
Development of a Novel Tissue Blot Hybridization Chain Reaction for the Identification of Plant Viruses

Abstract
:1. Introduction
2. Results
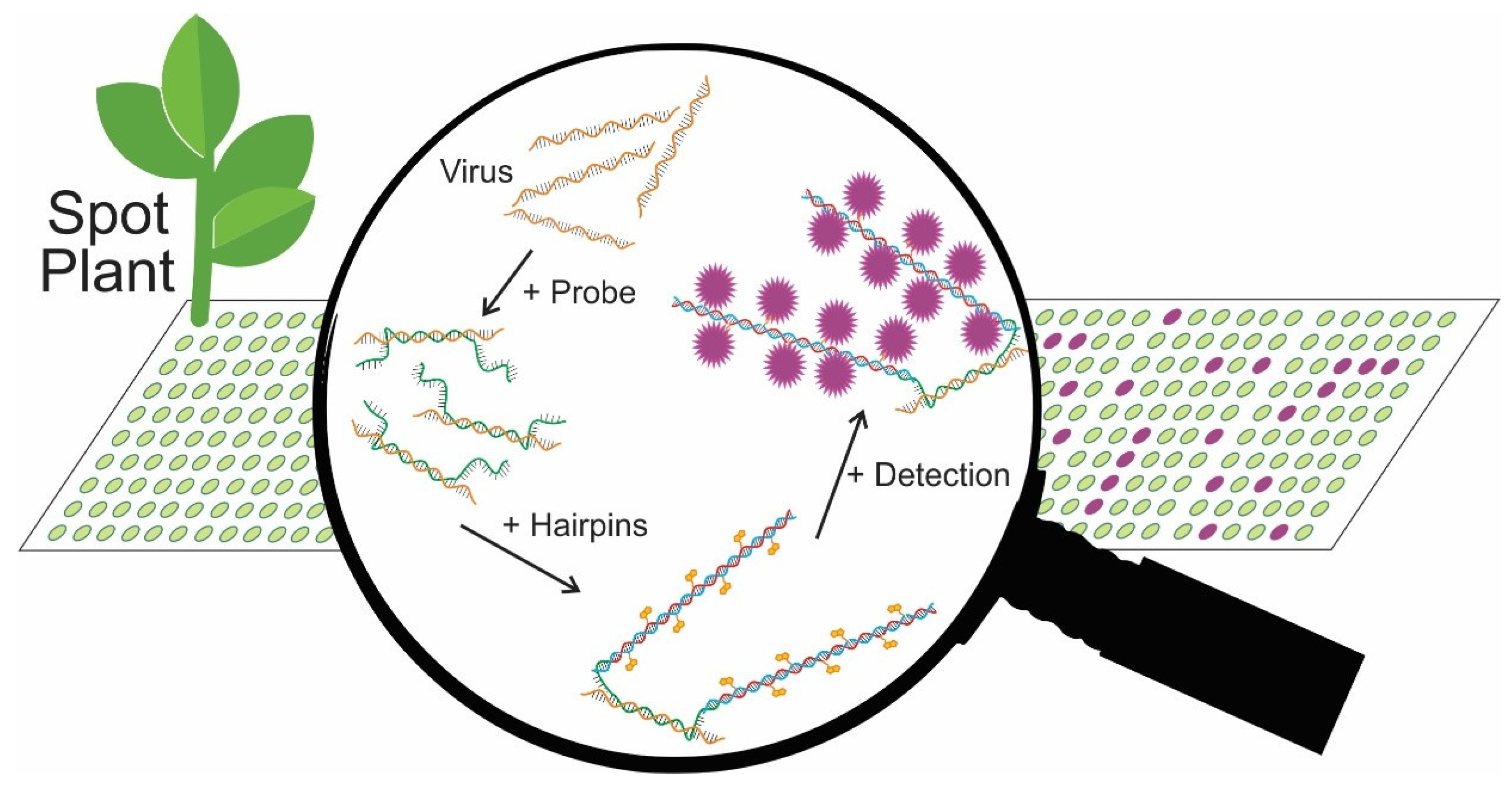
2.1. Principles of TB-HCR Method and Probe Design
2.1.1. Principles of TB-HCR
2.1.2. Probe and Hairpin Design
2.2. TB-HCR for TYLCV
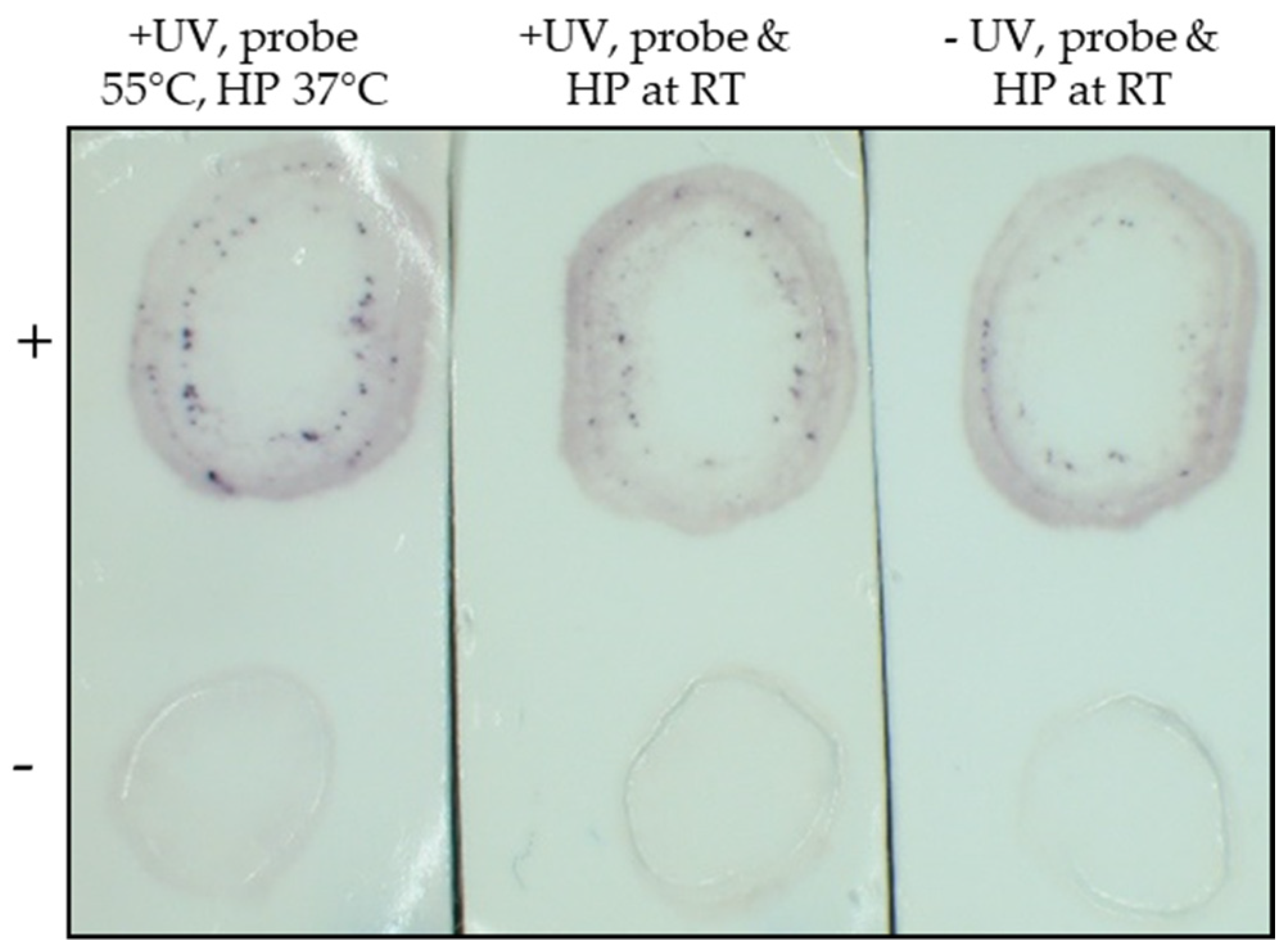
2.2.1. Optimization of TB-HCR Procedure
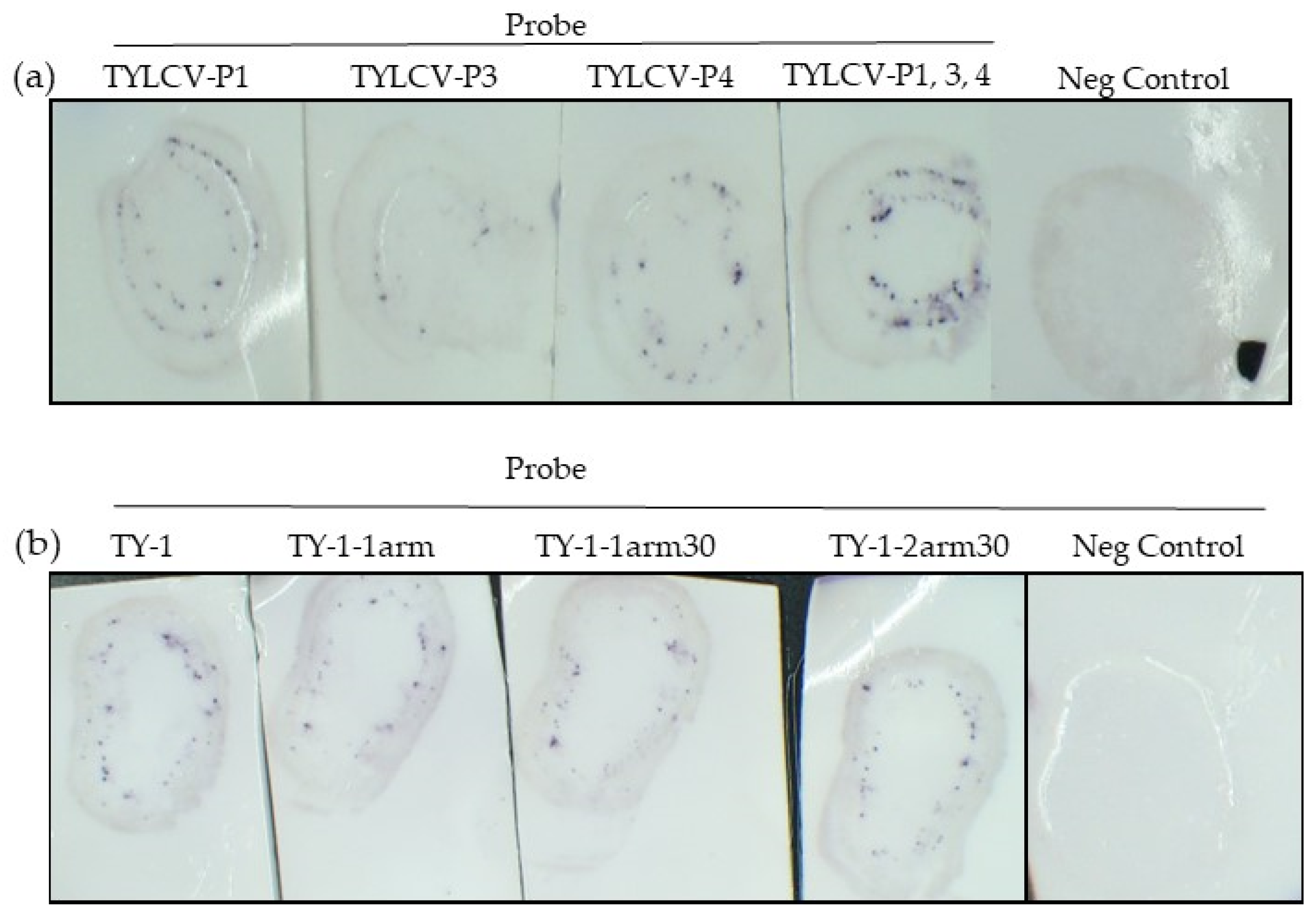
2.2.2. Using Multiple TYLCV Probes Improves the Signal
2.2.3. Different TYLCV-P1 Probe Arrangements
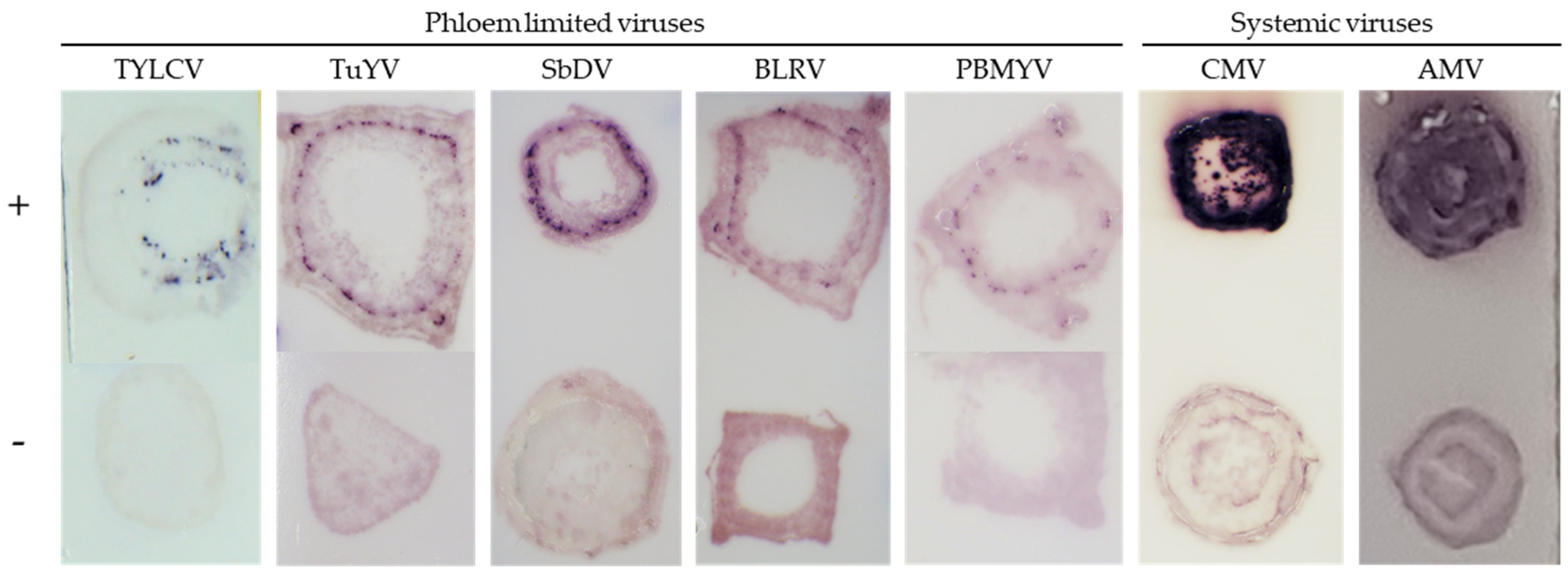
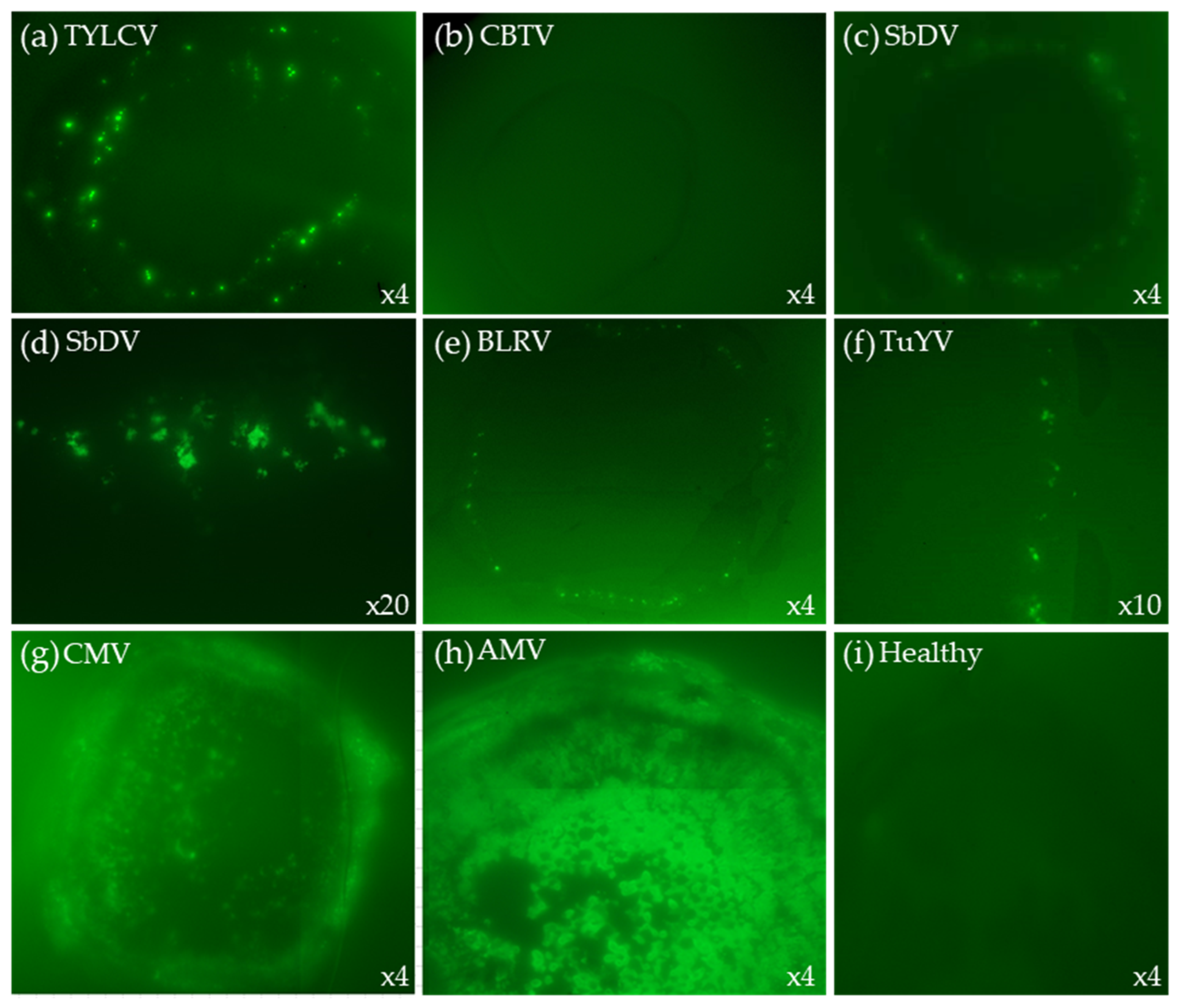
2.3. TB-HCR with Different Viruses
2.3.1. Phloem Limited Viruses
2.3.2. Systemic Viruses
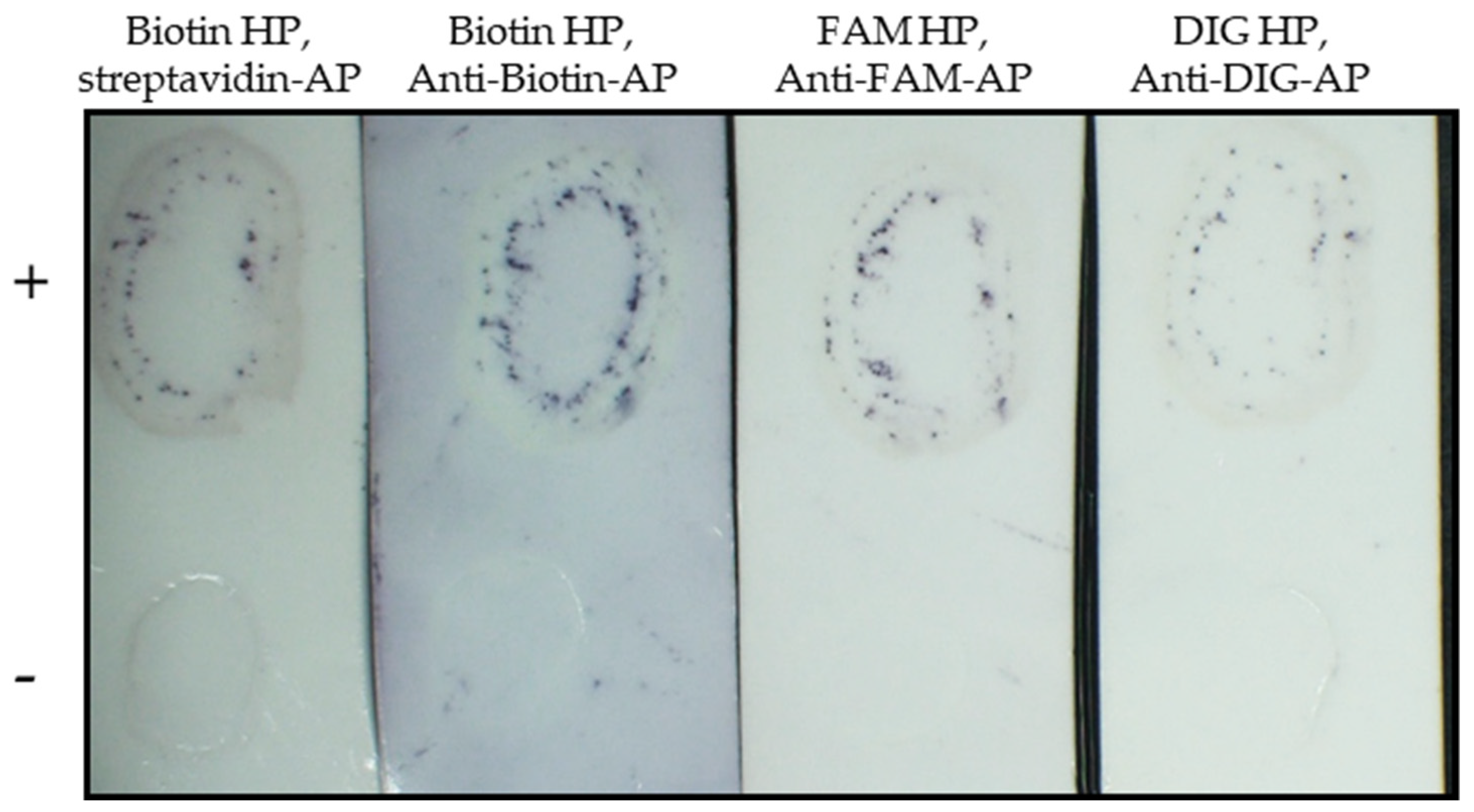
2.4. Different Reporter Methods
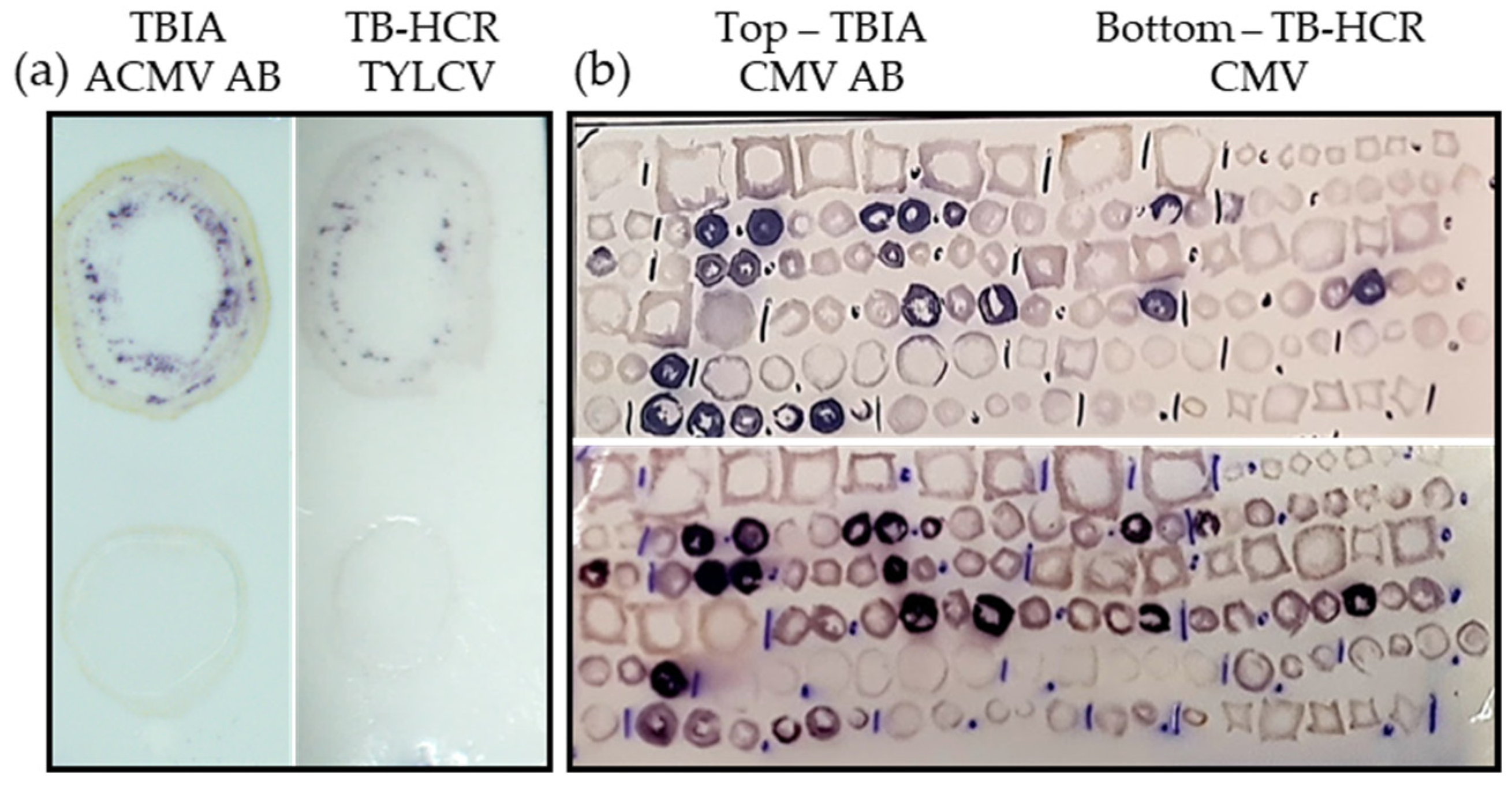
2.5. Comparison of TBIA and TB-HCR
3. Discussion
4. Materials and Methods
4.1. Plant Collection and Tissue Blots
4.2. DNA Probes and HCR Hairpins
4.3. Tissue Blot–Hybridization Chain Reaction
4.4. Tissue Blot Immunoassay
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Makkouk, K.M.; Comeau, A. Evaluation of various methods for the detection of barley yellow dwarf virus by the tissue-blot immunoassay and its use for virus detection in cereals inoculated at different growth stages. Eur. J. Plant Pathol. 1994, 100, 71–80. [Google Scholar] [CrossRef]
- Asaad, N.Y.; Kumari, S.G.; Haj-Kassem, A.A.; Shalaby, A.-B.A.; Al-Shaabi, S.; Malhotra, R.S. Detection and Characterization of Chickpea Chlorotic Stunt Virus in Syria. J. Phytopathol. 2009, 157, 756–761. [Google Scholar] [CrossRef]
- Katul, L. Characterization by Serology and Molecular Biology of Bean Leaf Roll Virus and Faba Bean Necrotic Yellows Virus. Ph.D. Thesis, University of Göttingen, Göttingen, Germany, 1992. [Google Scholar]
- Choi, H.M.T.; Beck, V.A.; Pierce, N.A. Next-Generation in Situ Hybridization Chain Reaction: Higher Gain, Lower Cost, Greater Durability. ACS Nano 2014, 8, 4284–4294. [Google Scholar] [CrossRef] [PubMed]
- Dirks, R.M.; Pierce, N.A. Triggered amplification by hybridization chain reaction. Proc. Natl. Acad. Sci. USA 2004, 101, 15275–15278. [Google Scholar] [CrossRef] [PubMed]
- Yin, P.; Choi, H.M.T.; Calvert, C.R.; Pierce, N.A. Programming biomolecular self-assembly pathways. Nature 2008, 451, 318–322. [Google Scholar] [CrossRef] [PubMed]
- Choi, H.M.T.; Chang, J.Y.; Trinh, L.A.; Padilla, J.E.; Fraser, S.E.; Pierce, N.A. Programmable in situ amplification for multiplexed imaging of mRNA expression. Nat. Biotechnol. 2010, 28, 1208–1212. [Google Scholar] [CrossRef] [PubMed]
- Zhao, J.; Chen, C.; Zhang, L.; Jiang, J.; Yu, R. An electrochemical aptasensor based on hybridization chain reaction with enzyme-signal amplification for interferon-gamma detection. Biosens. Bioelectron. 2012, 36, 129–134. [Google Scholar] [CrossRef] [PubMed]
- Song, W.; Zhu, K.; Cao, Z.; Lau, C.; Lu, J. Hybridization chain reaction-based aptameric system for the highly selective and sensitive detection of protein. Analyst 2012, 137, 1396–1401. [Google Scholar] [CrossRef] [PubMed]
- Zhu, G.; Zhang, S.; Song, E.; Zheng, J.; Hu, R.; Fang, X.; Tan, W. Building Fluorescent DNA Nanodevices on Target Living Cell Surfaces. Angew. Chem. Int. Ed. 2013, 52, 5490–5496. [Google Scholar] [CrossRef] [PubMed]
- Shimron, S.; Wang, F.; Orbach, R.; Willner, I. Amplified Detection of DNA through the Enzyme-Free Autonomous Assembly of Hemin/G-Quadruplex DNAzyme Nanowires. Anal. Chem. 2012, 84, 1042–1048. [Google Scholar] [CrossRef] [PubMed]
- Zhuang, J.; Fu, L.; Xu, M.; Yang, H.; Chen, G.; Tang, D. Sensitive electrochemical monitoring of nucleic acids coupling DNA nanostructures with hybridization chain reaction. Anal. Chim. Acta 2013, 783, 17–23. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Lau, C.; Kai, M.; Lu, J. Hybridization chain reaction-based instantaneous derivatization technology for chemiluminescence detection of specific DNA sequences. Analyst 2013, 138, 2691–2697. [Google Scholar] [CrossRef] [PubMed]
- Dong, J.; Cui, X.; Deng, Y.; Tang, Z. Amplified detection of nucleic acid by G-quadruplex based hybridization chain reaction. Biosens. Bioelectron. 2012, 38, 258–263. [Google Scholar] [CrossRef] [PubMed]
- Choi, J.; Routenberg Love, K.; Gong, Y.; Gierahn, T.M.; Love, J.C. Immuno-Hybridization Chain Reaction for Enhancing Detection of Individual Cytokine-Secreting Human Peripheral Mononuclear Cells. Anal. Chem. 2011, 83, 6890–6895. [Google Scholar] [CrossRef] [PubMed]
- Jou, A.F.-J.; Chou, Y.-T.; Willner, I.; Ho, J.-a.A. Imaging of Cancer Cells and Dictated Cytotoxicity Using Aptamer-Guided Hybridization Chain Reaction (HCR)-Generated G-Quadruplex Chains. Angew. Chem. Int. Ed. 2021, 60, 21673–21678. [Google Scholar] [CrossRef] [PubMed]
- Kim, K.T.; Angerani, S.; Winssinger, N. A minimal hybridization chain reaction (HCR) system using peptide nucleic acids. Chem. Sci. 2021, 12, 8218–8223. [Google Scholar] [CrossRef] [PubMed]
- Schwarzkopf, M.; Pierce, N.A. Multiplexed miRNA northern blots via hybridization chain reaction. Nucleic Acids Res. 2016, 44, e129. [Google Scholar] [CrossRef] [PubMed]
- Idili, A.; Porchetta, A.; Amodio, A.; Vallée-Bélisle, A.; Ricci, F. Controlling Hybridization Chain Reactions with pH. Nano Lett. 2015, 15, 5539–5544. [Google Scholar] [CrossRef] [PubMed]
- Xu, G.; Lai, M.; Wilson, R.; Glidle, A.; Reboud, J.; Cooper, J.M. Branched hybridization chain reaction—Using highly dimensional DNA nanostructures for label-free, reagent-less, multiplexed molecular diagnostics. Microsyst. Nanoeng. 2019, 5, 37. [Google Scholar] [CrossRef] [PubMed]
- Thomas, J.E.; Massalski, P.R.; Harrison, B.D. Production of Monoclonal Antibodies to African Cassava Mosaic Virus and Differences in Their Reactivities with Other Whitefly-transmitted Geminiviruses. J. Gen. Virol. 1986, 67, 2739. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Name | Virus DNA Probe Sequence | Probe Start (nt Position) on Reference Genome |
|---|---|---|
| Basic probe design |  | |
| Initiator1(init1) | GAGGAGGGCAGCAAACGGGAAGAGTCTTCCTTTACG | |
| Initiator2(init2) | GCATTCTTTCTTGAGGAGGGCAGCAAACGGGAAGAG | |
| A1-Biotin + | biotin–GAGGAGGGCTCCTTTACG TATTCTTGAGYATTCCATTAGMGAATGCYGGGCAGTCTGATAGACTCGGCCCGA | NC_003743 (3783 nt) |
| AMV-CP-PR1 | init1 TATT ACACGCAGCCCGTCTGTGGCAGTATAGTATTCGTCGGTT ATAT init2 | NC_ 002025 (1355 nt) |
| AMV-CP-PR2 | init1 TATT TTCTCTCGACCCAAACTTCGTTGAATCGGTATGAGGGA ATAT init2 | NC_ 002025 (1775 nt) |
| AMV-MP-PR1 | init1 TATT ACTTCATCAGCTAGTAACATTTCCTCAGCATAAGACACT ATAT init2 | NC_ 002025 (338 nt) |
| BLRV_PR3 | init1 TATT ATCCTGAATTGGTCCTCTGAAGTGTCGTGCCATGCCTGTCCATTGATA ATAT init2 | NC_003369 (3601 nt) |
| CMV-SG1_PR | init1 TATT TCGGCAAAGGATTAACTCGAATTTGAATGCGCGAAACAAGYTTCTTAT ATAT init2 | NC_001440 (1602 nt) |
| CMV-SG1_PR2 | init1 TATT ACCAGTACCGGTGAGGCTCCGTCCGCRAACATAGCAGAGATGGCGG ATAT init2 | NC_001440 (1711 nt) |
| CMV-SG1_PR3 | init1 TATT AACGTCTTRTTRAGTCGCGAAAGYTGYTGCGACARGACTCTAAAG ATAT init2 | NC_001440 (1393 nt) |
| CMV-SG2_PR | init1 TATT CGACGGAAAGATCGGATGATGAAGGYACTTTCCGAACTGTAACCC ATAT init2 | AJ585519 (1598 nt) |
| CMV-SG2_PR2 | init1 TATT TAGGAATTCGTTGATGCTCGACGTCGACATGAAGTACAATCTCGTCCT ATAT init2 | AJ585519 (1820 nt) |
| CMV-SG2_PR3 | init1 TATT GCATCCGCACCAGAAGCGGACCGAGAACCTCTACGCGGGCGACGA ATAT init2 | AJ585519 (1278 nt) |
| HCR-A1-TuYV | init1 TATT CTTGAGYATTCCATTAGMGAATGCYGGGCAGTCTGATAGACTCGGCCCGA ATAT init2 | NC_003743 (3783 nt) |
| HCR-A2 * | init1 TATT CTTGAGTATKCCATCWGMGARYGGCTTGCAYTCTGATARAGACGGCCCGA ATAT init2 | NC_003056 (3328 nt) |
| HCR-A3 * | init1 TATT ATATTCATGGTAGGCCTTGAGTATTCCAGAACTGAAYTCWGGCTTCTCTG ATAT init2 | NC_028793 (4017 nt) |
| PBMYV-PR2 | init1 TATT AGGGAGCTATATTTGCAATGGGGGTCCAGCTCATAAGCGATGGAACC ATAT init2 | MK955806 (4045 nt) |
| PBMYV-PR3 | init1 TATT GCCCCGTTTCCTTTGTATAGGATTCGGAACTGATCCTCGGATGCGTCG ATAT init2 | NC_028793 (4250 nt) |
| SbDV-PR2 | init1 TATT TCATAAGCGATGGARCCTGASGAGGTGGAAGAGGCCTCGGTGATG ATAT init2 | NC_003056 (3414 nt) |
| SbDV-PR3 | init1 TATT CCTCCTCTATTATTTCTCCTTCGTCGTCGTTGTCGTCCTCTGCGTTGTG ATAT init2 | NC_003056 (3189 nt) |
| TuYV-PR2 | init1 TATT GAAAGGGAGTTGAGTTTACAGTGTGGGTCCAGCTCGTAAGCGATGGAA ATAT init2 | NC_003743 (3901 nt) |
| TuYV-PR3 | init1 TATT TTGCCTTTGTAGAGGATCCTGAATTGGTCCTCGGCAACGTCGTGCCAT ATAT init2 | NC_003743 (4024 nt) |
| TY1-1arm | init1 ATAT GTACTGGGCTCATTATCGAACATATTAAAAACCTGTCCAAAATCCATT | GU178814 (807 nt) |
| TY1-1arm30 | init1 ATAT GCTCATTATCGAACATATTAAAAACCTGTCC | GU178814 (800 nt) |
| TY1-2arm30 | init1 ATAT GCTCATTATCGAACATATTAAAAACCTGTCC ATAT init2 | GU178814 (800 nt) |
| TYLCV-P1 | init1 ATAT GTACTGGGCTCATTATCGAACATATTAAAAACCTGTCCAAAATCCATT ATAT init2 | GU178814 (807 nt) |
| TYLCV-P2 | init1 ATAT CCCAAACAGGTCAGCACATTTCCATCCGAACATTCAGGCAGCTAAGAG ATAT init2 | GU178814 (2382 nt) |
| TYLCV-P3 | init1 ATAT TGCAAATATTTAATAGCTAACATACAACGAAATCCGTGAACAGATTCAGG ATAT init2 | GU178814 (224 nt) |
| TYLCV-P4 | init1 ATAT GCCATATACAATAACAAGGCGTTTTCAGTATGGTTCTCATACTTGGCTGC ATAT init2 | GU178814 (1008 nt) |
| Name | Hairpin Sequences | |
| Hpin-B1-1T (Choi et al. 2014) | CGTAAAGGAAGACTCTTCCCGTTTGCTGCCCTCCTCGCATTCTTTCTTGAGGAGGGCAGCAAACGGGAAGAG | |
| Hpin-B1-2 (Choi et al. 2014) | /5′–Biotin-C6/GAGGAGGGCAGCAAACGGGAAGAGTCTTCCTTTACGCTCTTCCCGTTTGCTGCCCTCCTCAAG AAAGAATGC | |
| HCR B1 H1 488 | CGTAAAGGAAGACTCTTCCCGTTTGCTGCCCTCCTCGCATTCTTTCTTGAGGAGGGCAGCAAACGGGAAGAG/iSp9/3AlexaF488N/ | |
| HCR B1 H2 488 | /5ATTO488N/iSp18/GAGGAGGGCAGCAAACGGGAAGAGTCTTCCTTTACGCTCTTCCCGTTTGCTGCCCTCCTCAAGAAAGAATGC | |
| Hpin B1-2D | DIG-GAGGAGGGCAGCAAACGGGAAGAGTCTTCCTTTACGCTCTTCCCGTTTGCTGCCCTCCTCAAGAAAGAATGC | |
| Hpin B1 H2-FAM | FAM-GAGGAGGGCAGCAAACGGGAAGAGTCTTCCTTTACGCTCTTCCCGTTTGCTGCCCTCCTCAAGAAAGAATGC |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Filardo, F.; Vukovic, P.; Sharman, M.; Gambley, C.; Campbell, P. Development of a Novel Tissue Blot Hybridization Chain Reaction for the Identification of Plant Viruses. Plants 2022, 11, 2325. https://doi.org/10.3390/plants11172325
Filardo F, Vukovic P, Sharman M, Gambley C, Campbell P. Development of a Novel Tissue Blot Hybridization Chain Reaction for the Identification of Plant Viruses. Plants. 2022; 11(17):2325. https://doi.org/10.3390/plants11172325
Chicago/Turabian StyleFilardo, Fiona, Peter Vukovic, Murray Sharman, Cherie Gambley, and Paul Campbell. 2022. "Development of a Novel Tissue Blot Hybridization Chain Reaction for the Identification of Plant Viruses" Plants 11, no. 17: 2325. https://doi.org/10.3390/plants11172325