
Selection of Sweetpotato Parental Genotypes Using Simple Sequence Repeat Markers


 , , and
, , and
Abstract
:1. Introduction
2. Material and Methods
2.1. Plant Material
2.2. DNA Extraction and SSR Genotyping
2.3. Data Analysis
3. Results
3.1. Genetic Diversity of the Selected Sweetpotato Genotypes
3.2. Within and among Population Variation
3.3. Gene Flow and Genetic Distances
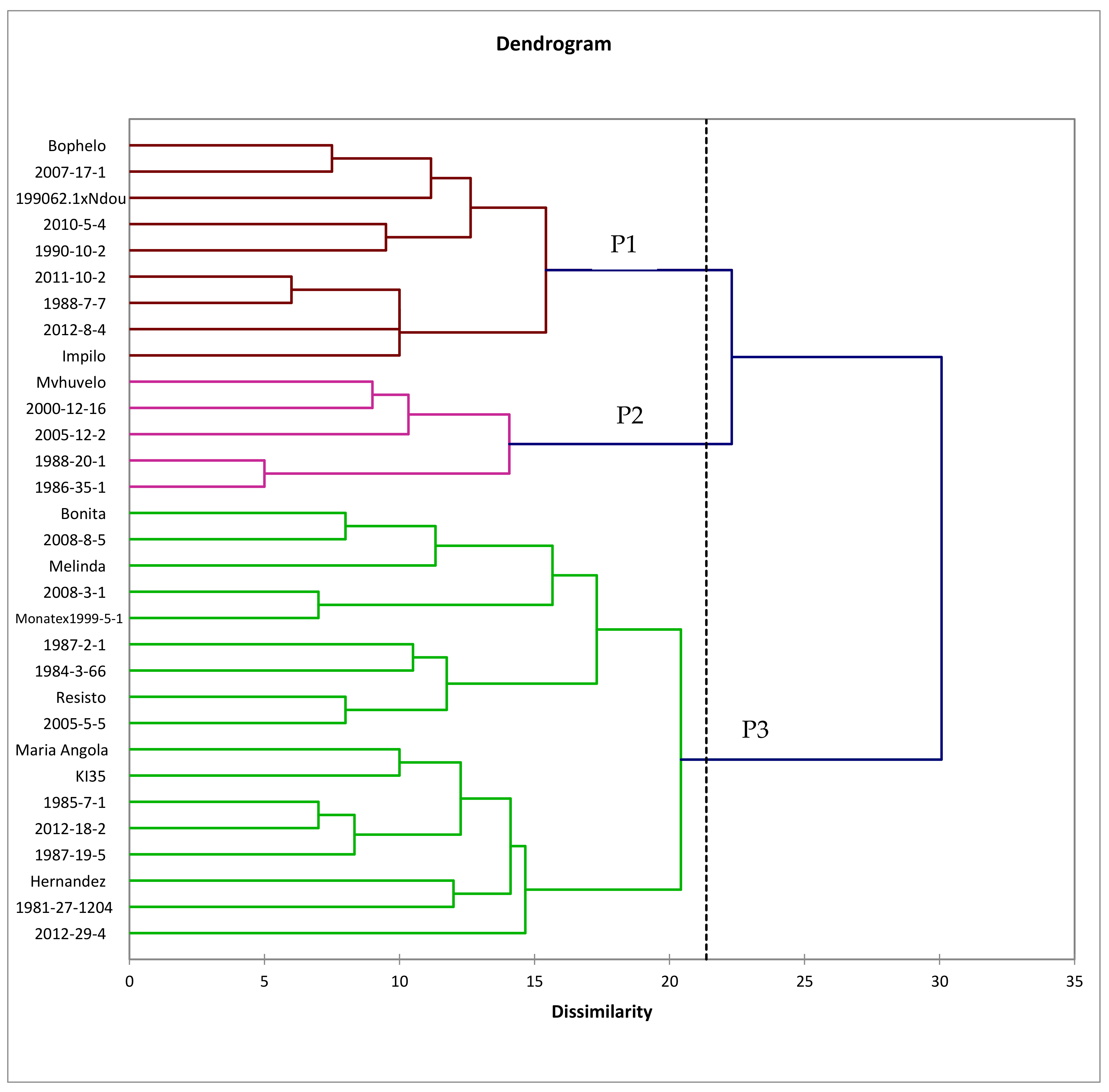
3.4. Genetic Relatedness of Genotypes
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Food and Agriculture Organization of the United Nations. FAOSTAT Statistical Database. FAO. 2019. Available online: http://www.fao.org/faostat/en/#home (accessed on 29 March 2019).
- Lebot, V. Tropical Root and Tuber Crops: Cassava, Sweet Potato, Yams and Aroids; Centre for Agriculture and Bioscience International: Wallingford, UK, 2009; p. 413. [Google Scholar]
- Grüneberg, W.J.; Ma, D.; Mwanga, R.O.M.; Carey, E.E.; Huamani, K.; Diaz, F.; Eyzaguirre, R.; Guaf, E.; Jusuf, M.; Karuniawan, A.; et al. Advances in Sweetpotato Breeding from 1993 to 2012. In Potato and Sweetpotato in Africa: Transforming the Value Chains for Food and Nutrition Security; Nyongesa, M., Quinn, S., Parker, M., Low, J., Eds.; Centre for Agriculture and Bioscience International (CABI): Wallingford, UK, 2015; pp. 1–77. [Google Scholar]
- Loebenstein, G. Origin, Distribution and Economic Importance. In The Sweet Potato Biomedical and Life Sciences; Loebenstein, G., Thottappilly, G., Eds.; Springer: Berlin/Heidelberg, Germany, 2009; pp. 9–12. [Google Scholar]
- Srisuwan, S.; Sihachakr, D.; Siljak-Yakovlev, S. The origin and evolution of sweet potato [Ipomoea batatas (L) Lam.] and its wild relatives through the cytogenetic approaches. Plant Sci. J. 2006, 171, 424–433. [Google Scholar] [CrossRef] [PubMed]
- Low, J.W.; Mwanga, R.O.M.; Andrade, M.; Carey, E.; Ball, A. Tackling vitamin A deficiency with biofortified sweetpotato in sub-Saharan Africa. Glob. Food Secur. 2017, 14, 23–30. [Google Scholar] [CrossRef] [PubMed]
- Zhang, D.; Carbajulca, D.; Ojeda, L.; Rossel, G.; Milla, S.; Herrera, C.; Ghislain, M. Microsatellite Analysis of Genetic Diversity in Sweetpotato Varieties from Latin America. In CIP Program Report 1999–2000; International Potato Center: Lima, Peru, 1999; pp. 295–301. [Google Scholar]
- Gichuki, S.T.; Berenyi, M.; Zhang, D.; Hermann, M.; Schmidt, J.; Glössl, J.; Burg, K. Genetic diversity in sweetpotato [Ipomoea batatas (L.) Lam.] in relationship to geographic sources as assessed with RAPD markers. Genet. Resour. Crop Evol. 2003, 50, 429–437. [Google Scholar] [CrossRef]
- Jata, S.K.; Nedunchezhian, M.; Misra, S.R. The Triple ‘f’ (food, fodder and fuel) crop sweet potato [Ipomoea batatas (L.) Lam.]. Orissa Rev. 2011, 1, 82–92. [Google Scholar]
- Monteros-Altamirano, A.; Paredes, D.; Buitrón-Bustamante, J.; Tapia, C.; Peña, G. Genetic diversity of sweet potatoes [Ipomoea batatas (L) Lam.] in Ecuador. Genet. Resour. Crop Evol. 2021, 68, 307–320. [Google Scholar] [CrossRef]
- Karuri, H.; Ateka, E.; Amata, R.; Nyende, A.; Muigai, A.; Mwasame, E.; Gichuki, S. Evaluating diversity among Kenyan sweet potato genotypes using morphological and SSR markers. Int. J. Agric. Biol. 2010, 12, 33–38. [Google Scholar]
- Low, J.; Lynam, J.; Lemaga, B.; Crissman, C.; Barker, I.; Thiele, G.; Namanda, S.; Wheatley, C.; Andrade, M. Sweetpotato in Sub-Saharan Africa. In The Sweet Potato; Loebenstein, G., Thottappilly, G., Eds.; Springer: Berlin/Heidelberg, Germany, 2009; pp. 359–390. [Google Scholar]
- Laurie, S.M.; Magoro, M.D. Evaluation and release of new sweetpotato varieties through farmer participatory selection. Afr. J. Agric. Res. 2008, 3, 672–676. [Google Scholar]
- Motsa, N.M.; Modi, A.T.; Mabhaudhi, T. Sweet potato (Ipomoea batatas L.) as a drought tolerant and food security crop. S. Afr. J. Sci. 2015, 111, 1–8. [Google Scholar]
- Naidoo, S.I.M.; Laurie, S.M.; Shimelis, H.; Laing, M.D. Morpho-agronomical characterisation of local and international sweetpotato germplasm from the South African collection. S. Afr. J. Plant Soil 2020, 37, 308–320. [Google Scholar] [CrossRef]
- Chipungu, F.; Changadeya, W.; Ambali, A.; Saka, J.; Mahungu, N.; Mkumbira, J. Characterization of sweetpotato accessions in Malawi using morphological markers and farmers indigenous knowledge system (IKS). Afr. J. Agric. Res. 2017, 12, 3088–3098. [Google Scholar]
- Yang, X.S.; Su, W.J.; Wang, L.J.; Lei, J.; Chai, S.S.; Liu, Q.C. Molecular diversity and genetic structure of 380 sweetpotato accessions as revealed by SSR markers. J. Integr. Agric. 2015, 14, 633–641. [Google Scholar] [CrossRef]
- Arif, I.A.; Bakir, M.A.; Khan, H.A.; Al Farhan, A.H.; Al Homaidan, A.A.; Bahkali, A.H.; AlSadoon, M.; Shobrak, M. A brief review of molecular techniques to assess plant diversity. Int. J. Mol. Sci. 2010, 1, 2079–2096. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tumwegamire, S.; Rubaihayo, P.; Labonte, D.; Diaz, F.; Kapinga, R.; Mwanga, R.; Grüneberg, W. Genetic diversity in white-and orange-fleshed sweetpotato farmer varieties from East Africa evaluated by simple sequence repeat markers. Crop Sci. 2011, 51, 1132–1142. [Google Scholar] [CrossRef]
- Gwandu, C.; Tairo, F.; Mneney, E.; Kullaya, A. Characterization of Tanzanian elite sweet potato genotypes for sweet potato virus disease (SPVD) resistance and high dry matter content using simple sequence repeat (SSR) markers. Afr. J. Biotechnol. 2012, 11, 9582–9590. [Google Scholar]
- Da Silva, A.V.C.; Andrade, L.N.T.; Rabbani, A.R.C.; Nunes, M.U.C.; Pinheiro, L.R. Genetic diversity of sweetpotato collection from North-eastern Brazil. Afr. J. Biotechnol. 2014, 13, 1109–1116. [Google Scholar]
- Palumbo, F.; Galvao, A.C.; Nicoletto, C.; Sambo, P.; Barcaccia, G. Diversity analysis of sweet potato genetic resources using morphological and qualitative traits and molecular markers. Genes 2019, 10, 840. [Google Scholar] [CrossRef] [Green Version]
- McGregor, C.; Greyling, M.; Banda, J.; Laurie, S. DNA fingerprinting of sweet potato [Ipomoea batatas (L.) Lam]: Two case studies in Africa. In: International Symposium on Molecular Markers for Characterizing Genotypes and Identifying Cultivars in Horticulture. Acta Hortic. 2001, 546, 243–248. [Google Scholar] [CrossRef]
- Hundayehu, M.C.; Du Toit, E.; Laurie, S.M.; Steyn, M.; Greyling, R.; Myeza, N. Effect of Long-Term in Vitro Sub-Culturing on Quality Degeneration of Sweetpotato Varieties: Morpho-Anatomic Assessment and Simple Sequence Repeat (SSR) Analysis. In Potato and Sweetpotato in Africa: Transforming the Value Chains for Food and Nutrition Security; Low, J., Nyongesa, M., Quinn, S., Parker, M., Eds.; Centre for Agriculture and Bioscience International (CABI): Wallingford, UK, 2015; pp. 311–321. [Google Scholar]
- Laurie, S.M.; Booyse, M.; Labuschagne, M.T.; Greyling, M.M. Multi-environment performance of new orange-fleshed sweetpotato varieties in South Africa. Crop Sci. 2015, 5, 1585–1595. [Google Scholar] [CrossRef]
- Naidoo, S.I.M.; Laurie, S.M.; Odeny, D.A.; Vorster, B.J.; Mphela, W.M.; Greyling, M.M.; Crampton, B.G. Genetic analysis of yield and flesh colour in sweetpotato. Afr. Crop Sci. J. 2016, 24, 61–73. [Google Scholar] [CrossRef] [Green Version]
- Selaocoe, M.E.; Adebola, P.; Pillay, M.; Laurie, S.M. Genetic diversity of South African sweetpotato germplasm using molecular markers. J. Crop Improv. 2019, 33, 814–823. [Google Scholar] [CrossRef]
- Edwards, K.; Johnstone, C.; Thompson, C. A simple and rapid method for the preparation of plant genomic DNA for PCR analysis. Nucleic Acids Res. 1991, 19, 1349. [Google Scholar] [CrossRef] [PubMed]
- Buteler, M.; Jarret, R.; Labonte, D. Sequence characterization of microsatellites in diploid and polyploid Ipomoea. Theor. Appl. Genet. 1999, 99, 123–132. [Google Scholar] [CrossRef]
- Veasey, E.A.; Borges, A.; Rosa, M.; Squeiroz-Silva, J.R.; Bressan, E.D.A.; Peroni, N. Genetic diversity in Brazilian sweet potato [Ipomoea batatas (L.) Lam., Solanales, Convolvulaceae] landraces assessed with microsatellite markers. Genet. Mol. Biol. 2008, 31, 725–733. [Google Scholar] [CrossRef]
- Nei, M.; Li, W.H. Mathematical model for studying genetic variation in terms of restriction endonucleases. Proc. Natl. Acad. Sci. USA 1979, 76, 5269–5273. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peakall, R.; Smouse, P.E. GENALEX 6: Genetic analysis in Excel. Population genetic software for teaching and research. Mol. Ecol. Notes 2007, 6, 288–295. [Google Scholar] [CrossRef]
- Weir, B.S. Genetic Data Analysis II; Sinauer Associates, Inc.: Sunderland, MA, USA, 1996; p. 445. [Google Scholar]
- Wright, S. The genetical structure of populations. Ann. Eugen. 1951, 15, 323–354. [Google Scholar] [CrossRef] [PubMed]
- Ward, J.H. Hierarchical grouping to optimize an objective function. J. Am. Stat. Assoc. 1963, 58, 236–244. [Google Scholar] [CrossRef]
- Hu, J.; Nakatani, M.; Lalusin, A.G.; Fujimura, T. New microsatellite markers developed from reported Ipomoea trifida sequences and their application to sweetpotato and its related wild species. Sci. Hortic. 2004, 102, 375–386. [Google Scholar] [CrossRef]
- Slatkin, M. Detecting small amounts of gene flow from phylogenies of alleles. Genetics 1989, 121, 609–612. [Google Scholar] [CrossRef]
- Morjan, C.L.; Rieseberg, L.H. How species evolve collectively: Implications of gene flow and selection for the spread of advantageous alleles. Mol. Ecol. 2004, 13, 1341–1356. [Google Scholar] [CrossRef]
- Wright, S. Evolution and the Genetics of Populations: Variability within and among Natural Populations; University of Chicago Press: Chicago, IL, USA, 1978. [Google Scholar]
- Rodriguez-Bonilla, L.; Cuevas, H.E.; Montero-Rojas, M.; Bird-Pico, F.; Luciano-Rosario, D.; Siritunga, D. Assessment of genetic diversity of sweet potato in Puerto Rico. PLoS ONE 2014, 9, e116184. [Google Scholar]
- Ngailo, S.; Shimelis, H.; Sibiya, J.; Amelework, B.; Mutunda, K. Genetic diversity assessment of Tanzanian sweetpotato genotypes using simple sequence repeat markers. S. Afr. J. Bot. 2016, 102, 40–45. [Google Scholar] [CrossRef]
- Meng, Y.S.; Ning, Z.H.A.O.; Hui, L.I.; Hong, Z.H.A.I.; He, S.Z.; Liu, Q.C. SSR fingerprinting of 203 sweetpotato [Ipomoea batatas (L.) Lam.] varieties. J. Integr. Agric. 2018, 17, 86–93. [Google Scholar] [CrossRef] [Green Version]
- Nair, A.G.; Vidya, P.; Ambu, V.; Sreekumar, J.; Mohan, C. Genetic diversity studies in cultivated sweetpotato [Ipomoea batatas (L.) Lam.] revealed by simple sequence repeat markers. Intent. J. Adv. Biotechnol. Res. 2017, 7, 33–48. [Google Scholar]
- Zawedde, B.; Ghislain, M.; Magembe, E.; Amaro, G.; Grumet, R.; Hancock, J. Characterization of the genetic diversity of Uganda’s sweet potato (Ipomoea batatas) germplasm using microsatellites markers. Genet. Resour. Crop Ecol. 2015, 62, 501–513. [Google Scholar] [CrossRef] [Green Version]
- Gichuru, V.; Aritua, V.; Lubega, G.W.; Edema, R.; Adipala, E.; Rubaihayo, P.R. A Preliminary Analysis of Diversity among East African Sweetpotato Landraces Using Morphological and Simple Sequence Repeats (SSR) Markers. In Proceedings of the II International Symposium on Sweetpotato and Cassava: Innovative Technologies for Commercialization, Kuala Lumpur, Malaysia, 14–17 June 2005; pp. 159–164. [Google Scholar]
- Botstein, D.; White, R.L.; Skolnick, M.; Davis, R.W. Construction of a genetic linkage map in man using restriction fragment length polymorphisms. Am. J. Hum. Genet. 1980, 32, 314–331. [Google Scholar]
- Hwang, S.Y.; Tseng, Y.T.; Lo, H.F. Application of simple sequence repeats in determining the genetic relationships of cultivars used in sweet potato polycross breeding in Taiwan. Sci. Hortic. 2002, 93, 215–224. [Google Scholar] [CrossRef]
- Koussao, S.; Gracen, V.; Asante, I.; Danquah, E.Y.; Ouedraogo, J.T.; Baptiste, T.J.; Vianney, T.M. Diversity analysis of sweet potato [Ipomoea batatas (L.) Lam.] germplasm from Burkina Faso using morphological and simple sequence repeats markers. Afr. J. Biotechnol. 2014, 13, 729–742. [Google Scholar]
- Ozias-Akins, P.; Jarret, R.L. Nuclear DNA content and ploidy levels in the genus Ipomoea. J. Am. Soc. Hortic. Sci. 1994, 119, 110–115. [Google Scholar] [CrossRef]
- He, G.; Prakash, C.S.; Jarret, R.L. Analysis of genetic diversity in sweetpotato (Ipomoea batatas) germplasm collection using DNA amplification fingerprint. Genome 1995, 38, 938–945. [Google Scholar] [CrossRef]
- Arizio, C.M.; Hompanera, N.; Suarez, E.Y.; Manifesto, M.M. Genotypic identification and diversity evaluation of a sweet potato [Ipomoea batatas (L). Lam.] collection using microsatellites. Plant Genet. Resour. C 2009, 7, 135–138. [Google Scholar] [CrossRef]
- Yada, B.; Tukamuhabwa, P.; Wanjala, B.; Kim, D.J.; Skilton, R.A.; Alajo, A.; Mwanga, R.O. Characterization of Ugandan sweetpotato germplasm using fluorescent labeled simple sequence repeat markers. HortScience 2010, 45, 225–230. [Google Scholar] [CrossRef] [Green Version]
- Whitlock, M.C.; Mccauley, D.E. Indirect measures of gene flow and migration: FST≠1⁄(4Nm+1)). J. Hered. 1999, 82, 117–125. [Google Scholar] [CrossRef]
- Govindaraju, D.R. Relationship between dispersal ability and levels of gene flow in plants. Oikos J. 1988, 52, 31–35. [Google Scholar] [CrossRef]
- Elameen, A.; Fjellheim, S.; Larsen, A.; Rognli, O.A.; Sundheim, L.; Msolla, S.; Masumba, E.; Mtunda, K.; Klemsdal, S.S. Analysis of genetic diversity in a sweet potato [Ipomoea batatas (L). Lam.] germplasm collection from Tanzania as revealed by AFLP. Genet. Resour. Crop Evol. 2008, 55, 397–408. [Google Scholar] [CrossRef]
- Naidoo, S.I.M.; Laurie, S.M.; Booyse, M.; Mphela, W.M.; Makunde, G.S.; Shimelis, H.; Laing, M.D. Combining ability, heterosis and heritability of sweetpotato root protein, β-carotene, sugars and mineral composition. Euphytica 2021, 6, 1–19. [Google Scholar] [CrossRef]
- Melchinger, A.E.; Lee, M.; Lamkey, K.R.; Hallauer, A.R.; Woodman, W.L. Genetic diversity for restriction fragment length polymorphisms and heterosis for two diallel sets of maize inbreds. Theor. Appl. Genet. 1990, 80, 488–496. [Google Scholar] [CrossRef] [Green Version]
- Nikiema, J. Exploitation of Heterosis in Sweetpotato [Ipomoea batatas (L.) LAM.] via Progeny Testing and Use of Molecular Markers. Bachelor’s Thesis, Kwame Nkrumah University of Science and Technology, Kumasi, Ghana, 2017. [Google Scholar]
- Shull, G.H. What is “heterosis”? Genetics 1948, 33, 439. [Google Scholar] [CrossRef]
- Moll, R.H.; Lonnquist, J.H.; Fortuna, J.V.; Johnson, E.C. The relation of heterosis and genetic divergence in maize. Genetics 1965, 52, 139–144. [Google Scholar] [CrossRef]
- Hallauer, A.R.; Russell, W.A.; Lamkey, K.R. Corn Breeding. In Corn and Corn Improvement, 3rd ed.; Agronomy Monographs; Sprague, G.F., Dudley, J.W., Eds.; American Society of Agronomy: Madison, WI, USA, 1988; pp. 463–564. [Google Scholar]


{kind=link}
| Accession | Pedigree | Origin | 1 Flowering Rate | 2 RPC | 3 RFC | 4 RY | 5 RDMC |
|---|---|---|---|---|---|---|---|
| Monate x 1999-5-1(1) | 1989-17-1 x 1999-5-1 (ex Excel/USA) | RSA x USA | Int | High | Pale-orange | Int-High | High |
| 1981-27-1204 | PN USA | USA | Int | Int-High | Orange | Low | Very low |
| 1984-3-66 | 1981-27-474 | RSA | Abundant | Int | Yellow | Int-Low | Int |
| 1985-7-1 | Blesbok | RSA | Abundant | Int | White | Int-High | Int-Low |
| 1986-35-1 | 1981-27-537 | RSA | Abundant | Int | White | Int-High | Int-Low |
| 1987-19-5 | Bosbok | RSA | Abundant | Int | Cream | Int | Int |
| 1987-2-1 | 1984-2-201 | RSA | Abundant | Int-High | Yellow | Int | Very low |
| 1988-20-1 | 1985-7-1 | RSA | Abundant | Int-High | Cream | Very high | Very low |
| 1988-7-7 | Bosbok | RSA | Abundant | Very low | White | Int-Low | Int |
| 1990-10-2 | 1985-10-5 | RSA | Abundant | Very low | White | Int | Int-Low |
| 199062.1 x Ndou | 199062.1 x Ndou; 1992-7-2 | Peru x RSA | Sparse | Low | Yellow/orange | Int-High | Int |
| 2000-12-16 | 1992-2-3 | RSA | Abundant | Very high | Cream | Int | Int-High |
| 2005-12-2 | Khano; 1984-2-201 | RSA | Abundant | High | White | High | Very low |
| 2005-5-5 | 2000-6-3 x Resisto * | RSA | Abundant | Very low | Orange | High | Int |
| 2007-17-1 | 2001-5-2; 1992-4-1 | RSA | Abundant | Int | White | Int-High | High |
| 2008-3-1 | 1985-6-3 x Rose Centennial * | RSA | Int | Int-Low | Orange | Int-High | Low |
| 2008-8-5 | Ndou | RSA | Abundant | Int | Orange | Int | Int |
| 2010-5-4 | 2004-11-8; 1997-14-18 | RSA | Int | High | Orange | High | Int |
| 2011-10-2 | 2001-5-2; 1992-4-1 | RSA | Abundant | Low | Orange | Int | Int-Low |
| 2012-18-2 | UW 250A 26-07-06 | Moz | Absent/Int | Int | Cream | Int | Int-High |
| 2012-29-4 | MUSQ 0674-22 | Moz | Int/Absent | High | Cream | Int | Int |
| 2012-8-4 | 08 ELITE 01-214 | USA | Abundant | Int | Orange | Int-High | Low |
| KI35 | - | Kenya | Sparse | Int | Pale orange | Very low | High |
| Bophelo | 1997-14-17 | RSA | Absent/Sparse/Abundant | Int | Orange | Int | Int |
| Hernandez | L70-323 | USA | Int | High | Orange | Int-Low | Int |
| Melinda | W-119 | Moz | Sparse/None | Int | Orange | Int-High | Int-Low |
| Maria Angola | - | Peru | - | High | White | Low | Int |
| Mvuvhelo | Rose Centennial * | RSA | Absent/Sparse/Abundant | Int | White | Int-High | Int-High |
| Resisto | W-56 | USA | Abundant | High | Orange | Low | High |
| Bonita | W-152; Excel | USA | Int | Int | White | Int-Low | Int |
| Impilo | 1985-6-3 x Rose Centennial * | RSA | Abundant | High | Orange | Int-Low | Int |
| Marker Name | Primer Sequences | Ta 1 (°C) | Expected Size | Motif | Reference |
|---|---|---|---|---|---|
| IBSSR04 | F: CTC CTT TGC CTC CTT TCA TGC | 60 | 160–216 | (GA)11 | [26,36] |
| R: CCT TGC TCC CCA TTT TCT TG | |||||
| IBSSR17 | F: ACG TGC AGA CTT AGC CAC AC | 56 | 201–245 | (AG)6N(AG)17 | [26,36] |
| R: AGG AAG CCA GAT GTT CAG ATG | |||||
| IBSSR18 | F: GAT CTT GAA TTA GCC CAC | 58 | 90–110 | (GA)7(AG)5(GA)4 | [26,36] |
| R: AGA TGG ATG ACC GTA TGC | |||||
| IBSSR19 | F: GCG AAT CAA GTC TTT TGT CCA C | 65 | 171–195 | (CA)25 | [26,36] |
| R: GGG ACT GTC CTT TGG GTA TG | |||||
| IB-242 | F: GCG GAA CGG ACG AGA AAA | 52 | 95–135 | (CT)3CA(CT)11 | [19,29] |
| R: ATG GCA GAG TGA AAA TGG AAC A | |||||
| IB-248 | F: GAG AGG CCA TTG AAG AGG AA | 62 | 164–177 | (CT)9(CT)8 | [19,29] |
| R: AAG GAC CAC CGT AAA TCC AA | |||||
| IB-286 | F: AGC CAC TCC AAC AGC ACA TA | 50 | 90–122 | (CT)12 | [19,29] |
| R: GGT TTC CCA ATC AGC AAT TC | |||||
| IB-297 | F: GCA ATT TCA CAC ACA AAC ACG | 58 | 130–196 | (CT)13 | [26,29] |
| R: CCC TTC TTC CAC TTT CA | |||||
| IB-316 | F: CAA ACG CAC AAC GCT GTC | 54 | 150 | (CT)3C(CT)8 | [26,29] |
| R: CGC GTC CCG CTT ATT TAA C | |||||
| 690524 | F: AAG GAA GGG CTA GTG GAG AAG GTC | 57 | 240–315 | (CT)13 | [20,26] |
| R: CAA GGC AAC AAA TAC ACA CAC ACG |
| Loci | Genetic Parameters | |||||||
|---|---|---|---|---|---|---|---|---|
| Expected Frag. Size Range | Na | Ne | Ho | He | FIS | PIC | MAF | |
| IBSSR04 | 204–226 | 10 | 8.25 | 0.97 | 0.89 | −0.10 | 0.88 | 0.15 |
| IBSSR17 | 210–235 | 11 | 5.34 | 0.77 | 0.83 | 0.05 | 0.82 | 0.29 |
| IBSSR18 | 81–113 | 14 | 7.45 | 0.90 | 0.88 | −0.04 | 0.87 | 0.21 |
| IBSSR19 | 195–247 | 15 | 7.34 | 0.59 | 0.88 | 0.32 | 0.86 | 0.22 |
| IB-242 | 137–160 | 6 | 3.67 | 1.00 | 0.74 | −0.37 | 0.73 | 0.34 |
| IB-248 | 157–204 | 14 | 10.33 | 1.00 | 0.92 | −0.11 | 0.90 | 0.11 |
| IB-286 | 105–158 | 13 | 11.37 | 1.00 | 0.93 | −0.10 | 0.91 | 0.13 |
| 690524 | 283–310 | 12 | 5.55 | 0.77 | 0.83 | 0.06 | 0.82 | 0.29 |
| Mean | - | 11.88 | 7.41 | 0.88 | 0.86 | −0.04 | 0.85 | 0.22 |
| SE | 1.03 | 0.91 | 0.05 | 0.02 | 0.07 | 0.02 | 0.03 | |
| Genetic Parameter | ||||||||
|---|---|---|---|---|---|---|---|---|
| RPC | ||||||||
| Population | N | Na | Ne | I | Ho | He | PA | %P |
| Clones with high RPC | 14.00 | 9.13 | 6.39 | 1.93 | 0.88 | 0.85 | 17 | 100 |
| Clones with intermediate RPC | 12.00 | 8.63 | 6.65 | 1.97 | 0.86 | 0.87 | 14 | 100 |
| Clones with low RPC | 5.00 | 5.00 | 4.19 | 1.48 | 0.91 | 0.84 | 5 | 100 |
| RFC | ||||||||
| Orange | 15.00 | 9.75 | 6.81 | 2.01 | 0.88 | 0.86 | 20 | 100 |
| White | 8.00 | 7.75 | 5.96 | 1.86 | 0.89 | 0.87 | 11 | 100 |
| Yellow and cream | 7.00 | 6.50 | 5.17 | 1.71 | 0.84 | 0.85 | 5 | 100 |
| Overall mean | 10.04 | 8.00 | 5.98 | 1.86 | 0.88 | 0.86 | - | 100 |
| SE | 0.84 | 0.55 | 0.44 | 0.08 | 0.04 | 0.02 | - | 0 |
| Nm | |||
| RFC | Orange | White | Yellow and cream |
| Orange | - | 8.39 | 9.57 |
| White | - | 6.57 | |
| Yellow and cream | - | ||
| RPC | High | Intermediate | Low |
| High | - | 9.14 | 4.72 |
| Intermediate | - | 5.33 | |
| Low | - | ||
| FST | |||
| RFC | Orange | White | Yellow and cream |
| Orange | - | 0.03 | 0.03 |
| White | - | 0.04 | |
| Yellow and cream | - | ||
| RPC | High | Intermediate | Low |
| High | - | 0.03 | 0.05 |
| Intermediate | - | 0.05 | |
| Low | - | ||
| GD | |||
| RFC | Orange | White | Yellow and cream |
| Orange | - | ||
| White | 0.05 | - | |
| Yellow and cream | 0.00 | 0.02 | - |
| RPC | High | Intermediate | Low |
| High | - | ||
| Intermediate | 0.08 | - | |
| Low | 0.14 | 0.05 | - |
| GI | |||
| RFC | Orange | White | Yellow and cream |
| Orange | - | ||
| White | 0.95 | - | |
| Yellow and cream | 1.00 | 0.98 | - |
| RPC | High | Intermediate | Low |
| High | - | ||
| Intermediate | 0.92 | - | |
| Low | 0.87 | 0.95 | - |
| Source | df | SS | MS | Est. Var. | Perc. Var | F-Statistics |
|---|---|---|---|---|---|---|
| Among the groups of individuals | 2 | 6.337 | 3.168 | 0.000 | 0% | FST = 0.782 |
| Among individual | 28 | 99.309 | 3.547 | 0.080 | 2% | FIS = 0.023 |
| Within individual | 31 | 105.000 | 3.387 | 3.387 | 98% | FIT = 0.001 |
| Total | 61 | 210.645 | 3.467 | 100% |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Naidoo, S.I.M.; Laurie, S.M.; Amelework, A.B.; Shimelis, H.; Laing, M. Selection of Sweetpotato Parental Genotypes Using Simple Sequence Repeat Markers. Plants 2022, 11, 1802. https://doi.org/10.3390/plants11141802
Naidoo SIM, Laurie SM, Amelework AB, Shimelis H, Laing M. Selection of Sweetpotato Parental Genotypes Using Simple Sequence Repeat Markers. Plants. 2022; 11(14):1802. https://doi.org/10.3390/plants11141802
Chicago/Turabian StyleNaidoo, Sonia I. M., Sunette M. Laurie, Assefa B. Amelework, Hussein Shimelis, and Mark Laing. 2022. "Selection of Sweetpotato Parental Genotypes Using Simple Sequence Repeat Markers" Plants 11, no. 14: 1802. https://doi.org/10.3390/plants11141802