
The Potential of Genome-Wide Prediction to Support Parental Selection, Evaluated with Data from a Commercial Barley Breeding Program
 ,
,  and
and
Abstract
:1. Introduction
2. Results
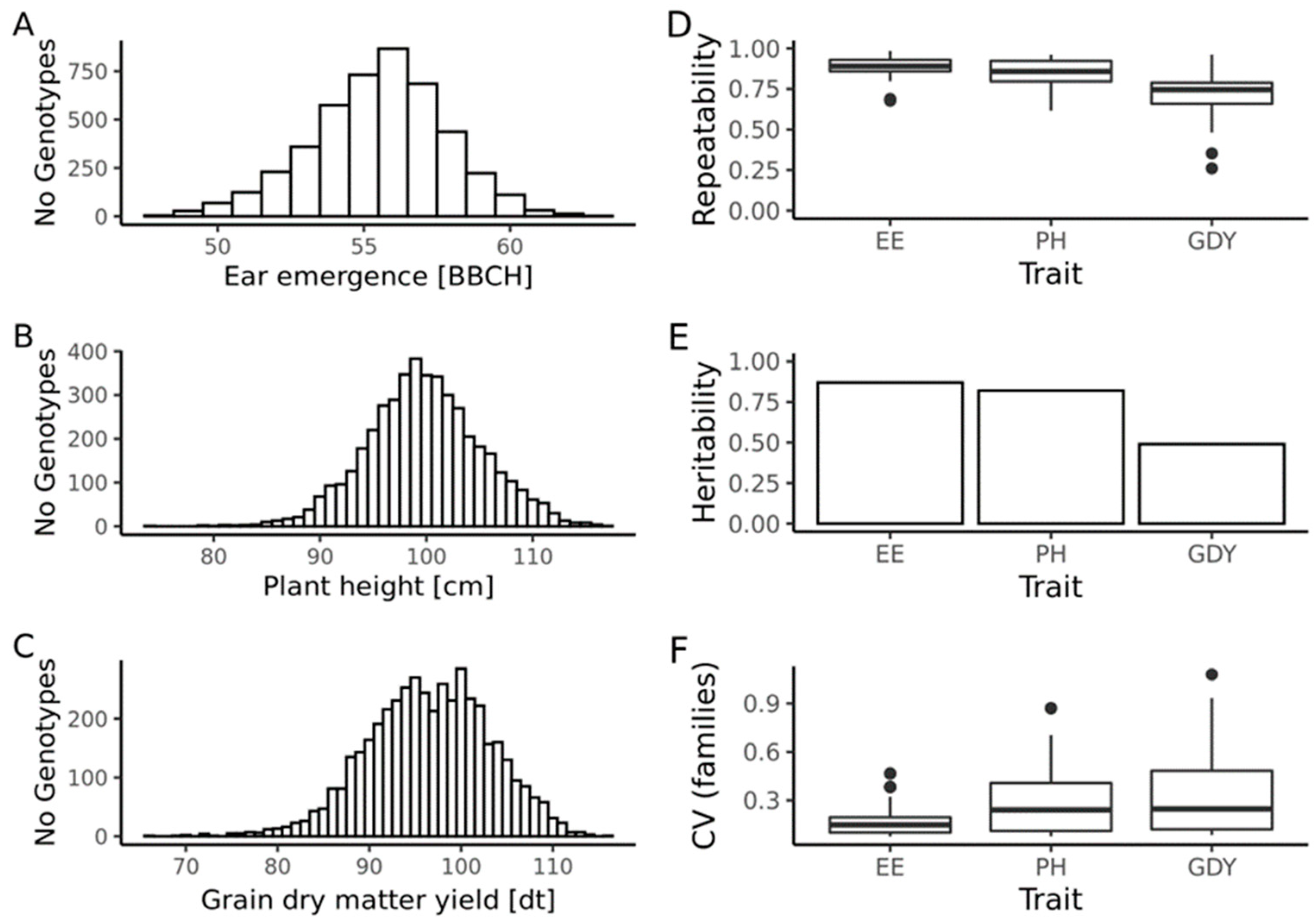
2.1. Quality of Phenotypic Data
2.2. Genome-Wide Prediction of Line Performances within and across Breeding Cycles
2.3. Prediction of the Family Mean, the Family Variance, and the Usefulness Criterion
3. Discussion
3.1. Leave-One-Cycle-Out Cross-Validations Revealed That Performances of Individual Genotypes Can Be Predicted across Breeding Cycles
3.2. Prediction Abilities of the Family Mean across Cycles Were Lower than Reported in Previous Simulation Studies
3.3. Low Ability to Predict the Genetic Variance of Families
3.4. Prediction of the Usefulness Criterion
4. Materials and Methods
4.1. Plant Material and Field Trials
4.2. Genomic Data
4.3. Phenotypic Data Analysis
4.4. Genome-Wide Prediction within and across Breeding Cycles
4.5. Prediction of the Family Mean
4.6. Prediction of the Family Variance
4.7. Prediction of the Usefulness Criterion
4.8. Validating Predictions of the Family Mean and the Family Variance
5. Conclusions
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
Appendix A

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Trait | Mean | Variance | UFCu | UFCµ | N° Families |
|---|---|---|---|---|---|
| Ear emergence | 0.64 | 0.44 | 0.67 | 0.68 | 21 |
| Plant height | 0.41 | 0.12 | 0.55 | 0.53 | 17 |
| Grain yield | 0.31 | 0.33 | −0.15 | −0.13 | 21 |
References
- Schnell, F.; Utz, H. Bericht über die Arbeitstagung der Vereinigung österreichischer Pflanzenzüchter; BAL Gumpenstein: Gumpenstein, Austria, 1975; pp. 243–248. Available online: https://scholar.google.com/scholar_lookup?title=Bericht+über+die+Arbeitstagung+der+Vereinigung+Österreichischer+Pflanzenzüchter&author=F.+W.+Schnell&author=H.+F.+Utz&publication_year=1975& (accessed on 26 September 2022).
- Condón, F.; Rasmusson, D.C.; Schiefelbein, E.; Velasquez, G.; Smith, K.P. Effect of Advanced Cycle Breeding on Genetic Gain and Phenotypic Diversity in Barley Breeding Germplasm. Crop Sci. 2009, 49, 1751–1761. [Google Scholar] [CrossRef]
- Melchinger, A.E.; Schmidt, W.; Geiger, H.H. Comparison of Testcrosses Produced from F2 and First Backcross Populations in Maize. Crop Sci. 1988, 28, 743–749. [Google Scholar] [CrossRef]
- Fountain, M.O.; Hallauer, A.R. Genetic Variation within Maize Breeding Populations. Crop Sci. 1996, 36, 26–32. [Google Scholar] [CrossRef]
- Utz, H.F.; Bohn, M.; Melchinger, A.E. Predicting Progeny Means and Variances of Winter Wheat Crosses from Phenotypic Values of Their Parents. Crop Sci. 2001, 41, 1470–1478. [Google Scholar] [CrossRef]
- Heffner, E.L.; Sorrells, M.E.; Jannink, J.-L. Genomic Selection for Crop Improvement. Crop Sci. 2009, 49, 1–12. [Google Scholar] [CrossRef]
- Jannink, J.-L. Dynamics of long-term genomic selection. Genet. Sel. Evol. 2010, 42, 35. [Google Scholar] [CrossRef]
- He, S.; Schulthess, A.W.; Mirdita, V.; Zhao, Y.; Korzun, V.; Bothe, R.; Ebmeyer, E.; Reif, J.C.; Jiang, Y. Genomic selection in a commercial winter wheat population. Theor. Appl. Genet. 2016, 129, 641–651. [Google Scholar] [CrossRef] [PubMed]
- Zhong, S.; Jannink, J.-L. Using Quantitative Trait Loci Results to Discriminate among Crosses on the Basis of Their Progeny Mean and Variance. Genetics 2007, 177, 567–576. [Google Scholar] [CrossRef]
- Bernardo, R. Genomewide Selection of Parental Inbreds: Classes of Loci and Virtual Biparental Populations. Crop Sci. 2014, 54, 2586–2595. [Google Scholar] [CrossRef]
- Mohammadi, M.; Tiede, T.; Smith, K.P. PopVar: A Genome-Wide Procedure for Predicting Genetic Variance and Correlated Response in Biparental Breeding Populations. Crop Sci. 2015, 55, 2068–2077. [Google Scholar] [CrossRef]
- Lado, B.; Battenfield, S.; Guzmán, C.; Quincke, M.; Singh, R.P.; Dreisigacker, S.; Peña, R.J.; Fritz, A.; Silva, P.; Poland, J.; et al. Strategies for Selecting Crosses Using Genomic Prediction in Two Wheat Breeding Programs. Plant Genome 2017, 10, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Yao, J.; Zhao, D.; Chen, X.; Zhang, Y.; Wang, J. Use of genomic selection and breeding simulation in cross prediction for improvement of yield and quality in wheat (Triticum aestivum L.). Crop J. 2018, 6, 353–365. [Google Scholar] [CrossRef]
- Lehermeier, C.; Campos, G.D.L.; Wimmer, V.; Schön, C.-C. Genomic variance estimates: With or without disequilibrium covariances? J. Anim. Breed. Genet. 2017, 134, 232–241. [Google Scholar] [CrossRef] [PubMed]
- Lehermeier, C.; Teyssèdre, S.; Schön, C.-C. Genetic Gain Increases by Applying the Usefulness Criterion with Improved Variance Prediction in Selection of Crosses. Genetics 2017, 207, 1651–1661. [Google Scholar] [CrossRef]
- Osthushenrich, T.; Frisch, M.; Herzog, E. Genomic selection of crossing partners on basis of the expected mean and variance of their derived lines. PLoS ONE 2017, 12, e0188839. [Google Scholar] [CrossRef]
- Osthushenrich, T.; Frisch, M.; Zenke-Philippi, C.; Jaiser, H.; Spiller, M.; Cselényi, L.; Krumnacker, K.; Boxberger, S.; Kopahnke, D.; Habekuß, A.; et al. Prediction of Means and Variances of Crosses with Genome-Wide Marker Effects in Barley. Front. Plant Sci. 2018, 871, 1899. [Google Scholar] [CrossRef]
- Wolfe, M.D.; Chan, A.W.; Kulakow, P.; Rabbi, I.; Jannink, J.-L. Genomic mating in outbred species: Predicting cross usefulness with additive and total genetic covariance matrices. Genetics 2021, 219, iyab122. [Google Scholar] [CrossRef]
- Zadoks, J.C.; Chang, T.T.; Konzak, C.F. A decimal code for the growth stages of cereals. Weed Res. 1974, 14, 415–421. [Google Scholar] [CrossRef]
- Thorwarth, P.; Ahlemeyer, J.; Bochard, A.-M.; Krumnacker, K.; Blümel, H.; Laubach, E.; Knöchel, N.; Cselényi, L.; Ordon, F.; Schmid, K.J. Genomic prediction ability for yield-related traits in German winter barley elite material. Theor. Appl. Genet. 2017, 130, 1669–1683. [Google Scholar] [CrossRef]
- Hofheinz, N.; Borchardt, D.; Weissleder, K.; Frisch, M. Genome-based prediction of test cross performance in two subsequent breeding cycles. Theor. Appl. Genet. 2012, 125, 1639–1645. [Google Scholar] [CrossRef]
- Michel, S.; Ametz, C.; Gungor, H.; Epure, D.; Grausgruber, H.; Löschenberger, F.; Buerstmayr, H. Genomic selection across multiple breeding cycles in applied bread wheat breeding. Theor. Appl. Genet. 2016, 129, 1179–1189. [Google Scholar] [CrossRef] [PubMed]
- Auinger, H.-J.; Schönleben, M.; Lehermeier, C.; Schmidt, M.; Korzun, V.; Geiger, H.H.; Piepho, H.-P.; Gordillo, A.; Wilde, P.; Bauer, E.; et al. Model training across multiple breeding cycles significantly improves genomic prediction accuracy in rye (Secale cereale L.). Theor. Appl. Genet. 2016, 129, 2043–2053. [Google Scholar] [CrossRef] [PubMed]
- Lin, Z.; Robinson, H.; Godoy, J.; Rattey, A.; Moody, D.; Mullan, D.; Keeble-Gagnere, G.; Forrest, K.; Tibbits, J.; Hayden, M.J.; et al. Genomic prediction for grain yield in a barley breeding program using genotype × environment interaction clusters. Crop Sci. 2021, 61, 2323–2335. [Google Scholar] [CrossRef]
- Hallauer, A.R.; Carena, M.J.; Filho, J.B.M. Quantitative Genetics in Maize Breeding, 3rd ed.; Springer: New York, NY, USA, 2010. [Google Scholar]
- Neyhart, J.L.; Smith, K.P. Validating Genomewide Predictions of Genetic Variance in a Contemporary Breeding Program. Crop Sci. 2019, 59, 1062–1072. [Google Scholar] [CrossRef]
- Adeyemo, E.; Bernardo, R. Predicting Genetic Variance from Genomewide Marker Effects Estimated from a Diverse Panel of Maize Inbreds. Crop Sci. 2019, 59, 583–590. [Google Scholar] [CrossRef]
- Zhang, H.; Yin, L.; Wang, M.; Yuan, X.; Liu, X. Factors Affecting the Accuracy of Genomic Selection for Agricultural Economic Traits in Maize, Cattle, and Pig Populations. Front. Genet. 2019, 10, 189. [Google Scholar] [CrossRef]
- Schmidt, M.; Kollers, S.; Maasberg-Prelle, A.; Großer, J.; Schinkel, B.; Tomerius, A.; Graner, A.; Korzun, V. Prediction of malting quality traits in barley based on genome-wide marker data to assess the potential of genomic selection. Theor. Appl. Genet. 2016, 129, 203–213. [Google Scholar] [CrossRef]
- Anscombe, F.J.; Tukey, J.W. The Examination and Analysis of Residuals. Qual. Assur. J. 1963, 5, 141–160. [Google Scholar] [CrossRef]
- Butler, D.G.; Cullis, B.R.; Gilmour, A.R.; Gogel, B.J.; Thompson, R. ASReml-R Reference Manual Version 4; VSN International Ltd.: Hemel Hempstead, UK, 2017. [Google Scholar]
- VanRaden, P. Efficient Methods to Compute Genomic Predictions. J. Dairy Sci. 2008, 91, 4414–4423. [Google Scholar] [CrossRef] [Green Version]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rembe, M.; Zhao, Y.; Wendler, N.; Oldach, K.; Korzun, V.; Reif, J.C. The Potential of Genome-Wide Prediction to Support Parental Selection, Evaluated with Data from a Commercial Barley Breeding Program. Plants 2022, 11, 2564. https://doi.org/10.3390/plants11192564
Rembe M, Zhao Y, Wendler N, Oldach K, Korzun V, Reif JC. The Potential of Genome-Wide Prediction to Support Parental Selection, Evaluated with Data from a Commercial Barley Breeding Program. Plants. 2022; 11(19):2564. https://doi.org/10.3390/plants11192564
Chicago/Turabian StyleRembe, Maximilian, Yusheng Zhao, Neele Wendler, Klaus Oldach, Viktor Korzun, and Jochen C. Reif. 2022. "The Potential of Genome-Wide Prediction to Support Parental Selection, Evaluated with Data from a Commercial Barley Breeding Program" Plants 11, no. 19: 2564. https://doi.org/10.3390/plants11192564