
Annotation of Siberian Larch (Larix sibirica Ledeb.) Nuclear Genome—One of the Most Cold-Resistant Tree Species in the Only Deciduous GENUS in Pinaceae

 ,
,
Abstract
:1. Introduction
2. Results
2.1. Transcriptome Assembly
2.2. Repeat Content
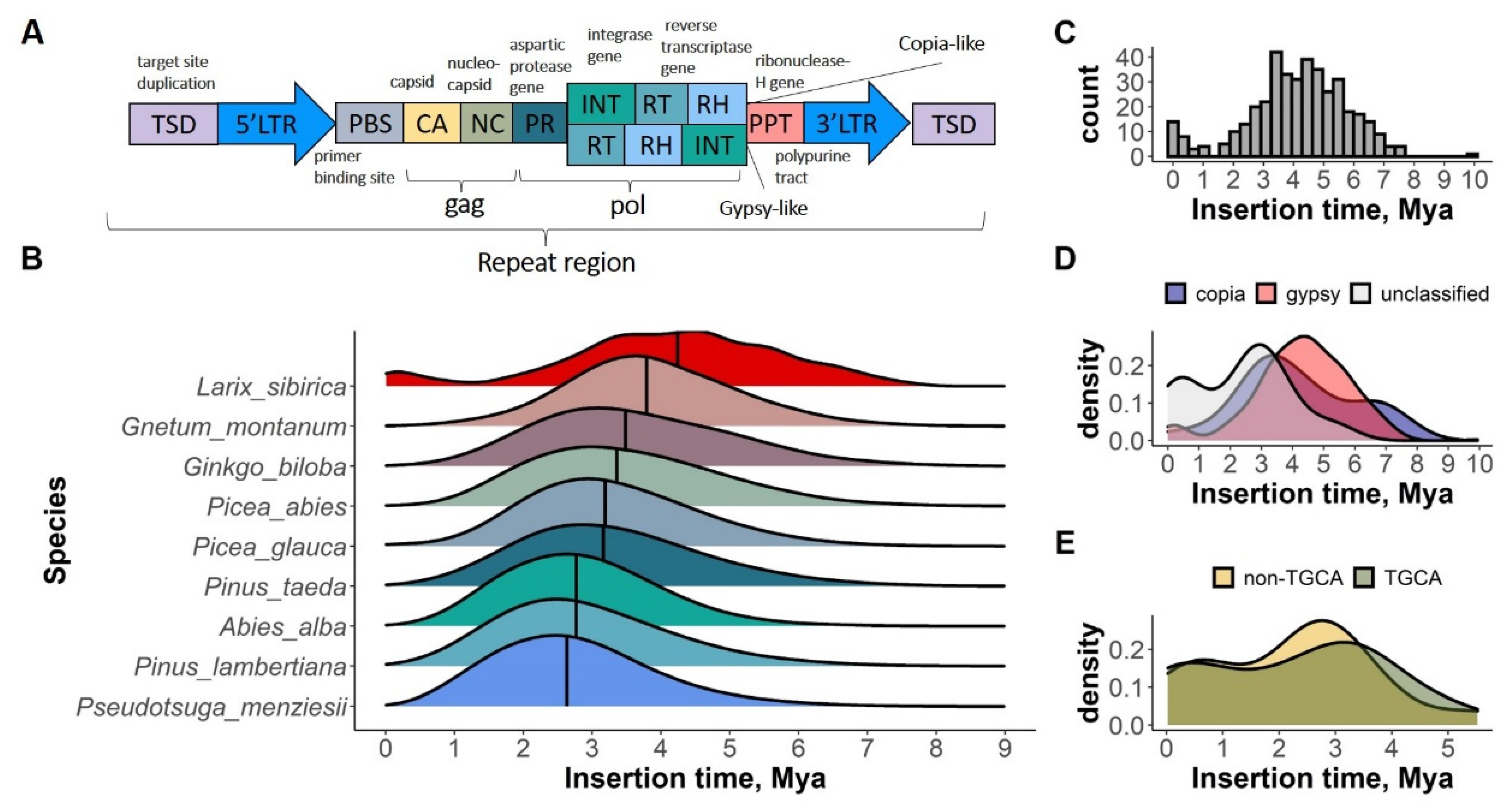
2.3. LTR-RT Insertion Time Estimate
2.4. Identification of LRR Genes
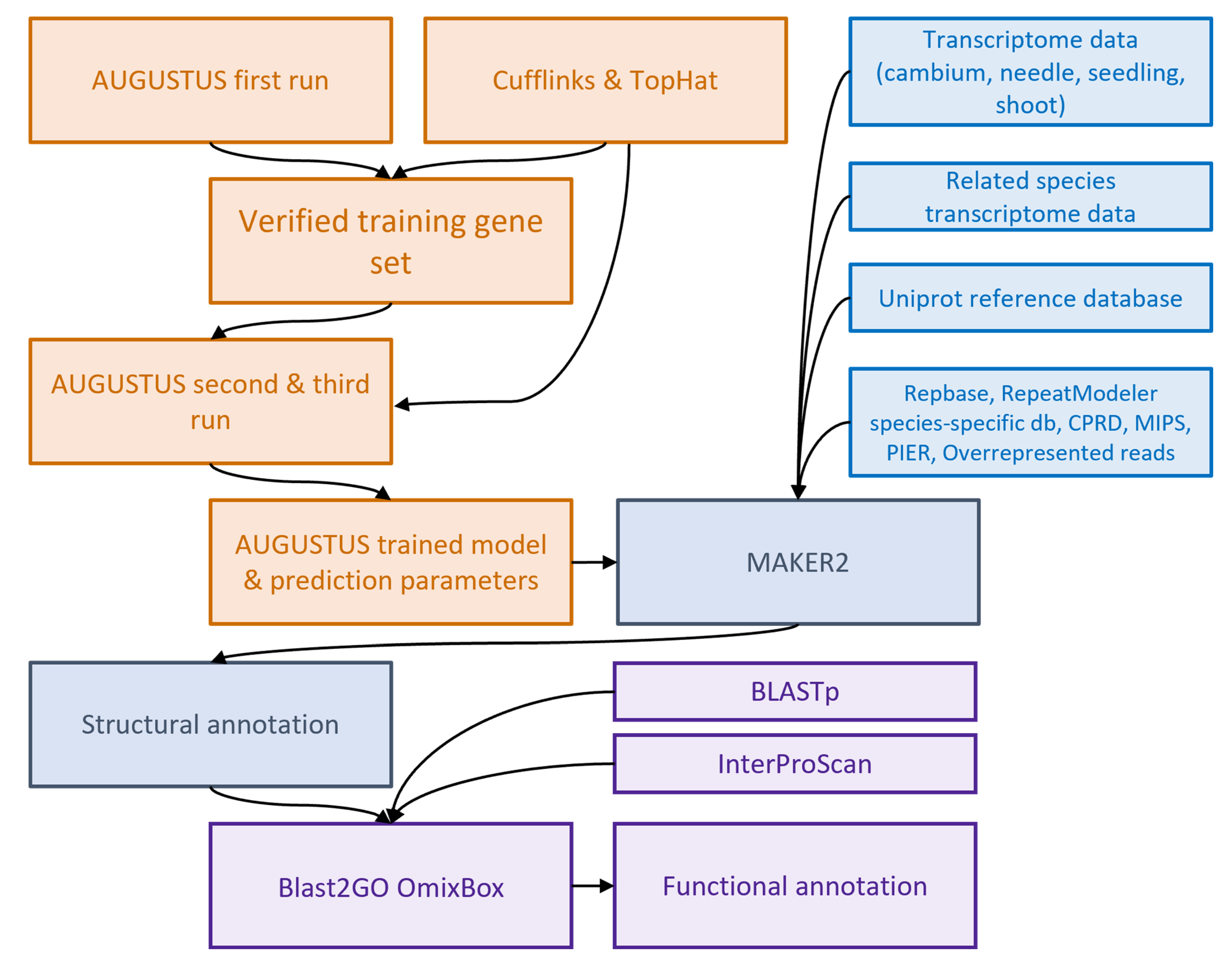
2.5. Structural Annotation Using MAKER2
2.6. Functional Annotation
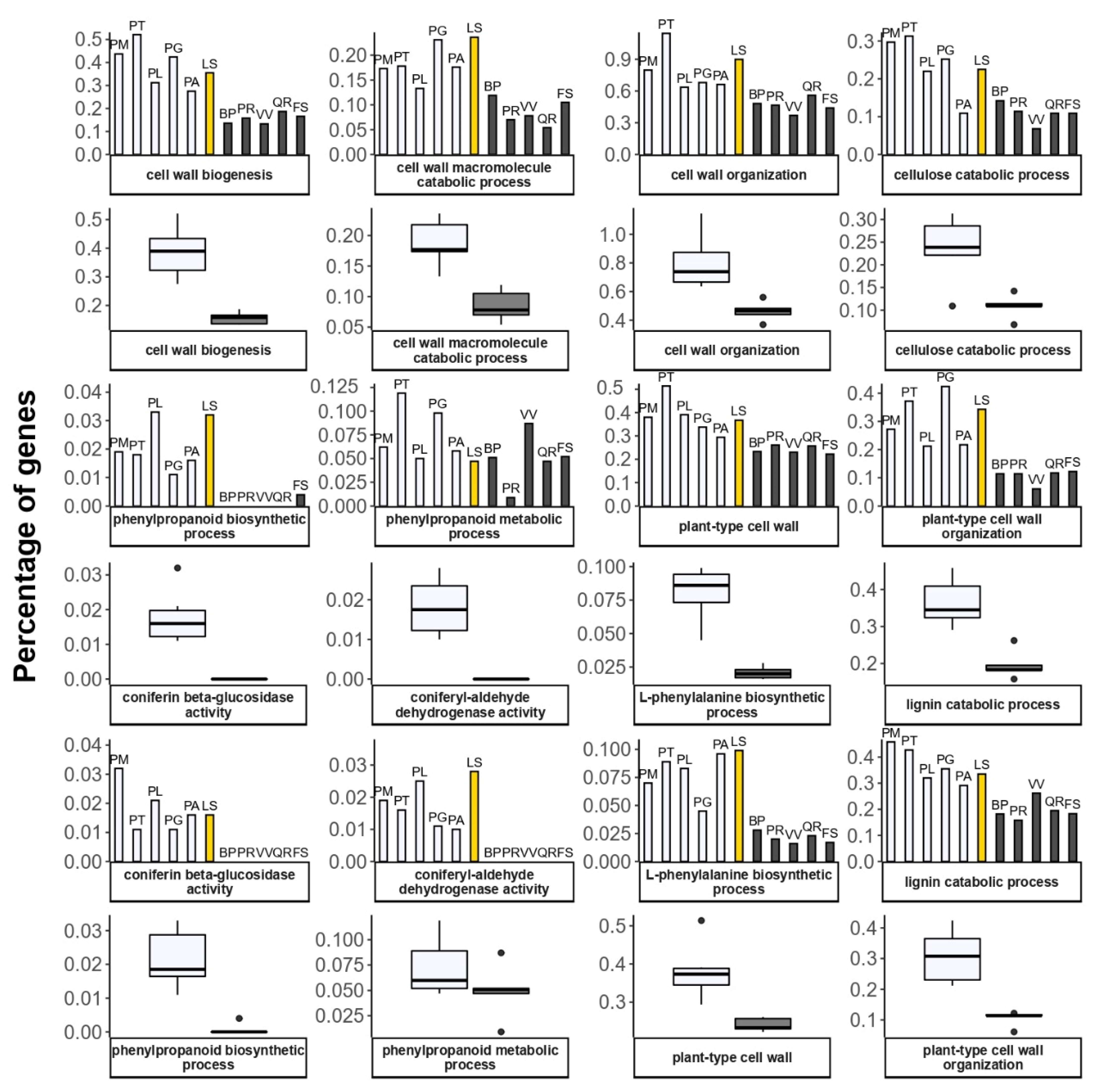
2.7. Comparing GO Annotations between Conifer and Angiosperm Species
3. Discussion
3.1. Repeat Content and LTR Insertion Time Estimate
3.2. Structural Annotation Using AUGUSTUS and MAKER2 Pipeline
3.3. Functional Annotation
3.3.1. Cell Wall and Phenylalanine Metabolism
3.3.2. Programmed Cell Death and Autophagy
3.3.3. Hormones
4. Materials and Methods
4.1. Genome Data
4.2. Transcriptome Sequencing and Assembly
4.3. Repetitive Elements (REs) Analysis and Masking
4.4. Identification of LRR Genes
4.5. AUGUSTUS Training
4.6. MAKER Annotation
4.7. Assembly Evaluation and Functional Annotation
4.8. LTR-RT Insertion Time Estimation
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- McLoughlin, S. Gymnosperms. In Encyclopedia of Geology, 2nd ed.; Alderton, D., Elias, S.A., Eds.; Elsevier: Amsterdam, The Netherlands, 2021; Volume 3, pp. 476–500. [Google Scholar] [CrossRef]
- Brenner, E.D.; Stevenson, D. Using Genomics to Study Evolutionary Origins of Seeds. In Landscapes, Genomics and Transgenic Conifers. Managing Forest Ecosystems; Williams, C.G., Ed.; Springer: Dordrecht, The Netherlands, 2006; Volume 9, pp. 85–106. [Google Scholar] [CrossRef]
- Soltis, P.S.; Soltis, D.E.; Savolainen, V.; Crane, P.R.; Barraclough, T.G. Rate heterogeneity among lineages of tracheophytes: Integration of molecular and fossil data and evidence for molecular living fossils. Proc. Natl. Acad. Sci. USA 2002, 99, 4430–4435. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wan, T.; Liu, Z.-M.; Li, L.-F.; Leitch, A.R.; Leitch, I.J.; Lohaus, R.; Liu, Z.-J.; Xin, H.-P.; Gong, Y.-B.; Liu, Y.; et al. A genome for gnetophytes and early evolution of seed plants. Nat. Plants 2018, 4, 82–89. [Google Scholar] [CrossRef] [PubMed]
- Stevens, K.A.; Wegrzyn, J.L.; Zimin, A.; Puiu, D.; Crepeau, M.; Cardeno, C.; Paul, R.; Gonzalez-Ibeas, D.; Koriabine, M.; Holtz-Morris, A.E.; et al. Sequence of the sugar pine megagenome. Genetics 2016, 204, 1613–1626. [Google Scholar] [CrossRef] [Green Version]
- Berardini, T.Z.; Reiser, L.; Li, D.; Mezheritsky, Y.; Muller, R.; Strait, E.; Huala, E. The Arabidopsis information resource: Making and mining the “gold standard” annotated reference plant genome. Genesis 2015, 53, 474–485. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Badouin, H.; Gouzy, J.; Grassa, C.J.; Murat, F.; Staton, S.E.; Cottret, L.; Lelandais-Brière, C.; Owens, G.L.; Carrère, S.; Mayjonade, B.; et al. The sunflower genome provides insights into oil metabolism, flowering and Asterid evolution. Nature 2017, 546, 148–152. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zimin, A.V.; Puiu, D.; Hall, R.; Kingan, S.; Clavijo, B.J.; Salzberg, S.L. The first near-complete assembly of the hexaploid bread wheat genome, Triticum aestivum. GigaScience 2017, 6, gix097. [Google Scholar] [CrossRef] [Green Version]
- Pellicer, J.; Fay, M.F.; Leitch, I.J. The largest eukaryotic genome of them all? Bot. J. Linn. Soc. 2010, 164, 10–15. [Google Scholar] [CrossRef] [Green Version]
- Neale, D.B.; Wegrzyn, J.L.; Stevens, K.A.; Zimin, A.V.; Puiu, D.; Crepeau, M.W.; Cardeno, C.; Koriabine, M.; Holtz-Morris, A.E.; Liechty, J.D.; et al. Decoding the massive genome of loblolly pine using haploid DNA and novel assembly strategies. Genome Biol. 2014, 15, R59. [Google Scholar] [CrossRef] [Green Version]
- Zimin, A.; Stevens, K.A.; Crepeau, M.W.; Holtz-Morris, A.; Koriabine, M.; Marçais, G.; Puiu, D.; Roberts, M.; Wegrzyn, J.L.; de Jong, P.J.; et al. Sequencing and assembly of the 22-Gb loblolly pine genome. Genetics 2014, 196, 875–890. [Google Scholar] [CrossRef] [Green Version]
- Warren, R.L.; Keeling, C.I.; Yuen, M.M.S.; Raymond, A.; Taylor, G.A.; Vandervalk, B.P.; Mohamadi, H.; Paulino, D.; Chiu, R.; Jackman, S.D.; et al. Improved white spruce (Picea glauca) genome assemblies and annotation of large gene families of conifer terpenoid and phenolic defense metabolism. Plant J. Cell Mol. Biol. 2015, 83, 189–212. [Google Scholar] [CrossRef]
- Li, Z.; Baniaga, A.E.; Sessa, E.B.; Scascitelli, M.; Graham, S.W.; Rieseberg, L.H.; Barker, M.S. Early genome duplications in conifers and other seed plants. Sci. Adv. 2015, 1, e1501084. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qiao, X.; Li, Q.; Yin, H.; Qi, K.; Li, L.; Wang, R.; Zhang, S.; Paterson, A.H. Gene duplication and evolution in recurring polyploidization–diploidization cycles in plants. Genome Biol. 2019, 20, 38. [Google Scholar] [CrossRef] [Green Version]
- Perera, D.; Magbanua, Z.V.; Thummasuwan, S.; Mukherjee, D.; Arick, M.; Chouvarine, P.; Nairn, C.J.; Schmutz, J.; Grimwood, J.; Dean, J.F.D.; et al. Exploring the loblolly pine (Pinus taeda L.) genome by BAC sequencing and Cot analysis. Gene 2018, 663, 165–177. [Google Scholar] [CrossRef] [PubMed]
- Neale, D.B.; McGuire, P.E.; Wheeler, N.C.; Stevens, K.A.; Crepeau, M.W.; Cardeno, C.; Zimin, A.V.; Puiu, D.; Pertea, G.M.; Sezen, U.U.; et al. The Douglas-fir genome sequence reveals specialization of the photosynthetic apparatus in Pinaceae. G3 Genes Genomes Genet. 2017, 7, 3157–3167. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wegrzyn, J.L.; Liechty, J.D.; Stevens, K.A.; Wu, L.-S.; Loopstra, C.A.; Vasquez-Gross, H.A.; Dougherty, W.M.; Lin, B.Y.; Zieve, J.J.; Martínez-García, P.J.; et al. Unique features of the loblolly pine (Pinus taeda L.) megagenome revealed through sequence annotation. Genetics 2014, 196, 891–909. [Google Scholar] [CrossRef] [Green Version]
- Pellicer, J.; Hidalgo, O.; Dodsworth, S.; Leitch, I.J. Genome size diversity and its impact on the evolution of land plants. Genes 2018, 9, 88. [Google Scholar] [CrossRef] [Green Version]
- Nystedt, B.; Street, N.R.; Wetterbom, A.; Zuccolo, A.; Lin, Y.-C.; Scofield, D.G.; Vezzi, F.; Delhomme, N.; Giacomello, S.; Alexeyenko, A.; et al. The Norway Spruce genome sequence and conifer genome evolution. Nature 2013, 497, 579–584. [Google Scholar] [CrossRef] [Green Version]
- Mosca, E.; Cruz, F.; Gómez-Garrido, J.; Bianco, L.; Rellstab, C.; Brodbeck, S.; Csilléry, K.; Fady, B.; Fladung, M.; Fussi, B.; et al. A reference genome sequence for the European silver fir (Abies alba Mill.): A community-generated genomic resource. G3 Genes Genomes Genet. 2019, 9, 2039–2049. [Google Scholar] [CrossRef] [Green Version]
- Kuzmin, D.A.; Feranchuk, S.I.; Sharov, V.V.; Cybin, A.N.; Makolov, S.V.; Putintseva, Y.A.; Oreshkova, N.V.; Krutovsky, K.V. Stepwise large genome assembly approach: A case of Siberian larch (Larix sibirica Ledeb). BMC Bioinform. 2019, 20, 37. [Google Scholar] [CrossRef] [Green Version]
- Sun, C.; Xie, Y.-H.; Li, Z.; Liu, Y.-J.; Sun, X.-M.; Li, J.-J.; Quan, W.-P.; Zeng, Q.-Y.; Van de Peer, Y.; Zhang, S.-G. The Larix kaempferi genome reveals new insights into wood properties. J. Integr. Plant Biol. 2022, 64, 1364–1373. [Google Scholar] [CrossRef]
- Niu, S.; Li, J.; Bo, W.; Yang, W.; Zuccolo, A.; Giacomello, S.; Chen, X.; Han, F.; Yang, J.; Song, Y.; et al. The Chinese pine genome and methylome unveil key features of conifer evolution. Cell 2022, 185, 204–217.e14. [Google Scholar] [CrossRef] [PubMed]
- Semerikov, V.L.; Lascoux, M. Nuclear and cytoplasmic variation within and between Eurasian Larix (Pinaceae) species. Am. J. Bot. 2003, 90, 1113–1123. [Google Scholar] [CrossRef] [PubMed]
- Tumenjargal, B.; Ishiguri, F.; Aiso, H.; Takahashi, Y.; Nezu, I.; Takashima, Y.; Baasan, B.; Chultem, G.; Ohshima, J.; Yokota, S. Physical and mechanical properties of wood and their geographic variations in Larix sibirica trees naturally grown in Mongolia. Sci. Rep. 2020, 10, 12936. [Google Scholar] [CrossRef] [PubMed]
- Semerikov, V.L.; Semerikova, S.A.; Polezhaeva, M.A.; Kosintsev, P.A.; Lascoux, M. Southern montane populations did not contribute to the recolonization of West Siberian Plain by Siberian larch (Larix sibirica): A range-wide analysis of cytoplasmic markers. Mol. Ecol. 2013, 22, 4958–4971. [Google Scholar] [CrossRef] [PubMed]
- Dulamsuren, C.; Hauck, M.; Khishigjargal, M.; Leuschner, H.H.; Leuschner, C. Diverging climate trends in Mongolian taiga forests influence growth and regeneration of Larix sibirica. Oecologia 2010, 163, 1091–1102. [Google Scholar] [CrossRef] [Green Version]
- Babushkina, E.A.; Vaganov, E.A.; Grachev, A.M.; Oreshkova, N.V.; Belokopytova, L.V.; Kostyakova, T.V.; Krutovsky, K.V. The effect of individual genetic heterozygosity on general homeostasis, heterosis and resilience in Siberian larch (Larix sibirica Ledeb.) using dendrochronology and microsatellite loci genotyping. Dendrochronologia 2016, 38, 26–37. [Google Scholar] [CrossRef] [Green Version]
- Oreshkova, N.V.; Belokon’, M.M.; Zham’’iansuren, S. Genetic diversity, population structure, and differentiation of Siberian larch, Gmelin larch and Cajander larch on SSR-markers data. Genetika 2013, 49, 204–213. [Google Scholar] [CrossRef]
- Oreshkova, N.V.; Putintseva, Y.A.; Sharov, V.V.; Kuzmin, D.A.; Krutovsky, K.V. Development of microsatellite genetic markers in Siberian larch (Larix sibirica Ledeb.) based on the de novo whole genome sequencing. Russ. J. Genet. 2017, 53, 1194–1199. [Google Scholar] [CrossRef] [Green Version]
- Oreshkova, N.V.; Bondar, E.I.; Putintseva, Y.A.; Sharov, V.V.; Kuzmin, D.A.; Krutovsky, K.V. Development of nuclear microsatellite markers with long (tri-, tetra-, penta-, and hexanucleotide) motifs for three larch species based on the de novo whole genome sequencing of Siberian larch (Larix sibirica Ledeb.). Russ. J. Genet. 2019, 55, 444–450. [Google Scholar] [CrossRef]
- Krutovsky, K.V.; Putintseva, Y.A.; Oreshkova, N.V.; Bondar, E.I.; Sharov, V.V.; Kuzmin, D.A. Postgenomic technologies in practical forestry: Development of genome-wide markers for timber origin identification and other applications. For. Eng. J. 2019, 9, 9–16. [Google Scholar] [CrossRef]
- Bondar, E.I.; Putintseva, Y.A.; Oreshkova, N.V.; Krutovsky, K.V. Siberian larch (Larix sibirica Ledeb.) chloroplast genome and development of polymorphic chloroplast markers. BMC Bioinform. 2019, 20, 38. [Google Scholar] [CrossRef] [PubMed]
- Putintseva, Y.A.; Bondar, E.I.; Simonov, E.P.; Sharov, V.V.; Oreshkova, N.V.; Kuzmin, D.A.; Konstantinov, Y.M.; Shmakov, V.N.; Belkov, V.I.; Sadovsky, M.G.; et al. Siberian larch (Larix sibirica Ledeb.) mitochondrial genome assembled using both short and long nucleotide sequence reads is currently the largest known mitogenome. BMC Genom. 2020, 21, 654. [Google Scholar] [CrossRef] [PubMed]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grabherr, M.G.; Haas, B.J.; Yassour, M.; Levin, J.Z.; Thompson, D.A.; Amit, I.; Adiconis, X.; Fan, L.; Raychowdhury, R.; Zeng, Q.; et al. Full-length transcriptome assembly from RNA-seq data without a reference genome. Nat. Biotechnol. 2011, 29, 644–652. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smit, A.; Hubley, R. RepeatModeler Open-1.0. 2008. Available online: https://www.repeatmasker.org/RepeatModeler (accessed on 6 June 2018).
- Smit, A.; Hubley, R.; Green, P. RepeatMasker Open-4.0. 2013. Available online: https://www.repeatmasker.org/RepeatMasker (accessed on 23 January 2016).
- Wegrzyn, J.L.; Lin, B.Y.; Zieve, J.J.; Dougherty, W.M.; Martínez-García, P.J.; Koriabine, M.; Holtz-Morris, A.; deJong, P.; Crepeau, M.; Langley, C.H.; et al. Insights into the loblolly pine genome: Characterization of BAC and fosmid sequences. PLoS ONE 2013, 8, e72439. [Google Scholar] [CrossRef] [Green Version]
- Magbanua, Z.V.; Ozkan, S.; Bartlett, B.D.; Chouvarine, P.; Saski, C.A.; Liston, A.; Cronn, R.C.; Nelson, C.D.; Peterson, D.G. Adventures in the enormous: A 1.8 million clone BAC library for the 21.7 Gb genome of loblolly pine. PLoS ONE 2011, 6, e16214. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.; Lu, P.; Luo, Z. GMATo: A novel tool for the identification and analysis of microsatellites in large genomes. Bioinformation 2013, 9, 541–544. [Google Scholar] [CrossRef] [Green Version]
- Benson, G. Tandem repeats finder: A program to analyze DNA sequences. Nucleic Acids Res. 1999, 27, 573–580. [Google Scholar] [CrossRef] [Green Version]
- Ellinghaus, D.; Kurtz, S.; Willhoeft, U. LTRharvest, an efficient and flexible software for de novo detection of LTR retrotransposons. BMC Bioinform. 2008, 9, 18. [Google Scholar] [CrossRef] [Green Version]
- Ou, S.; Jiang, N. LTR_retriever: A highly accurate and sensitive program for identification of long terminal repeat retrotransposons. Plant Physiol. 2018, 176, 1410–1422. [Google Scholar] [CrossRef] [Green Version]
- Mistry, J.; Finn, R.D.; Eddy, S.R.; Bateman, A.; Punta, M. Challenges in homology search: HMMER3 and convergent evolution of coiled-coil regions. Nucleic Acids Res. 2013, 41, e121. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Conesa, A.; Götz, S. Blast2GO: A comprehensive suite for functional analysis in plant genomics. Int. J. Plant Genom. 2008, 2008, 619832. [Google Scholar] [CrossRef] [PubMed]
- Götz, S.; García-Gómez, J.M.; Terol, J.; Williams, T.D.; Nagaraj, S.H.; Nueda, M.J.; Robles, M.; Talón, M.; Dopazo, J.; Conesa, A. High-throughput functional annotation and data mining with the Blast2GO suite. Nucleic Acids Res. 2008, 36, 3420–3435. [Google Scholar] [CrossRef] [PubMed]
- Jones, P.; Binns, D.; Chang, H.-Y.; Fraser, M.; Li, W.; McAnulla, C.; McWilliam, H.; Maslen, J.; Mitchell, A.; Nuka, G.; et al. InterProScan 5: Genome-scale protein function classification. Bioinformatics 2014, 30, 1236–1240. [Google Scholar] [CrossRef] [Green Version]
- Seppey, M.; Manni, M.; Zdobnov, E.M. BUSCO: Assessing Genome Assembly and Annotation Completeness. In Gene Prediction. Methods in Molecular Biology; Kollmar, M., Ed.; Humana: New York, NY, USA, 2019; Volume 1962, pp. 227–245. [Google Scholar] [CrossRef]
- Holt, C.; Yandell, M. MAKER2: An annotation pipeline and genome-database management tool for second-generation genome projects. BMC Bioinform. 2011, 12, 491. [Google Scholar] [CrossRef] [Green Version]
- Koralewski, T.E.; Krutovsky, K.V. Evolution of exon-intron structure and alternative splicing. PLoS ONE 2011, 6, e18055. [Google Scholar] [CrossRef] [Green Version]
- Eilbeck, K.; Moore, B.; Holt, C.; Yandell, M. Quantitative measures for the management and comparison of annotated genomes. BMC Bioinform. 2009, 10, 67. [Google Scholar] [CrossRef] [Green Version]
- Eilbeck, K.; Lewis, S.E.; Mungall, C.J.; Yandell, M.; Stein, L.; Durbin, R.; Ashburner, M. The sequence ontology: A tool for the unification of genome annotations. Genome Biol. 2005, 6, R44. [Google Scholar] [CrossRef] [Green Version]
- Sena, J.S.; Giguère, I.; Boyle, B.; Rigault, P.; Birol, I.; Zuccolo, A.; Ritland, K.; Ritland, C.; Bohlmann, J.; Jones, S.; et al. Evolution of gene structure in the conifer Picea glauca: A comparative analysis of the impact of intron size. BMC Plant Biol. 2014, 14, 95. [Google Scholar] [CrossRef] [Green Version]
- Batalova, A.Y.; Putintseva, Y.A.; Sadovsky, M.G.; Krutovsky, K.V. Comparative genomics of seasonal senescence in forest trees. Int. J. Mol. Sci. 2022, 23, 3761. [Google Scholar] [CrossRef]
- Naville, M.; Henriet, S.; Warren, I.; Sumic, S.; Reeve, M.; Volff, J.-N.; Chourrout, D. Massive changes of genome size driven by expansions of non-autonomous transposable elements. Curr. Biol. 2019, 29, 1161–1168.e6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Belyayev, A. Bursts of transposable elements as an evolutionary driving force. J. Evol. Biol. 2014, 27, 2573–2584. [Google Scholar] [CrossRef] [PubMed]
- Zeh, D.W.; Zeh, J.A.; Ishida, Y. Transposable elements and an epigenetic basis for punctuated equilibria. BioEssays 2009, 31, 715–726. [Google Scholar] [CrossRef] [PubMed]
- Tsukahara, S.; Kobayashi, A.; Kawabe, A.; Mathieu, O.; Miura, A.; Kakutani, T. Bursts of retrotransposition reproduced in Arabidopsis. Nature 2009, 461, 423–426. [Google Scholar] [CrossRef]
- Piegu, B.; Guyot, R.; Picault, N.; Roulin, A.; Saniyal, A.; Kim, H.; Collura, K.; Brar, D.S.; Jackson, S.; Wing, R.A.; et al. Doubling genome size without polyploidization: Dynamics of retrotransposition-driven genomic expansions in Oryza australiensis, a wild relative of rice. Genome Res. 2006, 16, 1262–1269. [Google Scholar] [CrossRef] [Green Version]
- Wang, W.; Ma, L.; Becher, H.; Garcia, S.; Kovarikova, A.; Leitch, I.J.; Leitch, A.R.; Kovarik, A. Astonishing 35S rDNA diversity in the gymnosperm species Cycas revoluta Thunb. Chromosoma 2016, 125, 683–699. [Google Scholar] [CrossRef] [Green Version]
- Kelly, L.J.; Renny-Byfield, S.; Pellicer, J.; Macas, J.; Novák, P.; Neumann, P.; Lysak, M.A.; Day, P.D.; Berger, M.; Fay, M.F.; et al. Analysis of the giant genomes of Fritillaria (Liliaceae) indicates that a lack of DNA removal characterizes extreme expansions in genome size. New Phytol. 2015, 208, 596–607. [Google Scholar] [CrossRef] [Green Version]
- Civáň, P.; Švec, M.; Hauptvogel, P. On the coevolution of transposable elements and plant genomes. J. Bot. 2011, 2011, e893546. [Google Scholar] [CrossRef] [Green Version]
- Arkhipova, I.R. Distribution and phylogeny of Penelope-like elements in eukaryotes. Syst. Biol. 2006, 55, 875–885. [Google Scholar] [CrossRef] [Green Version]
- Evgen’ev, M.B.; Arkhipova, I.R. Penelope-like elements—A new class of retroelements: Distribution, function and possible evolutionary significance. Cytogenet. Genome Res. 2005, 110, 510–521. [Google Scholar] [CrossRef]
- Lin, X.; Faridi, N.; Casola, C. An ancient transkingdom horizontal transfer of Penelope-like retroelements from arthropods to conifers. Genome Biol. Evol. 2016, 8, 1252–1266. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gao, D.; Chu, Y.; Xia, H.; Xu, C.; Heyduk, K.; Abernathy, B.; Ozias-Akins, P.; Leebens-Mack, J.H.; Jackson, S.A. Horizontal transfer of non-LTR retrotransposons from arthropods to flowering plants. Mol. Biol. Evol. 2018, 35, 354–364. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, L.; Yan, L.; Jiang, J.; Wang, Y.; Jiang, Y.; Yan, T.; Cao, Y. The structure and retrotransposition mechanism of LTR-retrotransposons in the asexual yeast Candida albicans. Virulence 2014, 5, 655–664. [Google Scholar] [CrossRef] [Green Version]
- Wicker, T.; Sabot, F.; Hua-Van, A.; Bennetzen, J.L.; Capy, P.; Chalhoub, B.; Flavell, A.; Leroy, P.; Morgante, M.; Panaud, O.; et al. A unified classification system for eukaryotic transposable elements. Nat. Rev. Genet. 2007, 8, 973–982. [Google Scholar] [CrossRef]
- Aroh, O.; Halanych, K.M. Genome-wide characterization of LTR retrotransposons in the non-model deep-sea annelid Lamellibrachia luymesi. BMC Genom. 2021, 22, 466. [Google Scholar] [CrossRef]
- Mascagni, F.; Usai, G.; Natali, L.; Cavallini, A.; Giordani, T. A Comparison of methods for LTR-retrotransposon insertion time profiling in the Populus trichocarpa genome. Caryologia 2018, 71, 85–92. [Google Scholar] [CrossRef]
- Barghini, E.; Mascagni, F.; Natali, L.; Giordani, T.; Cavallini, A. Identification and characterisation of short interspersed nuclear elements in the olive tree (Olea europaea l.) genome. Mol. Genet. Genom. 2017, 292, 53–61. [Google Scholar] [CrossRef]
- Kumar, A.; Bennetzen, J.L. Plant retrotransposons. Annu. Rev. Genet. 1999, 33, 479–532. [Google Scholar] [CrossRef] [Green Version]
- Yin, H.; Du, J.; Wu, J.; Wei, S.; Xu, Y.; Tao, S.; Wu, J.; Zhang, S. Genome-wide annotation and comparative analysis of long terminal repeat retrotransposons between pear species of P. bretschneideri and P. communis. Sci. Rep. 2015, 5, 17644. [Google Scholar] [CrossRef] [Green Version]
- Yin, H.; Liu, J.; Xu, Y.; Liu, X.; Zhang, S.; Ma, J.; Du, J. TARE1, a mutated Copia-like LTR retrotransposon followed by recent massive amplification in tomato. PLoS ONE 2013, 8, e68587. [Google Scholar] [CrossRef] [Green Version]
- Zhao, M.; Du, J.; Lin, F.; Tong, C.; Yu, J.; Huang, S.; Wang, X.; Liu, S.; Ma, J. Shifts in the evolutionary rate and intensity of purifying selection between two Brassica genomes revealed by analyses of orthologous transposons and relics of a whole genome triplication. Plant J. 2013, 76, 211–222. [Google Scholar] [CrossRef]
- Buti, M.; Giordani, T.; Cattonaro, F.; Cossu, R.M.; Pistelli, L.; Vukich, M.; Morgante, M.; Cavallini, A.; Natali, L. Temporal dynamics in the evolution of the sunflower genome as revealed by sequencing and annotation of three large genomic regions. Theor. Appl. Genet. 2011, 123, 779. [Google Scholar] [CrossRef] [PubMed]
- Paterson, A.H.; Bowers, J.E.; Bruggmann, R.; Dubchak, I.; Grimwood, J.; Gundlach, H.; Haberer, G.; Hellsten, U.; Mitros, T.; Poliakov, A.; et al. The Sorghum bicolor genome and the diversification of grasses. Nature 2009, 457, 551–556. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brunner, S.; Fengler, K.; Morgante, M.; Tingey, S.; Rafalski, A. Evolution of DNA sequence nonhomologies among maize inbreds. Plant Cell 2005, 17, 343–360. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wan, T.; Liu, Z.; Leitch, I.J.; Xin, H.; Maggs-Kölling, G.; Gong, Y.; Li, Z.; Marais, E.; Liao, Y.; Dai, C.; et al. The Welwitschia genome reveals a unique biology underpinning extreme longevity in deserts. Nat. Commun. 2021, 12, 4247. [Google Scholar] [CrossRef] [PubMed]
- Zhou, S.-S.; Yan, X.-M.; Zhang, K.-F.; Liu, H.; Xu, J.; Nie, S.; Jia, K.-H.; Jiao, S.-Q.; Zhao, W.; Zhao, Y.-J.; et al. A comprehensive annotation dataset of intact LTR retrotransposons of 300 plant genomes. Sci. Data 2021, 8, 174. [Google Scholar] [CrossRef] [PubMed]
- Song, H.; Guo, Z.; Hu, X.; Qian, L.; Miao, F.; Zhang, X.; Chen, J. Evolutionary balance between LRR domain loss and young NBS–LRR genes production governs disease resistance in Arachis hypogaea cv. Tifrunner. BMC Genom. 2019, 20, 844. [Google Scholar] [CrossRef] [PubMed]
- Schaper, E.; Anisimova, M. The evolution and function of protein tandem repeats in plants. New Phytol. 2015, 206, 397–410. [Google Scholar] [CrossRef]
- Jones, J.D.G.; Dangl, J.L. The plant immune system. Nature 2006, 444, 323–329. [Google Scholar] [CrossRef] [Green Version]
- Kobe, B.; Kajava, A.V. The leucine-rich repeat as a protein recognition motif. Curr. Opin. Struct. Biol. 2001, 11, 725–732. [Google Scholar] [CrossRef]
- Niklas, K.J. The cell walls that bind the tree of life. BioScience 2004, 54, 831–841. [Google Scholar] [CrossRef] [Green Version]
- Sarkar, P.; Bosneaga, E.; Auer, M. Plant cell walls throughout evolution: Towards a molecular understanding of their design principles. J. Exp. Bot. 2009, 60, 3615–3635. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, L.; Cheng, X.F.; Leshkevich, J.; Umezawa, T.; Harding, S.A.; Chiang, V.L. The last step of syringyl monolignol biosynthesis in angiosperms is regulated by a novel gene encoding sinapyl alcohol dehydrogenase. Plant Cell 2001, 13, 1567–1586. [Google Scholar] [CrossRef] [Green Version]
- Hatfield, R.; Vermerris, W. Lignin formation in plants. The dilemma of linkage specificity. Plant Physiol. 2001, 126, 1351–1357. [Google Scholar] [CrossRef] [Green Version]
- Wagner, A.; Donaldson, L.; Ralph, J. Chapter 2—Lignification and Lignin Manipulations in Conifers. In Advances in Botanical Research; Jouanin, L., Lapierre, C., Eds.; Elsevier: Amsterdam, The Netherlands, 2012; Volume 61, pp. 37–76. [Google Scholar] [CrossRef]
- Pascual, M.B.; El-Azaz, J.; de la Torre, F.N.; Cañas, R.A.; Avila, C.; Cánovas, F.M. Biosynthesis and metabolic fate of phenylalanine in conifers. Front. Plant Sci. 2016, 7, 1030. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yadav, V.; Wang, Z.; Wei, C.; Amo, A.; Ahmed, B.; Yang, X.; Zhang, X. Phenylpropanoid pathway engineering: An emerging approach towards plant defense. Pathogens 2020, 9, 312. [Google Scholar] [CrossRef] [Green Version]
- Porth, I.; Hamberger, B.; White, R.; Ritland, K. Defense mechanisms against herbivory in Picea: Sequence evolution and expression regulation of gene family members in the phenylpropanoid pathway. BMC Genom. 2011, 12, 608. [Google Scholar] [CrossRef] [Green Version]
- Vogt, T. Phenylpropanoid biosynthesis. Mol. Plant 2010, 3, 2–20. [Google Scholar] [CrossRef] [Green Version]
- El-Azaz, J.; de la Torre, F.; Ávila, C.; Cánovas, F.M. Identification of a small protein domain present in all plant lineages that confers high prephenate dehydratase activity. Plant J. Cell Mol. Biol. 2016, 87, 215–229. [Google Scholar] [CrossRef] [Green Version]
- Bagal, U.R.; Leebens-Mack, J.H.; Lorenz, W.W.; Dean, J.F. The phenylalanine ammonia lyase (PAL) gene family shows a gymnosperm-specific lineage. BMC Genom. 2012, 13, S1. [Google Scholar] [CrossRef]
- van Doorn, W.G.; Beers, E.P.; Dangl, J.L.; Franklin-Tong, V.E.; Gallois, P.; Hara-Nishimura, I.; Jones, A.M.; Kawai-Yamada, M.; Lam, E.; Mundy, J.; et al. Morphological classification of plant cell deaths. Cell Death Differ. 2011, 18, 1241–1246. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Klim, J.; Gładki, A.; Kucharczyk, R.; Zielenkiewicz, U.; Kaczanowski, S. Ancestral state reconstruction of the apoptosis machinery in the common ancestor of eukaryotes. G3 Genes Genomes Genet. 2018, 8, 2121–2134. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Minina, E.A.; Smertenko, A.P.; Bozhkov, P.V. Vacuolar cell death in plants: Metacaspase releases the brakes on autophagy. Autophagy 2014, 10, 928–929. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hara-Nishimura, I.; Hatsugai, N. The role of vacuole in plant cell death. Cell Death Differ. 2011, 18, 1298–1304. [Google Scholar] [CrossRef] [Green Version]
- Kalra, G.; Bhatla, S.C. Senescence and Programmed Cell Death. In Plant Physiology, Development and Metabolism; Bhatla, S.C., Lal, M.A., Eds.; Springer: Singapore, 2018; pp. 937–966. [Google Scholar] [CrossRef]
- van Doorn, W.G. Classes of programmed cell death in plants, compared to those in animals. J. Exp. Bot. 2011, 62, 4749–4761. [Google Scholar] [CrossRef] [Green Version]
- Reape, T.J.; Molony, E.M.; McCabe, P.F. Programmed cell death in plants: Distinguishing between different modes. J. Exp. Bot. 2008, 59, 435–444. [Google Scholar] [CrossRef] [Green Version]
- Valandro, F.; Menguer, P.K.; Cabreira-Cagliari, C.; Margis-Pinheiro, M.; Cagliari, A. Programmed cell death (PCD) control in plants: New insights from the Arabidopsis thaliana deathosome. Plant Sci. 2020, 299, 110603. [Google Scholar] [CrossRef]
- Delorme, V.G.; McCabe, P.F.; Kim, D.J.; Leaver, C.J. A matrix metalloproteinase gene is expressed at the boundary of senescence and programmed cell death in cucumber. Plant Physiol. 2000, 123, 917–927. [Google Scholar] [CrossRef] [Green Version]
- Van Hautegem, T.; Waters, A.J.; Goodrich, J.; Nowack, M.K. Only in dying, life: Programmed cell death during plant development. Trends Plant Sci. 2015, 20, 102–113. [Google Scholar] [CrossRef]
- Koyama, T. The roles of ethylene and transcription factors in the regulation of onset of leaf senescence. Front. Plant Sci. 2014, 5, 650. [Google Scholar] [CrossRef]
- Daneva, A.; Gao, Z.; Van Durme, M.; Nowack, M.K. Functions and regulation of programmed cell death in plant development. Annu. Rev. Cell Dev. Biol. 2016, 32, 441–468. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.-H.; Kwon, C.; Lee, J.-H.; Chung, T. Genes for plant autophagy: Functions and interactions. Mol. Cells 2012, 34, 413–423. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, J.-K. Salt and drought stress signal transduction in plants. Annu. Rev. Plant Biol. 2002, 53, 247–273. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xue-Xuan, X.; Hong-Bo, S.; Yuan-Yuan, M.; Gang, X.; Jun-Na, S.; Dong-Gang, G.; Cheng-Jiang, R. Biotechnological implications from abscisic acid (ABA) roles in cold stress and leaf senescence as an important signal for improving plant sustainable survival under abiotic-stressed conditions. Crit. Rev. Biotechnol. 2010, 30, 222–230. [Google Scholar] [CrossRef]
- Costa-Broseta, Á.; Perea-Resa, C.; Castillo, M.-C.; Ruíz, M.F.; Salinas, J.; León, J. Nitric oxide controls constitutive freezing tolerance in Arabidopsis by attenuating the levels of osmoprotectants, stress-related hormones and anthocyanins. Sci. Rep. 2018, 8, 9268. [Google Scholar] [CrossRef]
- Wu, J.; Zhang, Y.; Yin, L.; Qu, J.; Lu, J. Linkage of cold acclimation and disease resistance through plant–pathogen interaction pathway in Vitis amurensis grapevine. Funct. Integr. Genom. 2014, 14, 741–755. [Google Scholar] [CrossRef]
- Preston, J.; Sandve, S. Adaptation to seasonality and the winter freeze. Front. Plant Sci. 2013, 4, 167. [Google Scholar] [CrossRef] [Green Version]
- Song, Y.; Xiang, F.; Zhang, G.; Miao, Y.; Miao, C.; Song, C.-P. Abscisic acid as an internal integrator of multiple physiological processes modulates leaf senescence onset in Arabidopsis thaliana. Front. Plant Sci. 2016, 7, 181. [Google Scholar] [CrossRef] [Green Version]
- Xu, P.; Chen, H.; Cai, W. Transcription factor CDF4 promotes leaf senescence and floral organ abscission by regulating abscisic acid and reactive oxygen species pathways in Arabidopsis. EMBO Rep. 2020, 21, e48967. [Google Scholar] [CrossRef]
- Lee, I.C.; Hong, S.W.; Whang, S.S.; Lim, P.O.; Nam, H.G.; Koo, J.C. Age-Dependent Action of an ABA-Inducible Receptor Kinase, RPK1, as a Positive Regulator of Senescence in Arabidopsis leaves. Plant Cell Physiol. 2011, 52, 651–662. [Google Scholar] [CrossRef] [Green Version]
- Raab, S.; Drechsel, G.; Zarepour, M.; Hartung, W.; Koshiba, T.; Bittner, F.; Hoth, S. Identification of a novel E3 ubiquitin ligase that is required for suppression of premature senescence in Arabidopsis. Plant J. Cell Mol. Biol. 2009, 59, 39–51. [Google Scholar] [CrossRef]
- Yang, J.; Worley, E.; Udvardi, M. A NAP-AAO3 regulatory module promotes chlorophyll degradation via ABA biosynthesis in Arabidopsis leaves. Plant Cell 2014, 26, 4862–4874. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Breeze, E.; Harrison, E.; McHattie, S.; Hughes, L.; Hickman, R.; Hill, C.; Kiddle, S.; Kim, Y.-S.; Penfold, C.A.; Jenkins, D.; et al. High-resolution temporal profiling of transcripts during Arabidopsis leaf senescence reveals a distinct chronology of processes and regulation. Plant Cell 2011, 23, 873–894. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Han, G.-Z. Evolution of jasmonate biosynthesis and signaling mechanisms. J. Exp. Bot. 2017, 68, 1323–1331. [Google Scholar] [CrossRef]
- Ali, M.S.; Baek, K.-H. Jasmonic acid signaling pathway in response to abiotic stresses in plants. Int. J. Mol. Sci. 2020, 21, 621. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Truman, W.; Bennett, M.H.; Kubigsteltig, I.; Turnbull, C.; Grant, M. Arabidopsis systemic immunity uses conserved defense signaling pathways and is mediated by jasmonates. Proc. Natl. Acad. Sci. USA 2007, 104, 1075–1080. [Google Scholar] [CrossRef] [Green Version]
- Mohamed, H.I.; Latif, H.H. Improvement of drought tolerance of soybean plants by using methyl jasmonate. Physiol. Mol. Biol. Plants 2017, 23, 545–556. [Google Scholar] [CrossRef]
- Todaka, D.; Shinozaki, K.; Yamaguchi-Shinozaki, K. Recent advances in the dissection of drought-stress regulatory networks and strategies for development of drought-tolerant transgenic rice plants. Front. Plant Sci. 2015, 6, 84. [Google Scholar] [CrossRef] [Green Version]
- Qiu, Z.; Guo, J.; Zhu, A.; Zhang, L.; Zhang, M. Exogenous jasmonic acid can enhance tolerance of wheat seedlings to salt stress. Ecotoxicol. Environ. Saf. 2014, 104, 202–208. [Google Scholar] [CrossRef]
- Fan, L.; Wang, Q.; Lv, J.; Gao, L.; Zuo, J.; Shi, J. Amelioration of postharvest chilling injury in cowpea (Vigna sinensis) by methyl jasmonate (MeJA) treatments. Sci. Hortic. 2016, 203, 95–101. [Google Scholar] [CrossRef]
- Zhao, M.-L.; Wang, J.-N.; Shan, W.; Fan, J.-G.; Kuang, J.-F.; Wu, K.-Q.; Li, X.-P.; Chen, W.-X.; He, F.-Y.; Chen, J.-Y.; et al. Induction of jasmonate signalling regulators MaMYC2s and their physical interactions with MaICE1 in methyl jasmonate-induced chilling tolerance in banana fruit. Plant Cell Environ. 2013, 36, 30–51. [Google Scholar] [CrossRef] [PubMed]
- Mewis, I.; Schreiner, M.; Nguyen, C.N.; Krumbein, A.; Ulrichs, C.; Lohse, M.; Zrenner, R. UV-B Irradiation changes specifically the secondary metabolite profile in broccoli sprouts: Induced signaling overlaps with defense response to biotic stressors. Plant Cell Physiol. 2012, 53, 1546–1560. [Google Scholar] [CrossRef] [Green Version]
- Cerrudo, I.; Keller, M.M.; Cargnel, M.D.; Demkura, P.V.; de Wit, M.; Patitucci, M.S.; Pierik, R.; Pieterse, C.M.J.; Ballaré, C.L. Low red/far-red ratios reduce Arabidopsis resistance to Botrytis cinerea and jasmonate responses via a COI1-JAZ10-dependent, salicylic acid-independent mechanism. Plant Physiol. 2012, 158, 2042–2052. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Svyatyna, K.; Riemann, M. Light-dependent regulation of the jasmonate pathway. Protoplasma 2012, 249 (Suppl. S2), S137–S145. [Google Scholar] [CrossRef] [PubMed]
- Kozlowski, G.; Buchala, A.; Métraux, J.-P. Methyl jasmonate protects Norway spruce [Picea abies (L.) Karst.] seedlings against Pythium ultimum Trow. Physiol. Mol. Plant Pathol. 1999, 55, 53–58. [Google Scholar] [CrossRef]
- Franceschi, V.R.; Krekling, T.; Christiansen, E. Application of methyl jasmonate on Picea abies (Pinaceae) stems induces defense-related responses in phloem and xylem. Am. J. Bot. 2002, 89, 578–586. [Google Scholar] [CrossRef]
- Wang, C.; Liu, Y.; Li, S.-S.; Han, G.-Z. Insights into the origin and evolution of the plant hormone signaling machinery. Plant Physiol. 2015, 167, 872–886. [Google Scholar] [CrossRef] [Green Version]
- Groen, S.C.; Whiteman, N.K. The evolution of ethylene signaling in plant chemical ecology. J. Chem. Ecol. 2014, 40, 700–716. [Google Scholar] [CrossRef]
- Song, L.; Florea, L. Rcorrector: Efficient and accurate error correction for Illumina RNA-seq reads. GigaScience 2015, 4, 48. [Google Scholar] [CrossRef] [Green Version]
- Haas, B.J.; Papanicolaou, A.; Yassour, M.; Grabherr, M.; Blood, P.D.; Bowden, J.; Couger, M.B.; Eccles, D.; Li, B.; Lieber, M.; et al. De novo transcript sequence reconstruction from RNA-seq using the trinity platform for reference generation and analysis. Nat. Protoc. 2013, 8, 1494–1512. [Google Scholar] [CrossRef]
- Finn, R.D.; Coggill, P.; Eberhardt, R.Y.; Eddy, S.R.; Mistry, J.; Mitchell, A.L.; Potter, S.C.; Punta, M.; Qureshi, M.; Sangrador-Vegas, A.; et al. The Pfam protein families database: Towards a more sustainable future. Nucleic Acids Res. 2016, 44, D279–D285. [Google Scholar] [CrossRef] [PubMed]
- El-Gebali, S.; Mistry, J.; Bateman, A.; Eddy, S.R.; Luciani, A.; Potter, S.C.; Qureshi, M.; Richardson, L.J.; Salazar, G.A.; Smart, A.; et al. The Pfam protein families database in 2019. Nucleic Acids Res. 2019, 47, D427–D432. [Google Scholar] [CrossRef] [PubMed]
- Bao, Z.; Eddy, S.R. Automated de novo identification of repeat sequence families in sequenced genomes. Genome Res. 2002, 12, 1269–1276. [Google Scholar] [CrossRef] [Green Version]
- Price, A.L.; Jones, N.C.; Pevzner, P.A. De novo identification of repeat families in large genomes. Bioinformatics 2005, 21 (Suppl. S1), i351–i358. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bao, W.; Kojima, K.K.; Kohany, O. Repbase update, a database of repetitive elements in eukaryotic genomes. Mob. DNA 2015, 6, 11. [Google Scholar] [CrossRef] [Green Version]
- Abrusán, G.; Grundmann, N.; DeMester, L.; Makalowski, W. TEclass—a tool for automated classification of unknown eukaryotic transposable elements. Bioinformatics 2009, 25, 1329–1330. [Google Scholar] [CrossRef] [Green Version]
- Nussbaumer, T.; Martis, M.M.; Roessner, S.K.; Pfeifer, M.; Bader, K.C.; Sharma, S.; Gundlach, H.; Spannagl, M. MIPS PlantsDB: A database framework for comparative plant genome research. Nucleic Acids Res. 2013, 41, D1144–D1151. [Google Scholar] [CrossRef] [Green Version]
- Kojima, K.K. Human transposable elements in repbase: Genomic footprints from fish to humans. Mob. DNA 2018, 9, 2. [Google Scholar] [CrossRef] [Green Version]
- Stanke, M.; Schöffmann, O.; Morgenstern, B.; Waack, S. Gene prediction in eukaryotes with a generalized hidden Markov model that uses hints from external sources. BMC Bioinform. 2006, 7, 62. [Google Scholar] [CrossRef] [Green Version]
- Scalzitti, N.; Jeannin-Girardon, A.; Collet, P.; Poch, O.; Thompson, J.D. A benchmark study of ab initio gene prediction methods in diverse eukaryotic organisms. BMC Genomics 2020, 21, 293. [Google Scholar] [CrossRef] [Green Version]
- Stanke, M.; Keller, O.; Gunduz, I.; Hayes, A.; Waack, S.; Morgenstern, B. AUGUSTUS: Ab initio prediction of alternative transcripts. Nucleic Acids Res. 2006, 34, W435–W439. [Google Scholar] [CrossRef] [Green Version]
- Trapnell, C.; Pachter, L.; Salzberg, S.L. TopHat: Discovering splice junctions with RNA-seq. Bioinformatics 2009, 25, 1105–1111. [Google Scholar] [CrossRef] [PubMed]
- Trapnell, C.; Roberts, A.; Goff, L.; Pertea, G.; Kim, D.; Kelley, D.R.; Pimentel, H.; Salzberg, S.L.; Rinn, J.L.; Pachter, L. Differential gene and transcript expression analysis of RNA-seq experiments with TopHat and Cufflinks. Nat. Protoc. 2012, 7, 562–578. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pearson, W.R. An introduction to sequence similarity (“homology”) searching. Curr. Protoc. Bioinform. 2013, 42, 3.1.1–3.1.8. [Google Scholar] [CrossRef] [PubMed]
- Benjamini, Y.; Hochberg, Y. Controlling the false discovery rate: A practical and powerful approach to multiple testing. J. R. Stat. Soc. Ser. B Methodol. 1995, 57, 289–300. [Google Scholar] [CrossRef]
- Storey, J.D. A direct approach to false discovery rates. J. R. Stat. Soc. Ser. B Stat. Methodol. 2002, 64, 479–498. [Google Scholar] [CrossRef] [Green Version]
- De La Torre, A.R.; Li, Z.; Van de Peer, Y.; Ingvarsson, P.K. Contrasting rates of molecular evolution and patterns of selection among gymnosperms and flowering plants. Mol. Biol. Evol. 2017, 34, 1363–1377. [Google Scholar] [CrossRef] [Green Version]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Parameter | Larix sibirica |
|---|---|
| Number of chromosomes | 12 |
| Estimated genome size (1C), Gbp | 12.03 (12.30 pg) 1 |
| Assembly length, Gbp | 5.59 2/12.34 3 |
| Assembly N50, bp | 3098 2/6443 3 |
| GC content, % | 35.41 |
| Repeat content, % | 65.98 |
| Number of predicted gene models | 39,370 |
| Number of full-length gene models | 24,551 |
| Average CDS length, bp | 244.29 |
| Average intron length, bp | 360.93 |
| Longest intron length, bp | 10,153 |
| Assembly | Number, mln | N50, bp | Maximum Length, bp | Total Length, Gbp |
|---|---|---|---|---|
| Contigs | 12.40 | 1074 | 128,642 | 7.99 |
| Scaffolds | 11.33 | 6443 | 354,326 | 12.34 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bondar, E.I.; Feranchuk, S.I.; Miroshnikova, K.A.; Sharov, V.V.; Kuzmin, D.A.; Oreshkova, N.V.; Krutovsky, K.V. Annotation of Siberian Larch (Larix sibirica Ledeb.) Nuclear Genome—One of the Most Cold-Resistant Tree Species in the Only Deciduous GENUS in Pinaceae. Plants 2022, 11, 2062. https://doi.org/10.3390/plants11152062
Bondar EI, Feranchuk SI, Miroshnikova KA, Sharov VV, Kuzmin DA, Oreshkova NV, Krutovsky KV. Annotation of Siberian Larch (Larix sibirica Ledeb.) Nuclear Genome—One of the Most Cold-Resistant Tree Species in the Only Deciduous GENUS in Pinaceae. Plants. 2022; 11(15):2062. https://doi.org/10.3390/plants11152062
Chicago/Turabian StyleBondar, Eugenia I., Sergey I. Feranchuk, Ksenia A. Miroshnikova, Vadim V. Sharov, Dmitry A. Kuzmin, Natalya V. Oreshkova, and Konstantin V. Krutovsky. 2022. "Annotation of Siberian Larch (Larix sibirica Ledeb.) Nuclear Genome—One of the Most Cold-Resistant Tree Species in the Only Deciduous GENUS in Pinaceae" Plants 11, no. 15: 2062. https://doi.org/10.3390/plants11152062