
Identification of the Potential Genes Regulating Seed Germination Speed in Maize

 , ,
, ,
Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
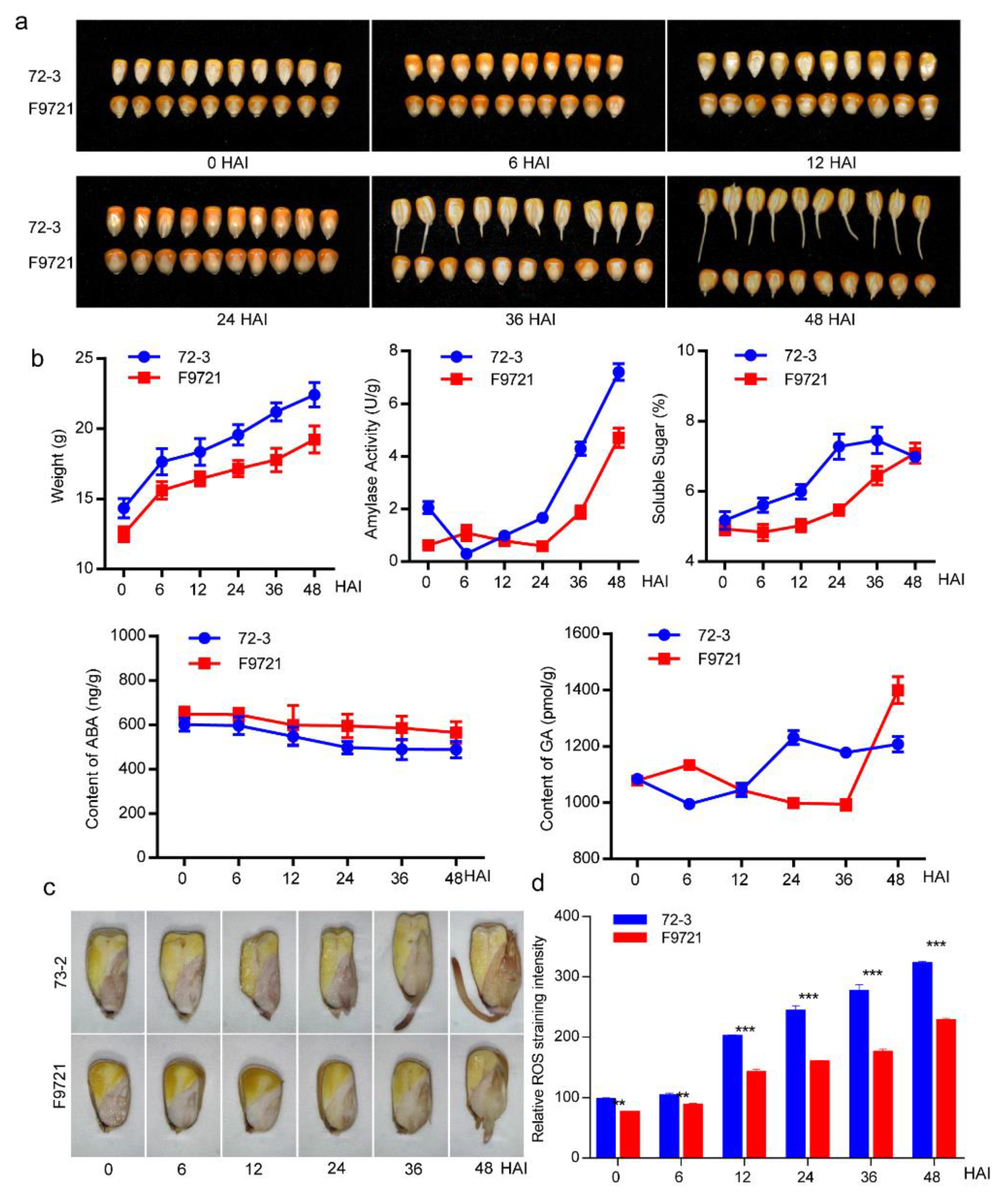
2.1. Materials and Germination Assays
2.2. Measurement of Total Soluble Sugar and Amylase Activity in Germinating Seeds
2.3. ABA, GA, and ROS Measurement
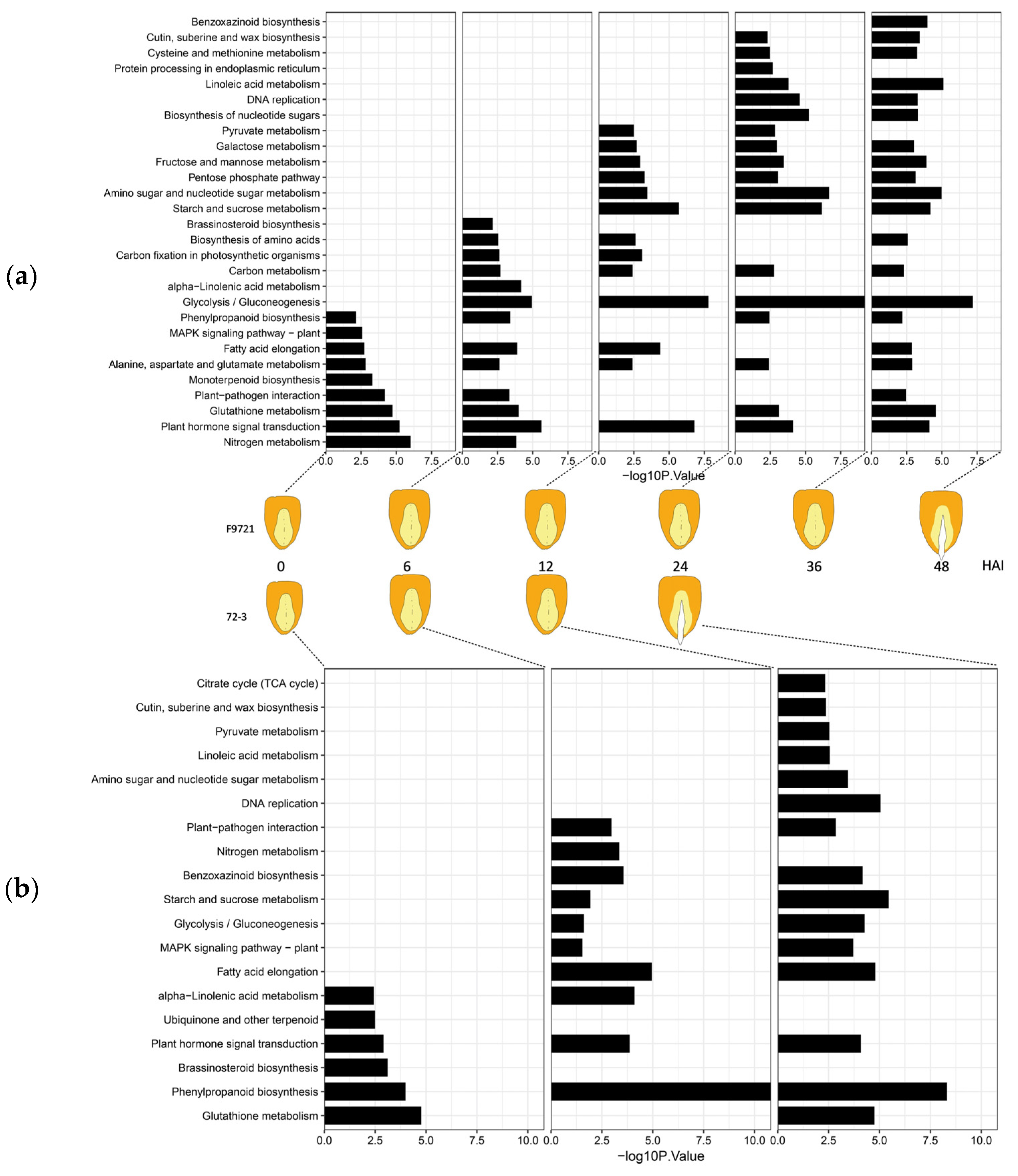
2.4. RNA Sequencing, Differential Gene Expression Analysis, and Enrichment Analysis
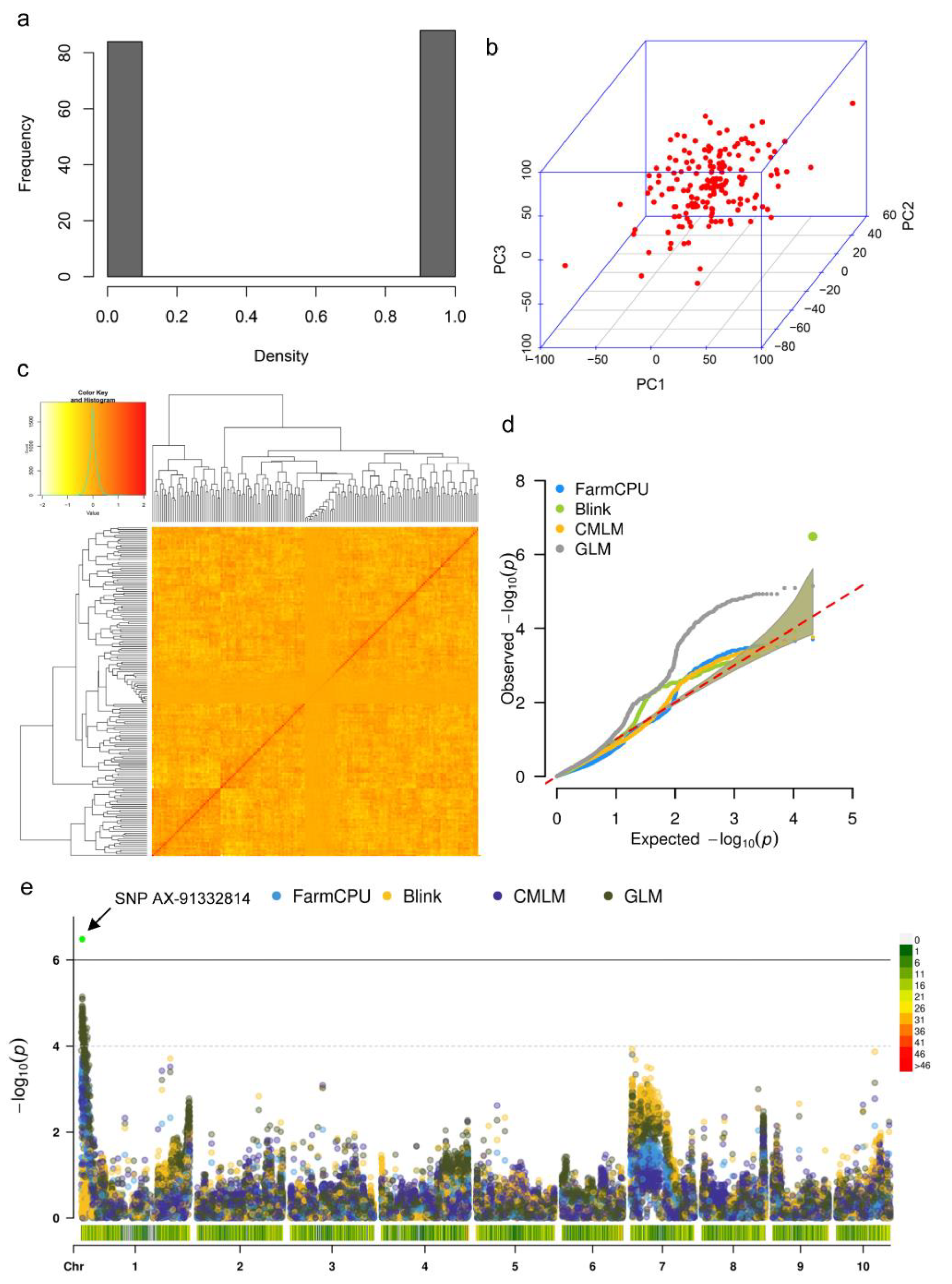
2.5. SNP Genotyping and GWAS of Genomic Locus for Germination Speed Variation
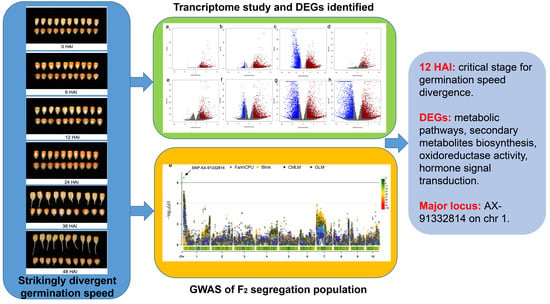
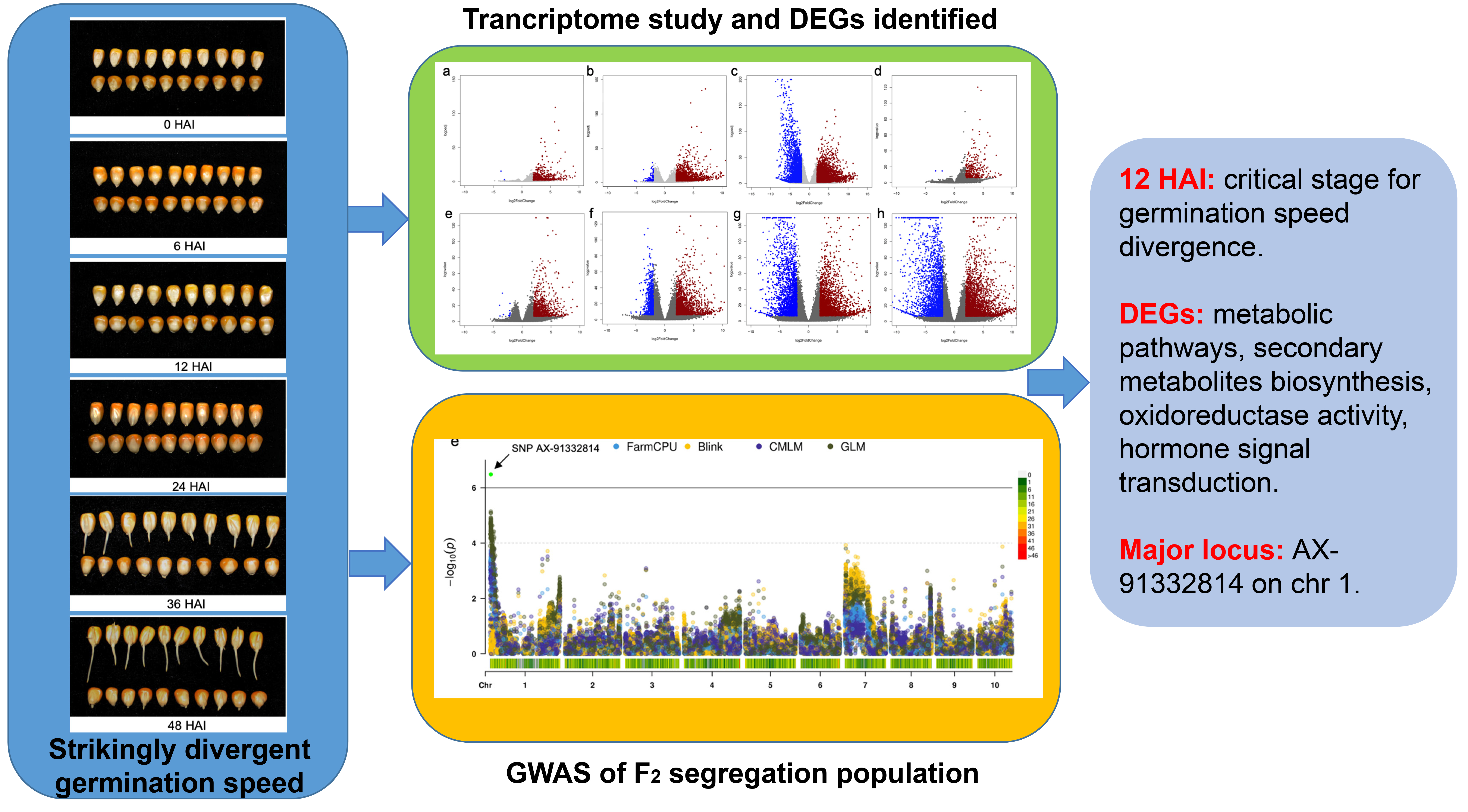
3. Results
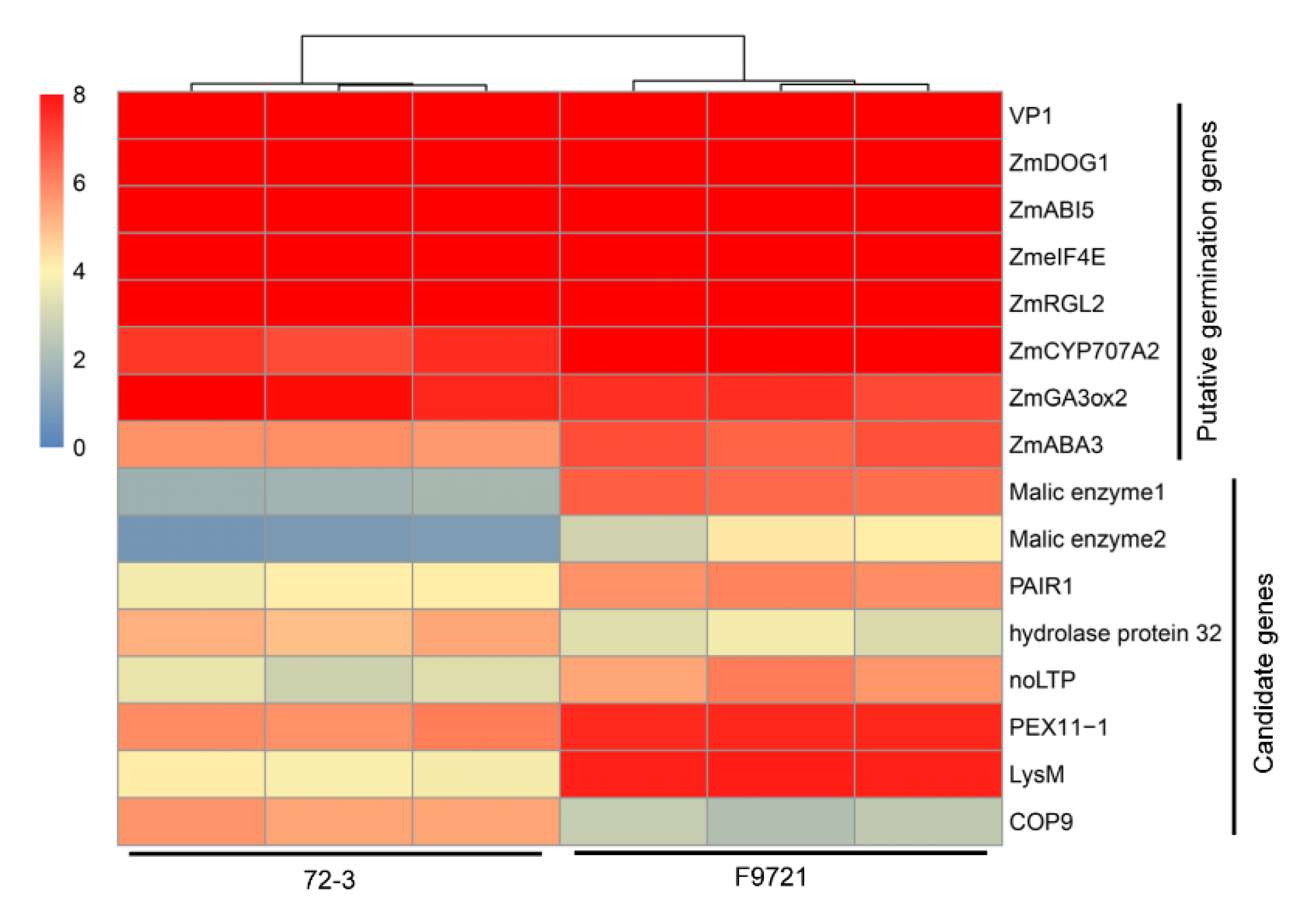
3.1. Characterization of Seed Germination Status in the Two Inbred Maize Lines
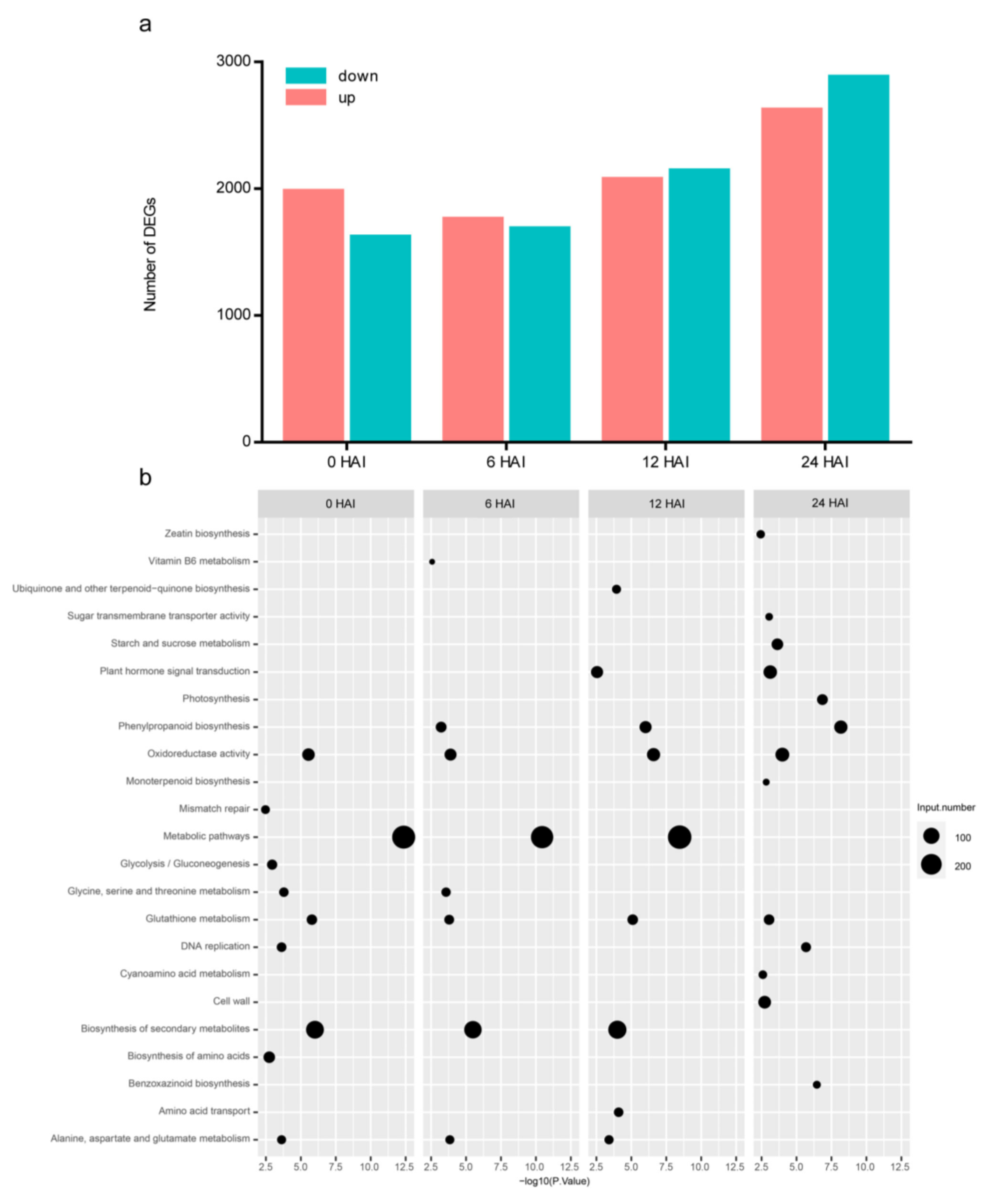
3.2. Transcriptome Dynamics during the Germination Process of 72-3 and F9721
3.3. Time-Series Analysis of DEGs between 72-3 and F9721
3.4. GWAS of Candidate Locus Responsible for Seed Germination Variation
4. Discussion
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| SNP | Single nucleotide polymorphism |
| QTL | Quantitative traits loci |
| ROS | Reactive oxygen species |
| PCA | Principal components analysis |
| FDR | False discovery rate |
| GLM | General linear model |
| CMLM | Compressed mixed linear model |
| FarmCPU | Fixed and random model circulating probability unification |
| BLINK | Bayesian-information and linkage-disequilibrium iteratively nested keyway |
| GWAS | Genome-wide association analysis |
| GAPIT | Genomic association and prediction integrated tool |
| LD | Linkage disequilibrium |
References
- Bewley, J.D. Seed germination and dormancy. Plant Cell 1997, 9, 1055–1066. [Google Scholar] [CrossRef] [Green Version]
- Finch-Savage, W.; Leubner-Metzger, G. Seed dormancy and the control of germination. New Phytol. 2006, 171, 501–523. [Google Scholar] [CrossRef]
- Carrera-Castaño, G.; Calleja-Cabrera, J.; Pernas, M.; Gómez, L.; Oñate-Sánchez, L. An updated overview on the regulation of seed germination. Plants 2020, 9, 703. [Google Scholar] [CrossRef]
- Tognacca, R.S.; Botto, J.F. Post-transcriptional regulation of seed dormancy and germination: Current understanding and future directions. Plant Commun. 2021, 2, 100169. [Google Scholar] [CrossRef]
- Klupczyńska, E.A.; Pawłowski, T.A. Regulation of seed dormancy and germination mechanisms in a changing environment. Int. J. Mol. Sci. 2021, 22, 1357. [Google Scholar] [CrossRef]
- He, D.L.; Han, C.; Yang, P.F. Gene expression profile changes in germinating rice. J. Integr. Plant Biol. 2011, 53, 835–844. [Google Scholar] [CrossRef]
- Bailly, C. The signalling role of ROS in the regulation of seed germination and dormancy. Biochem. J. 2019, 476, 3019–3032. [Google Scholar] [CrossRef]
- Nonogaki, H. Seed germination and dormancy: The classic story, new puzzles, and evolution. J. Integr. Plant Biol. 2019, 61, 541–563. [Google Scholar] [CrossRef] [Green Version]
- Ravindran, P.; Kumar, P.P. Regulation of seed germination: The involvement of multiple forces exerted via gibberellic acid signaling. Mol. Plant 2019, 12, 24–26. [Google Scholar] [CrossRef] [Green Version]
- Xu, F.; Tang, J.Y.; Gao, S.P.; Cheng, X.; Du, L.; Chu, C.C. Control of rice pre-harvest sprouting by glutaredoxin-mediated abscisic acid signaling. Plant J. 2019, 100, 1036–1051. [Google Scholar] [CrossRef]
- Zhao, L.; Wang, S.; Fu, Y.B.; Wang, H. Arabidopsis seed stored mRNAs are degraded constantly over aging time, as revealed by new quantification methods. Front. Plant Sci. 2020, 10, 1764. [Google Scholar] [CrossRef]
- Luo, X.F.; Dai, Y.J.; Zheng, C.; Yang, Y.Z.; Chen, W.; Wang, Q.C.; Chandrasekaran, U.; Du, J.B.; Liu, W.G.; Shu, K. The ABI4-RbohD/VTC2 regulatory module promotes Reactive Oxygen Species (ROS) accumulation to decrease seed germination under salinity stress. New Phytol. 2021, 229, 950–962. [Google Scholar] [CrossRef]
- Kaneko, M.; Itoh, H.; Ueguchi-Tanaka, M.; Ashikari, M.; Matsuoka, M. The alpha-amylase induction in endosperm during rice seed germination is caused by gibberellin synthesized in epithelium. Plant Physiol. 2002, 128, 1264–1270. [Google Scholar] [CrossRef] [Green Version]
- Li, Z.; Xu, J.G.; Gao, Y.; Wang, C.; Guo, G.Y.; Luo, Y.; Huang, Y.T.; Hu, W.M.; Sheteiwy, M.S.; Guan, Y.J.; et al. The synergistic priming effect of exogenous salicylic acid and H2O2 on chilling tolerance enhancement during maize (Zea mays L.) seed germination. Front. Plant Sci. 2017, 8, 1153. [Google Scholar] [CrossRef] [Green Version]
- Shu, K.; Liu, X.D.; Xie, Q.; He, Z.H. Two faces of one seed: Hormonal regulation of dormancy and germination. Mol. Plant 2016, 9, 34–45. [Google Scholar] [CrossRef] [Green Version]
- Rajjou, L.; Duval, M.; Gallardo, K.; Catusse, J.; Bally, J.; Job, C.; Job, D. Seed germination and vigor. Annu. Rev. Plant. Biol. 2012, 63, 507–533. [Google Scholar] [CrossRef] [Green Version]
- Shu, K.; Zhang, H.; Wang, S.; Chen, M.; Wu, Y.; Tang, S.; Liu, C.; Feng, Y.; Cao, X.; Xie, Q. ABI4 regulates primary seed dormancy by regulating the biogenesis of abscisic acid and gibberellins in Arabidopsis. PLoS Genet. 2013, 9, e1003577. [Google Scholar] [CrossRef] [Green Version]
- Tuan, P.A.; Kumar, R.; Rehal, P.K.; Toora, P.K.; Ayele, B.T. Molecular mechanisms underlying abscisic acid/gibberellin balance in the control of seed dormancy and germination in cereals. Front. Plant Sci. 2018, 9, 668. [Google Scholar] [CrossRef] [Green Version]
- Sano, N.; Marion-Poll, A. ABA metabolism and homeostasis in seed dormancy and germination. Int. J. Mol. Sci. 2021, 22, 5069. [Google Scholar] [CrossRef]
- Bailly, C.; Merendino, L. Oxidative signalling in seed germination and early seedling growth: An emerging role for ROS trafficking and inter-organelle communication. Biochem. J. 2021, 478, 1977–1984. [Google Scholar] [CrossRef]
- Krasuska, U.; Ciacka, K.; Bogatek, R.; Gniazdowska, A. Polyamines and nitric oxide link in regulation of dormancy removal and germination of Apple (Malus domestica Borkh.) embryos. J. Plant Growth Regul. 2014, 33, 590–601. [Google Scholar] [CrossRef] [Green Version]
- Ma, W.; Guan, X.; Li, J.; Pan, R.; Wang, L.; Liu, F.; Ma, H.; Zhu, S.; Hu, J.; Ruan, Y.L.; et al. Mitochondrial small heat shock protein mediates seed germination via thermal sensing. Proc. Natl. Acad. Sci. USA 2019, 116, 4716–4721. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lariguet, P.; Ranocha, P.; De Meyer, M.; Barbier, O.; Penel, C.; Dunand, C. Identification of a hydrogen peroxide signalling pathway in the control of light-dependent germination in Arabidopsis. Planta 2013, 238, 381–395. [Google Scholar] [CrossRef]
- Liu, Y.G.; Ye, N.H.; Liu, R.; Chen, M.X.; Zhang, J.H. H2O2 mediates the regulation of ABA catabolism and GA biosynthesis in Arabidopsis seed dormancy and germination. J. Exp. Bot. 2010, 61, 2979–2990. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ma, Z.; Bykova, N.V.; Igamberdiev, A.U. Cell signaling mechanisms and metabolic regulation of germination and dormancy in barley seeds. Crop J. 2017, 5, 459–477. [Google Scholar] [CrossRef]
- Leymarie, J.; Vitkauskaité, G.; Hoang, H.H.; Gendreau, E.; Chazoule, V.; Meimoun, P.; Corbineau, F.; El-Maarouf-Bouteau, H.; Bailly, C. Role of reactive oxygen species in the regulation of Arabidopsis seed dormancy. Plant Cell Physiol. 2012, 53, 96–106. [Google Scholar] [CrossRef] [Green Version]
- Han, Z.P.; Ku, L.X.; Zhang, Z.Z.; Zhang, J.; Guo, S.L.; Liu, H.Y.; Zhao, R.F.; Ren, Z.Z.; Zhang, L.K.; Su, H.K.; et al. QTLs for seed vigor-related traits identified in maize seeds germinated under artificial aging conditions. PLoS ONE 2014, 9, e92535. [Google Scholar] [CrossRef] [Green Version]
- Cao, Q.J.; Li, G.; Cui, Z.G.; Yang, F.T.; Jiang, X.L.; Diallo, L.; Kong, F.L. Seed priming with melatonin improves the seed germination of waxy maize under chilling stress via promoting the antioxidant system and starch metabolism. Sci. Rep. 2019, 9, 15044. [Google Scholar] [CrossRef] [Green Version]
- Wu, X.M.; Feng, F.Q.; Zhu, Y.Z.; Xie, F.G.; Yang, J.; Gong, J.; Liu, Y.; Zhu, W.; Gao, T.L.; Chen, D.Y.; et al. Construction of high-density genetic map and identification of QTLs associated with seed vigor after exposure to artificial aging conditions in sweet corn using SLAF-seq. Genes 2020, 11, 37. [Google Scholar] [CrossRef] [Green Version]
- Hu, S.D.; Lübberstedt, T.; Zhao, G.W.; Lee, M. QTL mapping of low-temperature germination ability in the maize IBM Syn4 RIL population. PLoS ONE 2016, 11, e0152795. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.C.; Liu, P.; Wang, C.; Zhang, N.; Zhu, Y.X.; Zou, C.Y.; Yuan, G.S.; Yang, C.; Gao, S.B.; Pan, G.T.; et al. Genome-wide association study uncovers new genetic loci and candidate genes underlying seed chilling-germination in maize. Peer J. 2021, 9, e11707. [Google Scholar] [CrossRef] [PubMed]
- Ren, R.C.; Lu, X.; Zhao, Y.J.; Wei, Y.M.; Wang, L.L.; Zhang, L.; Zhang, W.T.; Zhang, C.; Zhang, X.S.; Zhao, X.Y. Pentatricopeptide repeat protein DEK40 is required for mitochondrial function and kernel development in maize. J. Exp. Bot. 2019, 70, 6163–6179. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.F.; Zhou, Y.Q.; Chen, Y.R.; Gu, J. fastp: An ultra-fast all-in-one FASTQ preprocessor. Bioinformatics 2018, 34, i884–i890. [Google Scholar] [CrossRef]
- Kim, D.; Paggi, J.M.; Park, C.; Bennett, C.; Salzberg, S.L. Graph-based genome alignment and genotyping with HISAT2 and HISAT-genotype. Nat. Biotechnol. 2019, 37, 907–915. [Google Scholar] [CrossRef] [PubMed]
- Anders, S.; Pyl, P.T.; Huber, W. HTSeq-a Python framework to work with high-throughput sequencing data. Bioinformatics 2015, 31, 166–169. [Google Scholar] [CrossRef]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef] [Green Version]
- Bu, D.C.; Luo, H.T.; Huo, P.P.; Wang, Z.H.; Zhang, S.; He, Z.H.; Wu, Y.; Zhao, L.H.; Liu, J.J.; Guo, J.C.; et al. KOBAS-i: Intelligent prioritization and exploratory visualization of biological functions for gene enrichment analysis. Nucleic Acids Res. 2021, 49, W317–W325. [Google Scholar] [CrossRef]
- Purcell, S.; Neale, B.; Todd-Brown, K.; Thomas, L.; Ferreira, M.R.; Bender, D.; Maller, J.; Sklar, P.; De Bakker, P.I.W.; Daly, M.J.; et al. PLINK: A tool set for whole-genome association and population-based linkage analyses. Am. J. Hum. Genet. 2007, 81, 559–575. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.; Zhang, Z. GAPIT version 3: Boosting power and accuracy for genomic association and prediction. Genom Proteom Bioinf. 2021; in press. [Google Scholar] [CrossRef]
- Yin, L.; Zhang, H.; Tang, Z.; Xu, J.; Yin, D.; Zhang, Z.; Yuan, X.; Zhu, M.; Zhao, S.; Li, X.; et al. rMVP: A memory-efficient, visualization-enhanced, and parallel-accelerated tool for genome-wide association study. Genom Proteom Bioinf. 2021; in press. [Google Scholar] [CrossRef]
- Tian, Y.; Guan, B.; Zhou, D.W.; Yu, J.B.; Li, G.D.; Lou, Y.J. Responses of seed germination, seedling growth, and seed yield traits to seed pretreatment in maize (Zea mays L.). Sci. World J. 2014, 2014, 834630. [Google Scholar] [CrossRef] [Green Version]
- Han, C.; Yang, P.F. Studies on the molecular mechanisms of seed germination. Proteomics 2015, 15, 1671–1679. [Google Scholar] [CrossRef]
- Mhamdi, A.; Van Breusegem, F. Reactive oxygen species in plant development. Development 2018, 145, dev164376. [Google Scholar] [CrossRef] [Green Version]
- Katsuya-Gaviria, K.; Caro, E.; Carrillo-Barral, N.; Iglesias-Fernández, R. Reactive oxygen species (ROS) and nucleic acid modifications during seed dormancy. Plants 2020, 9, 679. [Google Scholar] [CrossRef]
- Mittler, R. ROS are good. Trends Plant Sci. 2017, 22, 11–19. [Google Scholar] [CrossRef] [Green Version]
- Verma, G.; Mishra, S.; Sangwan, N.; Sharma, S. Reactive oxygen species mediate axis-cotyledon signaling to induce reserve mobilization during germination and seedling establishment in Vigna radiata. J. Plant Physiol. 2015, 184, 79–88. [Google Scholar] [CrossRef]
- Diaz-Vivancos, P.; Barba-Espín, G.; Hernández, J.A. Elucidating hormonal/ROS networks during seed germination: Insights and perspectives. Plant Cell Rep. 2013, 32, 1491–1502. [Google Scholar] [CrossRef]
- Han, Z.P.; Wang, B.; Tian, L.; Wang, S.X.; Zhang, J.; Guo, S.L.; Zhang, H.C.; Xu, L.R.; Chen, Y.H. Comprehensive dynamic transcriptome analysis at two seed germination stages in maize (Zea mays L.). Physiol. Plant 2020, 168, 205–217. [Google Scholar]
- Carles, C.; Bies-Etheve, N.; Aspart, L.; Leon-Kloosterziel, K.M.; Koornneef, M.; Echeverria, M.; Delseny, M. Regulation of Arabidopsis thaliana Em genes: Role of ABI5. Plant J. 2002, 30, 373–383. [Google Scholar] [CrossRef] [Green Version]
- Skubacz, A.; Daszkowska-Golec, A.; Szarejko, I. The role and regulation of ABI5 (ABA-Insensitive 5) in plant development, abiotic stress responses and phytohormone crosstalk. Frontiers Plant Sci. 2016, 7, 1884. [Google Scholar] [CrossRef] [Green Version]
- Zhao, X.; Dou, L.R.; Gong, Z.Z.; Wang, X.F.; Mao, T.L. BES1 hinders ABSCISIC ACID INSENSITIVE5 and promotes seed germination in Arabidopsis. New Phytol. 2019, 221, 908–918. [Google Scholar] [CrossRef] [Green Version]
- Lee, S.; Cheng, H.; King, K.E.; Wang, W.; He, Y.; Hussain, A.; Lo, J.; Harberd, N.P.; Peng, J. Gibberellin regulates Arabidopsis seed germination via RGL2, a GAI/RGA-like gene whose expression is up-regulated following imbibition. Genes Dev. 2002, 16, 646–658. [Google Scholar] [CrossRef] [Green Version]
- Tyler, L.; Thomas, S.G.; Hu, J.; Dill, A.; Alonso, J.M.; Ecker, J.R.; Sun, T.P. Della proteins and gibberellin-regulated seed germination and floral development in Arabidopsis. Plant Physiol. 2004, 135, 1008–1019. [Google Scholar] [CrossRef] [Green Version]
- Cao, D.; Hussain, A.; Cheng, H.; Peng, J. Loss of function of four DELLA genes leads to light- and gibberellin-independent seed germination in Arabidopsis. Planta 2005, 223, 105–113. [Google Scholar] [CrossRef]
- Zhong, C.M.; Xu, H.; Ye, S.; Wang, S.Y.; Li, L.F.; Zhang, S.C.; Wang, X.J. Gibberellic acid-stimulated Arabidopsis6 serves as an integrator of gibberellin, abscisic acid, and glucose signaling during seed germination in Arabidopsis. Plant Physiol. 2015, 169, 2288–2303. [Google Scholar]
- Dinkova, T.D.; Márquez-Velázquez, N.A.; Aguilar, R.; Lázaro-Mixteco, P.E.; de Jiménez, E.S. Tight translational control by the initiation factors eIF4E and eIF(iso)4E is required for maize seed germination. Seed Sci. Res. 2011, 21, 85–93. [Google Scholar] [CrossRef]
- Nambara, E.; Suzuki, M.; Abrams, S.; McCarty, D.R.; Kamiya, Y.; McCourt, P. A screen for genes that function in abscisic acid signaling in Arabidopsis thaliana. Genetics 2002, 161, 1247–1255. [Google Scholar] [CrossRef]
- Suzuki, M.; Ketterling, M.G.; Li, Q.B.; McCarty, D.R. Viviparous1 alters global gene expression patterns through regulation of abscisic acid signaling. Plant Physiol. 2003, 132, 1664–1677. [Google Scholar] [CrossRef] [Green Version]
- Bentsink, L.; Jowett, J.; Hanhart, C.J.; Koornneef, M. Cloning of DOG1, a quantitative trait locus controlling seed dormancy in Arabidopsis. Proc. Natl. Acad. Sci. USA 2006, 103, 17042–17047. [Google Scholar] [CrossRef] [Green Version]
- Dekkers, B.J.W.; He, H.; Hanson, J.; Willems, L.A.J.; Jamar, D.C.L.; Cueff, G.; Rajjou, L.; Hilhorst, H.W.M.; Bentsink, L. The Arabidopsis Delay of Germination 1 gene affects ABSCISIC ACID INSENSITIVE 5 (ABI5) expression and genetically interacts with ABI3 during Arabidopsis seed development. Plant J. 2016, 85, 451–465. [Google Scholar] [CrossRef]
- Née, G.; Kramer, K.; Nakabayashi, K.; Yuan, B.; Xiang, Y.; Miatton, E.; Finkemeier, I.; Soppe, W.J.J. Delay Of GERMINATION1 requires PP2C phosphatases of the ABA signaling pathway to control seed dormancy. Nat. Commun. 2017, 8, 72. [Google Scholar] [CrossRef]
- Nishimura, N.; Tsuchiya, W.; Moresco, J.J.; Hayashi, Y.; Satoh, K.; Kaiwa, N.; Irisa, T.; Kinoshita, T.; Schroeder, J.I.; Yates, J.R. Control of seed dormancy and germination by DOG1-AHG1 PP2C phosphatase complex via binding to heme. Nat. Commun. 2018, 9, 2132. [Google Scholar] [CrossRef]
- Sall, K.; Dekkers, B.J.W.; Nonogaki, M.; Katsuragawa, Y.; Koyari, R.; Hendrix, D.; Willems, L.A.J.; Bentsink, L.; Nonogaki, H. DELAY OF GERMINATION 1-LIKE 4 acts as an inducer of seed reserve accumulation. Plant J. 2019, 100, 7–19. [Google Scholar] [CrossRef] [Green Version]
- Carrillo-Barral, N.; Rodríguez-Gacio, M.C.; Matilla, A.J. Delay of Germination-1 (DOG1): A key to understanding seed dormancy. Plants 2020, 9, 480. [Google Scholar] [CrossRef] [Green Version]
- Yazdanpanah, F.; Maurino, V.G.; Mettler-Altmann, T.; Buijs, G.; Bailly, M.; Jashni, M.K.; Willems, L.; Sergeeva, L.I.; Rajjou, L.; Hilhorst, H.W.M.; et al. NADP-MALIC ENZYME 1 affects germination after seed storage in Arabidopsis thaliana. Plant Cell Physiol. 2019, 60, 318–328. [Google Scholar] [CrossRef]
- Lau, O.S.; Deng, X.W. The photomorphogenic repressors COP1 and DET1: 20 years later. Trends Plant Sci. 2012, 17, 584–593. [Google Scholar] [CrossRef]
- Sano, N.; Ono, H.; Murata, K.; Yamada, T.; Hirasawa, T.; Kanekatsu, M. Accumulation of long-lived mRNAs associated with germination in embryos during seed development of rice. J. Exp. Bot. 2015, 66, 4035–4046. [Google Scholar] [CrossRef] [Green Version]
- Sajeev, N.; Bai, B.; Bentsink, L. Seeds: A unique system to study translational regulation. Trends Plant Sci. 2019, 24, 487–495. [Google Scholar] [CrossRef]
- Sano, N.; Permana, H.; Kumada, R.; Shinozaki, Y.; Tanabata, T.; Yamada, T.; Hirasawa, T.; Kanekatsu, M. Proteomic analysis of embryonic proteins synthesized from long-lived mRNAs during germination of rice seeds. Plant Cell Physiol. 2012, 53, 687–698. [Google Scholar] [CrossRef]
- Galland, M.; Huguet, R.; Arc, E.; Cueff, G.; Job, D.; Rajjou, L. Dynamic proteomics emphasizes the importance of selective mRNA translation and protein turnover during Arabidopsis seed germination. Mol Cell Protomics. 2014, 13, 252–268. [Google Scholar] [CrossRef] [Green Version]
- Bai, B.; Peviani, A.; van der Horst, S.; Gamm, M.; Snel, B.; Bentsink, L.; Hanson, J. Extensive translational regulation during seed germination revealed by polysomal profiling. New Phytol. 2017, 214, 233–244. [Google Scholar] [CrossRef] [PubMed]
- Nakabayashi, K.; Okamoto, M.; Koshiba, T.; Kamiya, Y.; Nambara, E. Genome-wide profiling of stored mRNA in Arabidopsis thaliana seed germination: Epigenetic and genetic regulation of transcription in seed. Plant J. 2005, 41, 697–709. [Google Scholar] [CrossRef] [PubMed]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, H.; Zang, J.; Huo, Y.; Zhang, Z.; Chen, H.; Chen, X.; Liu, J. Identification of the Potential Genes Regulating Seed Germination Speed in Maize. Plants 2022, 11, 556. https://doi.org/10.3390/plants11040556
Zhang H, Zang J, Huo Y, Zhang Z, Chen H, Chen X, Liu J. Identification of the Potential Genes Regulating Seed Germination Speed in Maize. Plants. 2022; 11(4):556. https://doi.org/10.3390/plants11040556
Chicago/Turabian StyleZhang, Huairen, Jie Zang, Yanqing Huo, Zhaogui Zhang, Huabang Chen, Xunji Chen, and Juan Liu. 2022. "Identification of the Potential Genes Regulating Seed Germination Speed in Maize" Plants 11, no. 4: 556. https://doi.org/10.3390/plants11040556