. 2012, 4, 359–361. [Google Scholar] [CrossRef]
- Zhang, H.; Wang, M.L.; Schaefer, R.; Dang, P.; Jiang, T.; Chen, C. GWAS and Coexpression Network Reveal Ionomic Variation in Cultivated Peanut. J. Agric. Food Chem. 2019, 67, 12026–12036. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.; Sun, Y.; Stang, P.; Berlin, J.A.; Wilcox, M.A.; Li, Q. Comparison of methods for correcting population stratification in a genome-wide association study of rheumatoid arthritis: Principal-component analysis versus multidimensional scaling. In BMC Proceedings; BioMed Central: London, UK, 2009; p. S109. [Google Scholar]
- Kang, H.M.; Sul, J.H.; Service, S.K.; Zaitlen, N.A.; Kong, S.-y.; Freimer, N.B.; Sabatti, C.; Eskin, E. Variance component model to account for sample structure in genome-wide association studies. Sci. Genet. 2010, 42, 348. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Turner, S.D. Qqman: An R package for visualizing GWAS results using QQ and manhattan plots. Biorxiv 2014, 005165. [Google Scholar] [CrossRef]
- Bertioli, D.J.; Cannon, S.B.; Froenicke, L.; Huang, G.; Farmer, A.D.; Cannon, E.K.; Liu, X.; Gao, D.; Clevenger, J.; Dash, S. The genome sequences of Arachis duranensis and Arachis ipaensis, the diploid ancestors of cultivated peanut. Sci. Genet. 2015, 47, 438. [Google Scholar] [CrossRef]
- Fonceka, D.; Tossim, H.-A.; Rivallan, R.; Vignes, H.; Faye, I.; Ndoye, O.; Moretzsohn, M.C.; Bertioli, D.J.; Glaszmann, J.-C.; Courtois, B. Fostered and left behind alleles in peanut: Interspecific QTL mapping reveals footprints of domestication and useful natural variation for breeding. BMC Plant Biol. 2012, 12, 26. [Google Scholar] [CrossRef] [Green Version]
- Chu, Y.; Chee, P.; Isleib, T.G.; Holbrook, C.C.; Ozias-Akins, P. Major seed size QTL on chromosome A05 of peanut (Arachis hypogaea) is conserved in the US mini-core germplasm collection. Mol. Breed. 2020, 40, 6. [Google Scholar] [CrossRef] [Green Version]
- Chavarro, C.; Chu, Y.; Holbrook, C.; Isleib, T.; Bertioli, D.; Hovav, R.; Butts, C.; Lamb, M.; Sorensen, R.; Jackson, S.A. Genetic analysis of seed and pod traits in a set of Recombinant Inbred Lines (RILs) in peanut (Arachis hypogaea L.). bioRxiv 2019, 738914. [Google Scholar] [CrossRef]
- Luo, H.; Ren, X.; Li, Z.; Xu, Z.; Li, X.; Huang, L.; Zhou, X.; Chen, Y.; Chen, W.; Lei, Y. Co-localization of major quantitative trait loci for pod size and weight to a 3.7 cM interval on chromosome A05 in cultivated peanut (Arachis hypogaea L.). BMC Genom. 2017, 18, 58. [Google Scholar] [CrossRef] [Green Version]
- Oracz, K.; El-Maarouf-Bouteau, H.; Kranner, I.; Bogatek, R.; Corbineau, F.; Bailly, C. The mechanisms involved in seed dormancy alleviation by hydrogen cyanide unravel the role of reactive oxygen species as key factors of cellular signaling during germination. Plant Physiol. 2009, 150, 494–505. [Google Scholar] [CrossRef] [Green Version]
- Chen, C.; Letnik, I.; Hacham, Y.; Dobrev, P.; Ben-Daniel, B.-H.; Vanková, R.; Amir, R.; Miller, G. Ascorbate Peroxidase6 protects Arabidopsis desiccating and germinating seeds from stress and mediates cross talk between reactive oxygen species, abscisic acid, and auxin. Plant Physiol. 2014, 166, 370–383. [Google Scholar] [CrossRef] [Green Version]
- Fu, W.; Shen, Y.; Hao, J.; Wu, J.; Ke, L.; Wu, C.; Huang, K.; Luo, B.; Xu, M.; Cheng, X.; et al. Acyl-CoA N-acyltransferase influences fertility by regulating lipid metabolism and jasmonic acid biogenesis in cotton. Sci. Rep. 2015, 5, 11790. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beisson, F.; Li, Y.; Bonaventure, G.; Pollard, M.; Ohlrogge, J.B. The acyltransferase GPAT5 is required for the synthesis of suberin in seed coat and root of Arabidopsis. Plant Cell 2007, 19, 351–368. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gao, Y.; Liu, J.; Chen, Y.; Tang, H.; Wang, Y.; He, Y.; Ou, Y.; Sun, X.; Wang, S.; Yao, Y. Tomato SlAN11 regulates flavonoid biosynthesis and seed dormancy by interaction with bHLH proteins but not with MYB proteins. Hortic. Res. 2018, 5, 27. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shah, F.A.; Ni, J.; Chen, J.; Wang, Q.; Liu, W.; Chen, X.; Tang, C.; Fu, S.; Wu, L. Proanthocyanidins in seed coat tegmen and endospermic cap inhibit seed germination in Sapium sebiferum. PeerJ 2018, 6, e4690. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wada, S.; Kennedy, J.A.; Reed, B.M. Seed-coat anatomy and proanthocyanidins contribute to the dormancy of Rubus seed. Sci. Hortic. 2011, 130, 762–768. [Google Scholar] [CrossRef]
- Kushiro, T.; Okamoto, M.; Nakabayashi, K.; Yamagishi, K.; Kitamura, S.; Asami, T.; Hirai, N.; Koshiba, T.; Kamiya, Y.; Nambara, E. The Arabidopsis cytochrome P450 CYP707A encodes ABA 8′-hydroxylases: Key enzymes in ABA catabolism. EMBO J. 2004, 23, 1647–1656. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Z.-L.; Xie, Z.; Zou, X.; Casaretto, J.; Ho, T.-H.D.; Shen, Q.J. A rice WRKY gene encodes a transcriptional repressor of the gibberellin signaling pathway in aleurone cells. Plant Physiol. 2004, 134, 1500–1513. [Google Scholar] [CrossRef] [Green Version]
- Zentella, R.; Zhang, Z.-L.; Park, M.; Thomas, S.G.; Endo, A.; Murase, K.; Fleet, C.M.; Jikumaru, Y.; Nambara, E.; Kamiya, Y. Global analysis of DELLA direct targets in early gibberellin signaling in Arabidopsis. Plant Cell 2007, 19, 3037–3057. [Google Scholar] [CrossRef]
- Sitrit, Y.; Downie, B.; Bennett, A.; Bradford, K. A novel exo-polygalacturonase is associated with radicle protrusion in tomato (Lycopersicon esculentum) seeds. In Plant Physiology; Amer Soc Plant Physiologists 15501 Monona Drive: Rockville, MD, USA, 1996; p. 752. [Google Scholar]
- Sitrit, Y.; Hadfield, K.A.; Bennett, A.B.; Bradford, K.J.; Downie, A.B. Expression of a polygalacturonase associated with tomato seed germination. Plant Physiol. 1999, 121, 419–428. [Google Scholar] [CrossRef] [Green Version]
- Liang, Y.; Baring, M.R.; Septiningsih, E.M. Mapping of Quantitative Trait Loci for Yield and Grade Related Traits in Peanut (Arachis hypogaea L.) Using High-Resolution SNP Markers. Plant Breed. Biotechnol. 2018, 6, 454–462. [Google Scholar] [CrossRef]
- Periappuram, C.; Steinhauer, L.; Barton, D.L.; Taylor, D.C.; Chatson, B.; Zou, J. The Plastidic Phosphoglucomutase from Arabidopsis. A Reversible Enzyme Reaction with an Important Role in Metabolic Control. Plant Physiol. 2000, 122, 1193. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, F.; Zhang, X.; Shen, Y.; Wang, H.; Liu, R.; Wang, X.; Gao, D.; Yang, Y.Z.; Liu, Y.; Tan, B.C. The pentatricopeptide repeat protein EMPTY PERICARP 8 is required for the splicing of three mitochondrial introns and seed development in maize. Plant J. 2018, 95, 919–932. [Google Scholar] [CrossRef] [PubMed]
- Geshi, N.; Johansen, J.N.; Dilokpimol, A.; Rolland, A.; Belcram, K.; Verger, S.; Kotake, T.; Tsumuraya, Y.; Kaneko, S.; Tryfona, T. A galactosyltransferase acting on arabinogalactan protein glycans is essential for embryo development in A rabidopsis. Plant J. 2013, 76, 128–137. [Google Scholar] [PubMed]
- Li, S.-J.; Hochstrasser, M. A new protease required for cell-cycle progression in yeast. Nature 1999, 398, 246–251. [Google Scholar] [CrossRef]
- Smith, M.A.; Moon, H.; Chowrira, G.; Kunst, L. Heterologous expression of a fatty acid hydroxylase gene in developing seeds of Arabidopsis thaliana. Planta 2003, 217, 507–516. [Google Scholar] [CrossRef]
- Petla, B.P.; Kamble, N.U.; Kumar, M.; Verma, P.; Ghosh, S.; Singh, A.; Rao, V.; Salvi, P.; Kaur, H.; Saxena, S.C. Rice Protein l-Isoaspartyl Methyltransferase isoforms differentially accumulate during seed maturation to restrict deleterious isoAsp and reactive oxygen species accumulation and are implicated in seed vigor and longevity. New Phytolog. 2016, 211, 627–645. [Google Scholar] [CrossRef] [Green Version]
- Pandey, M.K.; Upadhyaya, H.D.; Rathore, A.; Vadez, V.; Sheshshayee, M.; Sriswathi, M.; Govil, M.; Kumar, A.; Gowda, M.; Sharma, S. Genomewide association studies for 50 agronomic traits in peanut using the ‘reference set’comprising 300 genotypes from 48 countries of the semi-arid tropics of the world. PLoS ONE 2014, 9, e105228. [Google Scholar] [CrossRef] [Green Version]
- Gautami, B.; Foncéka, D.; Pandey, M.K.; Moretzsohn, M.C.; Sujay, V.; Qin, H.; Hong, Y.; Faye, I.; Chen, X.; BhanuPrakash, A. An international reference consensus genetic map with 897 marker loci based on 11 mapping populations for tetraploid groundnut (Arachis hypogaea L.). PLoS ONE 2012, 7, e41213. [Google Scholar] [CrossRef] [Green Version]
- Luo, M.; Dennis, E.S.; Berger, F.; Peacock, W.J.; Chaudhury, A. MINISEED3 (MINI3), a WRKY family gene, and HAIKU2 (IKU2), a leucine-rich repeat (LRR) KINASE gene, are regulators of seed size in Arabidopsis. Proc. Natl. Acad. Sci. USA 2005, 102, 17531–17536. [Google Scholar] [CrossRef] [Green Version]
- Orsi, C.H.; Tanksley, S.D. Natural variation in an ABC transporter gene associated with seed size evolution in tomato species. PLoS Genet. 2009, 5, e1000347. [Google Scholar] [CrossRef] [Green Version]
- Luo, H.; Xu, Z.; Li, Z.; Li, X.; Lv, J.; Ren, X.; Huang, L.; Zhou, X.; Chen, Y.; Yu, J.; et al. Development of SSR markers and identification of major quantitative trait loci controlling shelling percentage in cultivated peanut (Arachis hypogaea L.). Theor. Appl. Genet. 2017, 130, 1635–1648. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, L.; He, H.; Chen, W.; Ren, X.; Chen, Y.; Zhou, X.; Xia, Y.; Wang, X.; Jiang, X.; Liao, B.; et al. Quantitative trait locus analysis of agronomic and quality-related traits in cultivated peanut (Arachis hypogaea L.). Theor. Appl. Genet. 2015, 128, 1103–1115. [Google Scholar] [CrossRef] [PubMed] [Green Version]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| 7th Day | 14th Day | 21st Day | Seed Dormancy | Seed Weight | LSK | SMK | |
|---|---|---|---|---|---|---|---|
| 14th day | 0.99 * | ||||||
| 21st day | 0.98 * | 0.99 * | |||||
| Seed dormancy | −0.97 * | −0.98 * | −0.99 * | ||||
| Seed weight | −0.38 * | −0.33 * | −0.34 * | 0.35 * | |||
| LSK | 0.43 * | 0.41 * | 0.41 * | −0.4 * | −0.38 * | ||
| SMK | 0.02 | −0.01 | −0.01 | 0 | −0.6 | 0.23 | |
| Shelling % | 0.42 * | 0.45 * | 0.45 * | −0.43 * | 0.1 | 0.24 | −0.4 * |
| Variable | Min | Max | Mean | Std Dev | CV |
|---|---|---|---|---|---|
| SMK | 2.7 | 45.8 | 12.3 | 7.28 | 59 |
| LSK | 0.26 | 9.57 | 2.59 | 2.09 | 81 |
| seed weight | 27.55 | 95.68 | 48.49 | 12.87 | 27 |
| shelling percent | 64.11 | 78.08 | 72.79 | 2.62 | 3.6 |
| Traits | Number of Significant Associations | Number of Suggestive Associations |
|---|---|---|
| Germination rate at 7 days | 7 | 38 |
| Germination rate at 14 days | 4 | 92 |
| Germination rate at 21 days | 4 | 87 |
| Seed Dormancy | 4 | 246 |
| Gemination and Dormancy * | 11 | 266 |
| SMK | 2 | 6 |
| shelling percent | 10 | 39 |
| LSK | 0 | 17 |
| seed weight | 1 | 7 |
| Total | 32 | 532 |
| # | Trait | SNP Marker | Chr. | Position on Chr. | −log10 (p value) | PVE (%) | # of Genes in Vicinity |
|---|---|---|---|---|---|---|---|
| 1 | 7 days * | AX-177644204 | B09 | 51563617 | 5.74 | 20.6 | 16 |
| 2 | 7 days | AX-176814819 | A05 | 17060254 | 5.70 | 20.5 | 102 |
| 3 | 7 days | AX-176816015 | A04 | 11035231 | 4.58 | 16.4 | 86 |
| 4 | 7 days | AX-177643736 | B09 | 109630028 | 4.37 | 15.6 | 30 |
| 5 | 7 days | AX-176803643 | A08 | 28348986 | 4.33 | 15.5 | 142 |
| 6 | 7 days | AX-177642270 | B09 | 120713638 | 4.32 | 15.5 | 41 |
| 7 | 7 days | AX-147221650 | A05 | 7266706 | 4.31 | 15.4 | 126 |
| 1 | 14 days | AX-177644204 | B09 | 51563617 | 5.85 | 21 | 16 |
| 2 | 14 days | AX-176814819 | A05 | 17060254 | 5.64 | 20.3 | 102 |
| 3 | 14 days | AX-177643736 | B09 | 109630028 | 4.50 | 16.1 | 30 |
| 4 | 14 days | AX-176799628 | A06 | 110978824 | 4.32 | 15.4 | 155 |
| 1 | 21 days | AX-177644204 | B09 | 51563617 | 5.39 | 19.4 | 16 |
| 2 | 21 days | AX-176814819 | A05 | 17060254 | 5.10 | 18.3 | 102 |
| 3 | 21 days | AX-147258769 | B08 | 110623990 | 4.70 | 16.8 | 50 |
| 4 | 21 days | AX-176799628 | A06 | 110978824 | 4.32 | 15.5 | 155 |
| 1 | Seed Dormancy | AX-176814819 | A05 | 17060254 | 5.57 | 20 | 102 |
| 2 | Seed Dormancy | AX-177638083 | B09 | 125395376 | 4.97 | 17.8 | 61 |
| 3 | Seed Dormancy | AX-147258769 | B08 | 110623990 | 4.81 | 17.3 | 50 |
| 4 | Seed Dormancy | AX-176802837 | A02 | 28578269 | 4.40 | 15.7 | 23 |
| # | Trait | SNP Marker | Chr. | Position on Chr. | −log10 (p value) | PVE (%) | # of Genes in Vicinity |
|---|---|---|---|---|---|---|---|
| 1 | SMK | AX-176823847 | B05 | 118253381 | 5.27 | 17.0 | 29 |
| 2 | SMK | AX-176810056 | A07 | 75161338 | 4.57 | 14.7 | 73 |
| 1 | seed wt | AX-177638040 | A10 | 76559318 | 5.78 | 20.8 | 21 |
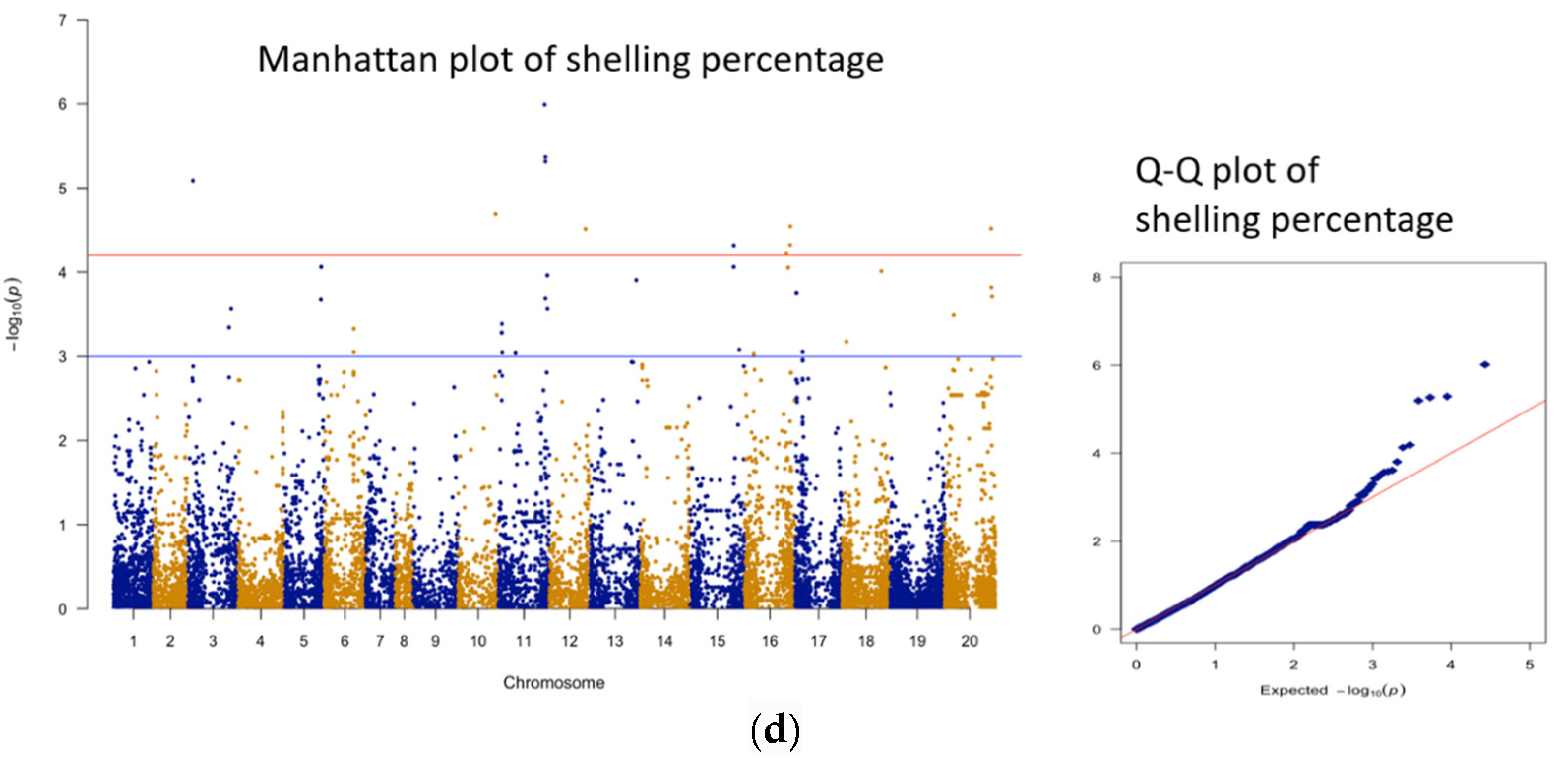
| 1 | shelling % | AX-176811900 | B01 | 123973617 | 5.99 | 19.4 | 113 |
| 2 | shelling % | AX-176806228 | B01 | 126274566 | 5.37 | 17.4 | 121 |
| 3 | shelling % | AX-176807776 | B01 | 126179070 | 5.32 | 17.2 | 121 |
| 4 | shelling % | AX-147216060 | A03 | 13426004 | 5.09 | 16.5 | 98 |
| 5 | shelling % | AX-147236668 | A10 | 101148536 | 4.69 | 15.2 | 95 |
| 6 | shelling % | AX-176808276 | B06 | 122427781 | 4.54 | 14.7 | 74 |
| 7 | shelling % | AX-177640468 | B10 | 124893402 | 4.52 | 14.6 | 49 |
| 8 | shelling % | AX-176813093 | B02 | 97213496 | 4.51 | 14.6 | 79 |
| 9 | shelling % | AX-176793701 | B06 | 121887242 | 4.33 | 13.9 | 71 |
| 10 | shelling % | AX-176802081 | B05 | 120108986 | 4.32 | 13.9 | 68 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Patel, J.D.; Wang, M.L.; Dang, P.; Butts, C.; Lamb, M.; Chen, C.Y. Insights into the Genomic Architecture of Seed and Pod Quality Traits in the U.S. Peanut Mini-Core Diversity Panel. Plants 2022, 11, 837. https://doi.org/10.3390/plants11070837
Patel JD, Wang ML, Dang P, Butts C, Lamb M, Chen CY. Insights into the Genomic Architecture of Seed and Pod Quality Traits in the U.S. Peanut Mini-Core Diversity Panel. Plants. 2022; 11(7):837. https://doi.org/10.3390/plants11070837
Chicago/Turabian StylePatel, Jinesh D., Ming Li Wang, Phat Dang, Chris Butts, Marshall Lamb, and Charles Y. Chen. 2022. "Insights into the Genomic Architecture of Seed and Pod Quality Traits in the U.S. Peanut Mini-Core Diversity Panel" Plants 11, no. 7: 837. https://doi.org/10.3390/plants11070837