
Transcriptome Analyses of Barley Roots Inoculated with Novel Paenibacillus sp. and Erwinia gerundensis Strains Reveal Beneficial Early-Stage Plant–Bacteria Interactions


Abstract
:1. Introduction
2. Results
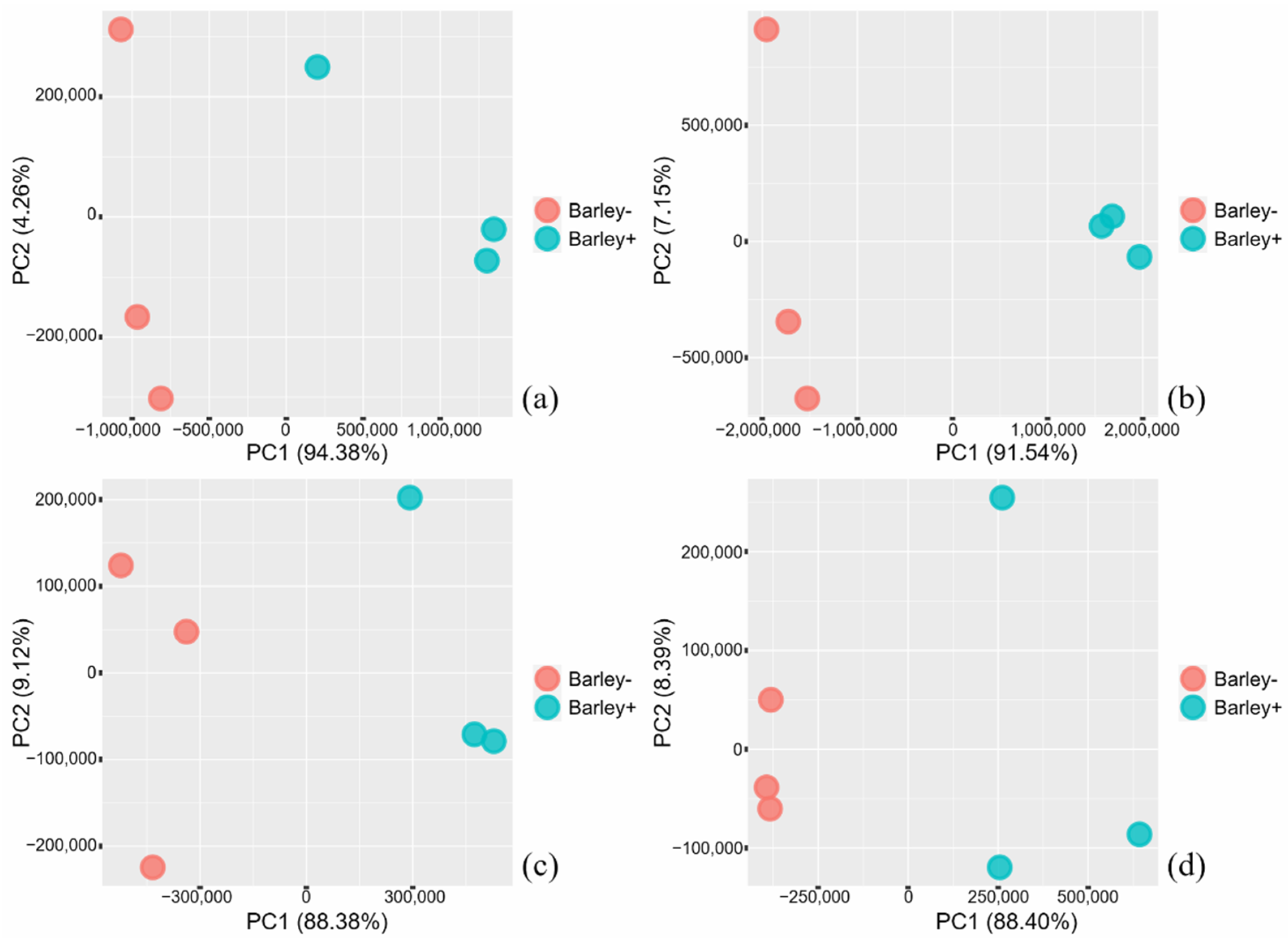
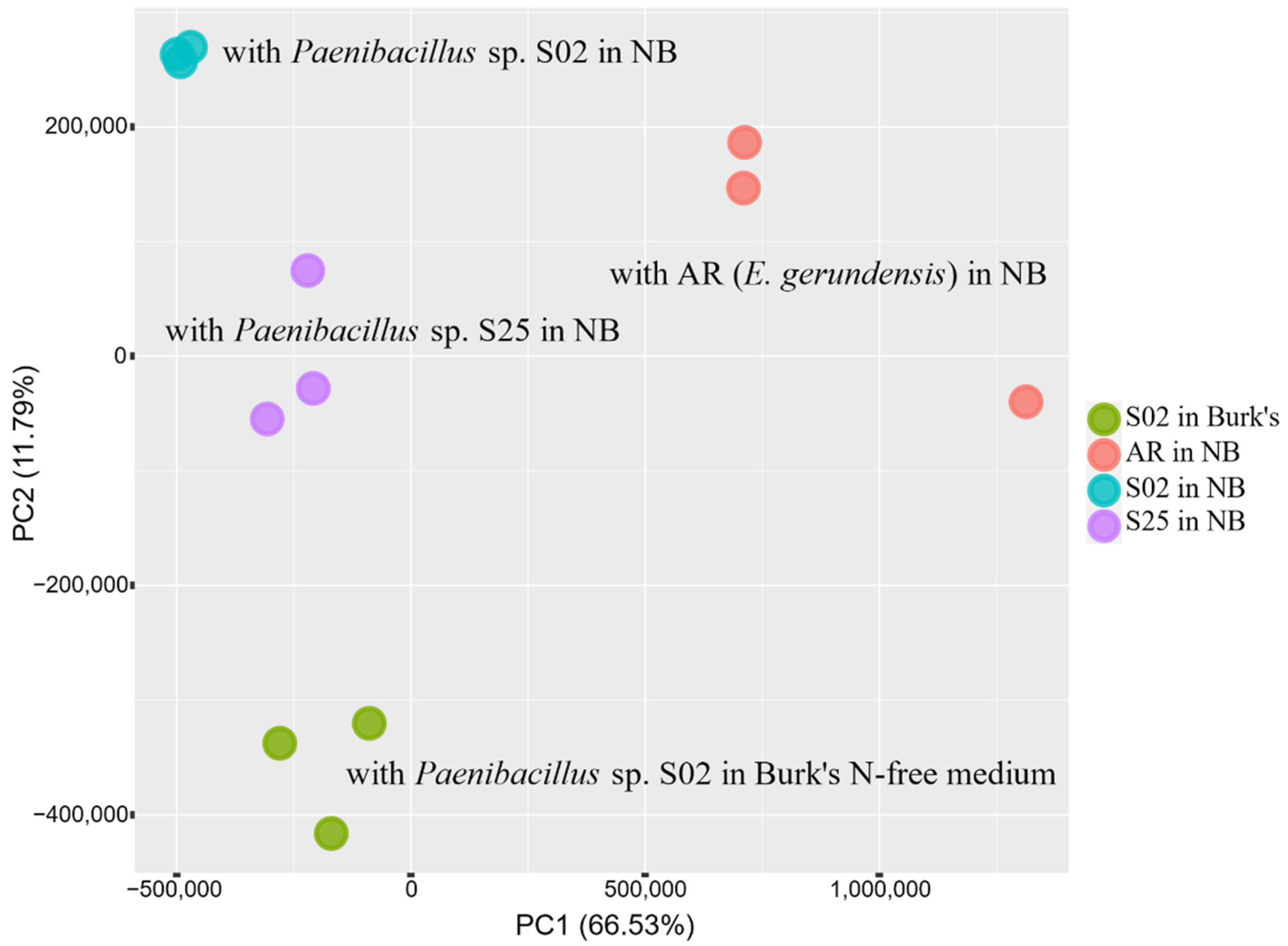
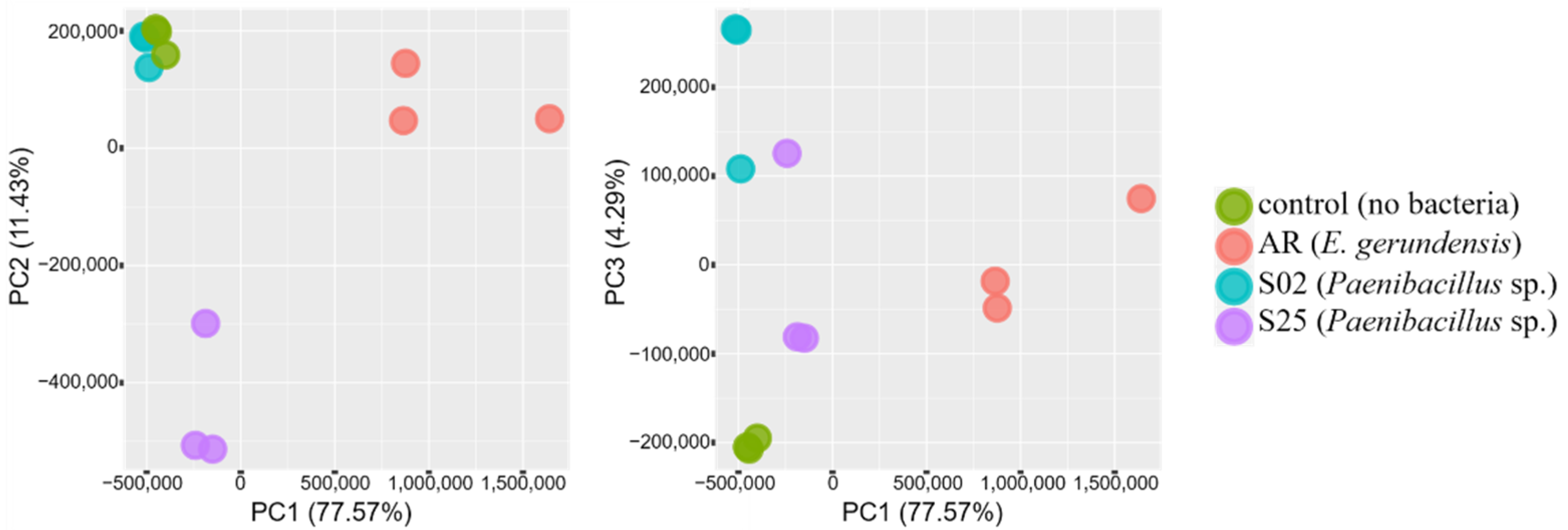
2.1. Transcriptome Sequencing—An Overview
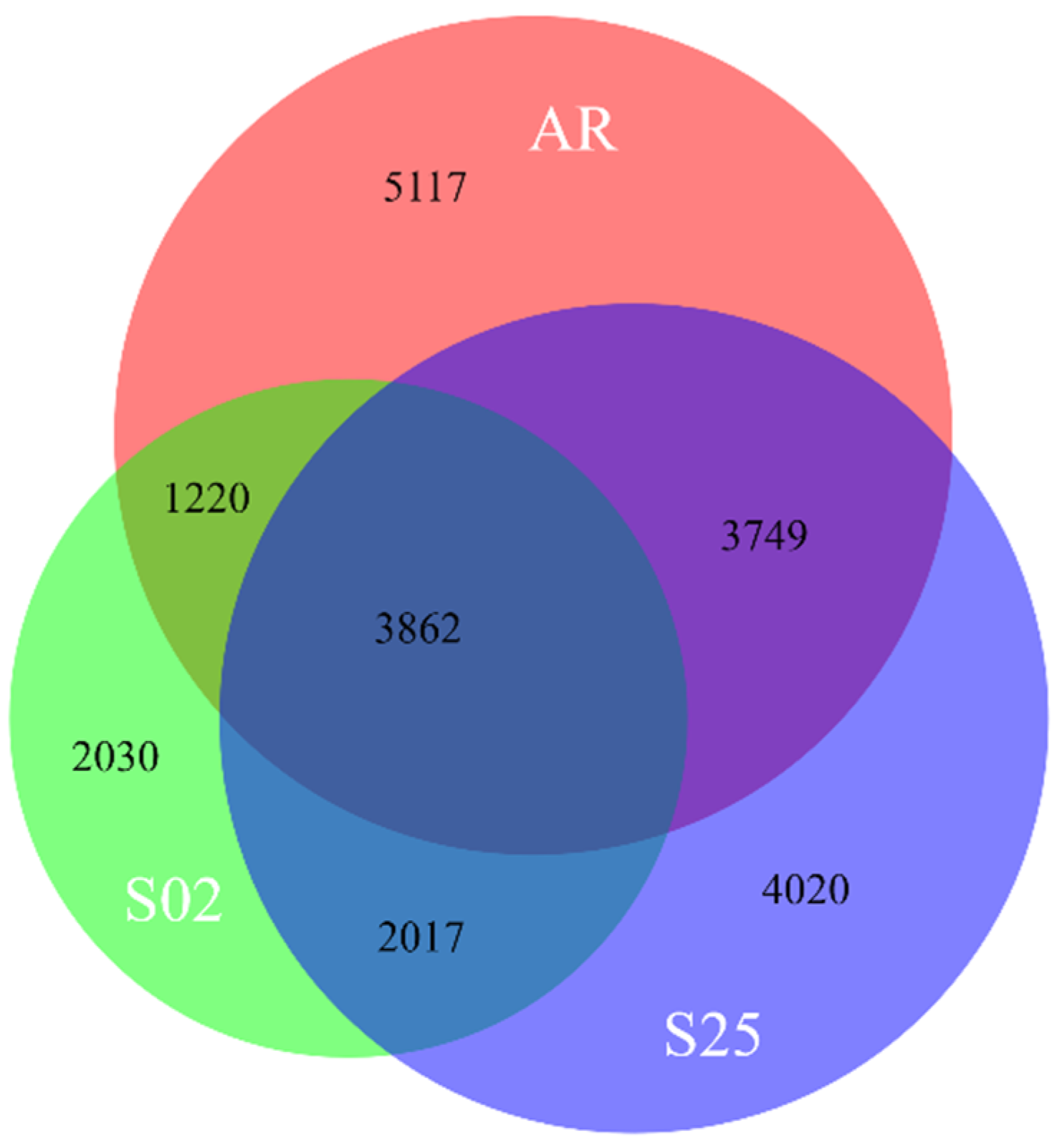
2.2. Transcriptome Analyses—An Overview
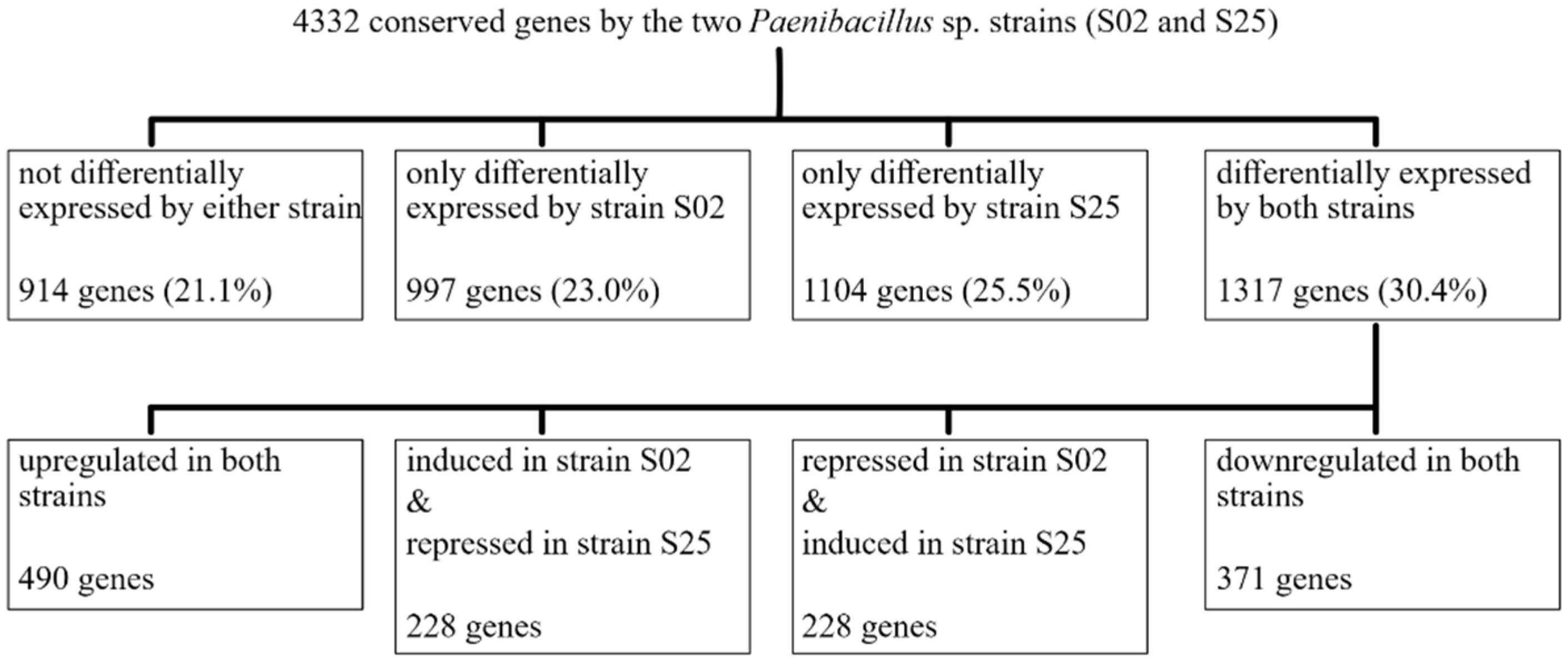
2.3. Transcriptome Analyses—Functional Genes Associated with Plant—Bacteria Interaction
2.3.1. Bacterial Initial Contact with Plants
2.3.2. Plant Growth-Promoting Genes
2.3.3. Secondary Metabolite Biosynthesis Gene Clusters
2.3.4. Defence and Stress Response Mechanisms Utilised by Barley Seedlings
2.3.5. Differentially Expressed Barley Transcripts Associated with Signal Transduction and Ethylene Biosynthesis
2.3.6. Differentially Expressed Barley Transcripts Associated with Nutrient Uptake and Metabolism
3. Discussion
3.1. Dual RNA-seq Analyses of Bacterial Strains and Barley Seedling Roots
3.2. Initial Contact Between Bacteria and Plants—Chemotaxis, Biofilm Formation and Plants’ Defence and Stress Response
3.3. Interactions between Bacteria and Plants—Plant Growth-Promoting Genes, Plant Nutrient Uptake and Metabolism, Signal Transduction and Ethylene Biosynthesis
3.4. Bacterial Secondary Metabolite
3.5. Specific Responses Associated with Bacterial Strains and Bacteria/Plant Species, and Their Implications for Enhanced Plant—Bacteria Interactions
4. Conclusions
5. Materials and Methods
5.1. Assay Design
5.2. Transcriptome Sequencing
5.3. Transcriptome Analyses
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Soto, M.J.; Nogales, J.; Pérez-Mendoza, D.; Gallegos, M.-T.; Olivares, J.; Sanjuán, J. Pathogenic and mutualistic plant-bacteria interactions: Ever increasing similarities. Cent. Eur. J. Biol. 2011, 6, 911–917. [Google Scholar] [CrossRef] [Green Version]
- Compant, S.; Clément, C.; Sessitsch, A. Plant growth-promoting bacteria in the rhizo- and endosphere of plants: Their role, colonization, mechanisms involved and prospects for utilization. Soil Biol. Biochem. 2010, 42, 669–678. [Google Scholar] [CrossRef] [Green Version]
- Carvalho, T.; Ballesteros, H.; Thiebaut, F.; Ferreira, P.; Hemerly, A. Nice to meet you: Genetic, epigenetic and metabolic controls of plant perception of beneficial associative and endophytic diazotrophic bacteria in non-leguminous plants. Plant Mol. Biol. 2016, 90, 561–574. [Google Scholar] [CrossRef]
- Xie, J.; Shi, H.; Du, Z.; Wang, T.; Liu, X.; Chen, S. Comparative genomic and functional analysis reveal conservation of plant growth promoting traits in Paenibacillus polymyxa and its closely related species. Sci. Rep. 2016, 6, 21329. [Google Scholar] [CrossRef]
- Wang, B.; Cheng, H.; Qian, W.; Zhao, W.; Liang, C.; Liu, C.; Cui, G.; Liu, H.; Zhang, L. Comparative genome analysis and mining of secondary metabolites of Paenibacillus polymyxa. Genes Genet. Syst. 2020, 95, 141–150. [Google Scholar] [CrossRef]
- Son, S.H.; Khan, Z.; Kim, S.G.; Kim, Y.H. Plant growth-promoting rhizobacteria, Paenibacillus polymyxa and Paenibacillus lentimorbus suppress disease complex caused by root-knot nematode and fusarium wilt fungus. J. Appl. Microbiol. 2009, 107, 524–532. [Google Scholar] [CrossRef]
- Rezzonico, F.; Smits, T.H.M.; Born, Y.; Blom, J.; Frey, J.E.; Goesmann, A.; Cleenwerck, I.; de Vos, P.; Bonaterra, A.; Duffy, B.; et al. Erwinia gerundensis sp. nov., a cosmopolitan epiphyte originally isolated from pome fruit trees. Int. J. Syst. Evol. Microbiol. 2016, 66, 1583–1592. [Google Scholar] [CrossRef]
- Udvardi, M.; Poole, P.S. Transport and metabolism in legume-rhizobia symbioses. Annu. Rev. Plant Biol. 2013, 64, 781–805. [Google Scholar] [CrossRef] [Green Version]
- Liu, A.; Contador, C.A.; Fan, K.; Lam, H.-M. Interaction and regulation of carbon, nitrogen, and phosphorus metabolisms in root nodules of legumes. Front. Plant Sci. 2018, 9, 1860. [Google Scholar] [CrossRef] [Green Version]
- Wolf, T.; Kämmer, P.; Brunke, S.; Linde, J. Two’s company: Studying interspecies relationships with dual RNA-seq. Curr. Opin. Microbiol. 2018, 42, 7–12. [Google Scholar] [CrossRef]
- Westermann, A.J.; Gorski, S.A.; Vogel, J. Dual RNA-seq of pathogen and host. Nat. Rev. Microbiol. 2012, 10, 618–630. [Google Scholar] [CrossRef] [PubMed]
- Hayden, K.J.; Garbelotto, M.; Knaus, B.J.; Cronn, R.C.; Rai, H.; Wright, J.W. Dual RNA-seq of the plant pathogen Phytophthora ramorum and its tanoak host. Tree Genet. Genom. 2014, 10, 489–502. [Google Scholar] [CrossRef] [Green Version]
- Liao, Z.-X.; Ni, Z.; Wei, X.-L.; Chen, L.; Li, J.-Y.; Yu, Y.-H.; Jiang, W.; Jiang, B.-L.; He, Y.-Q.; Huang, S. Dual RNA-seq of Xanthomonas oryzae pv. oryzicola infecting rice reveals novel insights into bacterial-plant interaction. PLoS ONE 2019, 14, e0215039. [Google Scholar] [CrossRef]
- Camilios-Neto, D.; Bonato, P.; Wassem, R.; Tadra-Sfeir, M.Z.; Brusamarello-Santos, L.C.C.; Valdameri, G.; Donatti, L.; Faoro, H.; Weiss, V.A.; Chubatsu, L.S.; et al. Dual RNA-seq transcriptional analysis of wheat roots colonized by Azospirillum brasilense reveals up-regulation of nutrient acquisition and cell cycle genes. BMC Genomics 2014, 15, 378. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, H.; Wang, J.; Sun, H.; Han, X.; Peng, Y.; Liu, J.; Liu, K.; Ding, Y.; Wang, C.; Du, B. Transcriptome profiles reveal the growth-promoting mechanisms of Paenibacillus polymyxa YC0136 on tobacco (Nicotiana tabacum L.). Front. Microbiol. 2020, 11, 2384. [Google Scholar] [CrossRef] [PubMed]
- Tannenbaum, I.; Kaur, J.; Mann, R.; Sawbridge, T.; Rodoni, B.; Spangenberg, G. Profiling the Lolium perenne microbiome: From seed to seed. Phytobiomes J. 2020, 4, 281–289. [Google Scholar] [CrossRef]
- Li, T.; Mann, R.; Kaur, J.; Spangenberg, G.; Sawbridge, T. Transcriptomics differentiate two novel bioactive strains of Paenibacillus sp. isolated from the perennial ryegrass seed microbiome. Sci. Rep. 2021, 11, 15545. [Google Scholar] [CrossRef]
- Li, T.; Tannenbaum, I.; Kaur, J.; Krill, C.; Sawbridge, T.; Mann, R.; Spangenberg, G. Novel Erwinia strains and related methods. WO Patent WO/2021/011998, 28 January 2021. [Google Scholar]
- Seemann, T. Prokka: Rapid prokaryotic genome annotation. Bioinformatics 2014, 30, 2068–2069. [Google Scholar] [CrossRef]
- Rapazote-Flores, P.; Bayer, M.; Milne, L.; Mayer, C.D.; Fuller, J.; Guo, W.; Hedley, P.E.; Morris, J.; Halpin, C.; Kam, J.; et al. BaRTv1.0: An improved barley reference transcript dataset to determine accurate changes in the barley transcriptome using RNA-seq. BMC Genom. 2019, 20, 968. [Google Scholar] [CrossRef] [Green Version]
- Salah Ud-Din, A.I.M.; Roujeinikova, A. Methyl-accepting chemotaxis proteins: A core sensing element in prokaryotes and archaea. Cell. Mol. Life Sci. 2017, 74, 3293–3303. [Google Scholar] [CrossRef]
- Minamino, T.; Kinoshita, M.; Namba, K. Directional switching mechanism of the bacterial flagellar motor. Comput. Struct. Biotechnol. J. 2019, 17, 1075–1081. [Google Scholar] [CrossRef] [PubMed]
- Chaparro, J.M.; Badri, D.V.; Bakker, M.G.; Sugiyama, A.; Manter, D.K.; Vivanco, J.M. Root exudation of phytochemicals in Arabidopsis follows specific patterns that are developmentally programmed and correlate with soil microbial functions. PLoS ONE 2013, 8, e55731. [Google Scholar] [CrossRef]
- Castiblanco, L.F.; Sundin, G.W. New insights on molecular regulation of biofilm formation in plant-associated bacteria. J. Integr. Plant Biol. 2016, 58, 362–372. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Zhang, L.; Liu, Z.; Zhao, D.; Liu, X.; Zhang, B.; Xie, J.; Hong, Y.; Li, P.; Chen, S.; et al. A minimal nitrogen fixation gene cluster from Paenibacillus sp. WLY78 enables expression of active nitrogenase in Escherichia coli. PLOS Genet. 2013, 9, e1003865. [Google Scholar] [CrossRef]
- Hernandez, J.A.; George, S.J.; Rubio, L.M. Molybdenum trafficking for nitrogen fixation. Biochemistry 2009, 48, 9711–9721. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Choi, S.K.; Park, S.Y.; Kim, R.; Kim, S.B.; Lee, C.H.; Kim, J.F.; Park, S.H. Identification of a polymyxin synthetase gene cluster of Paenibacillus polymyxa and heterologous expression of the gene in Bacillus subtilis. J. Bacteriol. 2009, 191, 3350–3358. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lohans, C.T.; van Belkum, M.J.; Cochrane, S.A.; Huang, Z.; Sit, C.S.; McMullen, L.M.; Vederas, J.C. Biochemical, structural, and genetic characterization of tridecaptin A1, an antagonist of Campylobacter jejuni. ChemBioChem 2014, 15, 243–249. [Google Scholar] [CrossRef]
- Park, J.E.; Kim, H.R.; Park, S.Y.; Choi, S.K.; Park, S.H. Identification of the biosynthesis gene cluster for the novel lantibiotic paenilan from Paenibacillus polymyxa E681 and characterization of its product. J. Appl. Microbiol. 2017, 123, 1133–1147. [Google Scholar] [CrossRef]
- Li, J.; Jensen, S.E. Nonribosomal biosynthesis of fusaricidins by Paenibacillus polymyxa PKB1 involves direct activation of a D-amino acid. Chem. Biol. 2008, 15, 118–127. [Google Scholar] [CrossRef] [Green Version]
- Park, C.-J.; Seo, Y.-S. Heat shock proteins: A review of the molecular chaperones for plant immunity. Plant Pathol. J. 2015, 31, 323–333. [Google Scholar] [CrossRef] [Green Version]
- Kourelis, J.; van der Hoorn, R.A.L. Defended to the nines: 25 years of resistance gene cloning identifies nine mechanisms for R protein function. Plant Cell 2018, 30, 285–299. [Google Scholar] [CrossRef] [Green Version]
- Bashi, Z.D.; Rimmer, S.R.; Khachatourians, G.G.; Hegedus, D.D. Brassica napus polygalacturonase inhibitor proteins inhibit Sclerotinia sclerotiorum polygalacturonase enzymatic and necrotizing activities and delay symptoms in transgenic plants. Can. J. Microbiol. 2012, 59, 79–86. [Google Scholar] [CrossRef]
- Moscetti, I.; Tundo, S.; Janni, M.; Sella, L.; Gazzetti, K.; Tauzin, A.; Giardina, T.; Masci, S.; Favaron, F.; D’Ovidio, R. Constitutive expression of the xylanase inhibitor TAXI-III delays Fusarium head blight symptoms in durum wheat transgenic plants. Mol. Plant-Microbe Interact. 2013, 26, 1464–1472. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rose, J.K.C.; Ham, K.-S.; Darvill, A.G.; Albersheim, P. Molecular cloning and characterization of glucanase inhibitor proteins: Coevolution of a counterdefense mechanism by plant pathogens. Plant Cell 2002, 14, 1329–1345. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, X.; Chen, W.; Zhao, Y.; Xiang, Y.; Jiang, H.; Zhu, S.; Cheng, B. Downregulation of caffeoyl-CoA O-methyltransferase (CCoAOMT) by RNA interference leads to reduced lignin production in maize straw. Genet. Mol. Biol. 2013, 36, 540–546. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mei, X.; Chen, Y.; Zhang, L.; Fu, X.; Wei, Q.; Grierson, D.; Zhou, Y.; Huang, Y.; Dong, F.; Yang, Z. Dual mechanisms regulating glutamate decarboxylases and accumulation of gamma-aminobutyric acid in tea (Camellia sinensis) leaves exposed to multiple stresses. Sci. Rep. 2016, 6, 23685. [Google Scholar] [CrossRef] [Green Version]
- Stone, S.L. The role of ubiquitin and the 26S proteasome in plant abiotic stress signalling. Front. Plant Sci. 2014, 5, 135. [Google Scholar] [CrossRef] [PubMed]
- Pandey, S.; Fartyal, D.; Agarwal, A.; Shukla, T.; James, D.; Kaul, T.; Negi, Y.K.; Arora, S.; Reddy, M.K. Abiotic stress tolerance in plants: Myriad roles of ascorbate peroxidase. Front. Plant Sci. 2017, 8, 581. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ku, Y.-S.; Sintaha, M.; Cheung, M.-Y.; Lam, H.-M. Plant hormone signalling crosstalks between biotic and abiotic stress responses. Int. J. Mol. Sci. 2018, 19, 3206. [Google Scholar] [CrossRef] [Green Version]
- Reyes, F.C.; Buono, R.; Otegui, M.S. Plant endosomal trafficking pathways. Curr. Opin. Plant Biol. 2011, 14, 666–673. [Google Scholar] [CrossRef]
- Houben, M.; Van de Poel, B. 1-aminocyclopropane-1-carboxylic acid oxidase (ACO): The enzyme that makes the plant hormone ethylene. Front. Plant Sci. 2019, 10, 695. [Google Scholar] [CrossRef] [Green Version]
- Howitt, S.M.; Udvardi, M.K. Structure, function and regulation of ammonium transporters in plants. Biochim. Biophys. Acta 2000, 1465, 152–170. [Google Scholar] [CrossRef] [Green Version]
- Zhou, Y.; Cai, H.; Xiao, J.; Li, X.; Zhang, Q.; Lian, X. Over-expression of aspartate aminotransferase genes in rice resulted in altered nitrogen metabolism and increased amino acid content in seeds. Theor. Appl. Genet. 2009, 118, 1381–1390. [Google Scholar] [CrossRef] [PubMed]
- Liang, J.; He, J. Protective role of anthocyanins in plants under low nitrogen stress. Biochem. Biophys. Res. Commun. 2018, 498, 946–953. [Google Scholar] [CrossRef]
- Kleczkowski, L.A.; Kunz, S.; Wilczynska, M. Mechanisms of UDP-glucose synthesis in plants. Crit. Rev. Plant Sci. 2010, 29, 191–203. [Google Scholar] [CrossRef]
- Fujii, S.; Hayashi, T.; Mizuno, K. Sucrose synthase is an integral component of the cellulose synthesis machinery. Plant Cell Physiol. 2010, 51, 294–301. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yuan, J.; Yang, F.; Wang, L.; Ma, J.; Li, J.; Pu, X.; Raza, W.; Huang, Q.; Shen, Q. PGPR strain Paenibacillus polymyxa SQR-21 potentially benefits watermelon growth by re-shaping root protein expression. AMB Express. 2017, 7, 104. [Google Scholar] [CrossRef] [Green Version]
- Zhou, C.; Guo, J.; Zhu, L.; Xiao, X.; Xie, Y.; Zhu, J.; Ma, Z.; Wang, J. Paenibacillus polymyxa BFKC01 enhances plant iron absorption via improved root systems and activated iron acquisition mechanisms. Plant Physiol. Biochem. 2016, 105, 162–173. [Google Scholar] [CrossRef]
- Kwon, Y.S.; Lee, D.Y.; Rakwal, R.; Baek, S.-B.; Lee, J.H.; Kwak, Y.-S.; Seo, J.-S.; Chung, W.S.; Bae, D.-W.; Kim, S.G. Proteomic analyses of the interaction between the plant-growth promoting rhizobacterium Paenibacillus polymyxa E681 and Arabidopsis thaliana. Proteomics 2016, 16, 122–135. [Google Scholar] [CrossRef]
- Metzker, M.L. Sequencing technologies—The next generation. Nat. Rev. Genet. 2010, 11, 31–46. [Google Scholar] [CrossRef] [Green Version]
- Koren, S.; Walenz, B.P.; Berlin, K.; Miller, J.R.; Bergman, N.H.; Phillippy, A.M. Canu: Scalable and accurate long-read assembly via adaptive k-mer weighting and repeat separation. Genome Res. 2017, 27, 722–736. [Google Scholar] [CrossRef] [Green Version]
- Brown, J.W.S.; Calixto, C.P.G.; Zhang, R. High-quality reference transcript datasets hold the key to transcript-specific RNA-sequencing analysis in plants. New Phytol. 2017, 213, 525–530. [Google Scholar] [CrossRef]
- Mascher, M.; Gundlach, H.; Himmelbach, A.; Beier, S.; Twardziok, S.O.; Wicker, T.; Radchuk, V.; Dockter, C.; Hedley, P.E.; Russell, J.; et al. A chromosome conformation capture ordered sequence of the barley genome. Nature 2017, 544, 427–433. [Google Scholar] [CrossRef] [Green Version]
- Bais, H.P.; Weir, T.L.; Perry, L.G.; Gilroy, S.; Vivanco, J.M. The role of root exudates in rhizosphere interactions with plants and other organisms. Annu. Rev. Plant Biol. 2006, 57, 233–266. [Google Scholar] [CrossRef] [Green Version]
- Shi, S.; Richardson, A.E.; O’Callaghan, M.; DeAngelis, K.M.; Jones, E.E.; Stewart, A.; Firestone, M.K.; Condron, L.M. Effects of selected root exudate components on soil bacterial communities. FEMS Microbiol. Ecol. 2011, 77, 600–610. [Google Scholar] [CrossRef] [Green Version]
- O’Neal, L.; Vo, L.; Alexandre, G. Specific root exudate compounds sensed by dedicated chemoreceptors shape Azospirillum brasilense chemotaxis in the rhizosphere. Appl. Environ. Microbiol. 2020, 86, e01026-20. [Google Scholar] [CrossRef]
- Feng, H.; Zhang, N.; Du, W.; Zhang, H.; Liu, Y.; Fu, R.; Shao, J.; Zhang, G.; Shen, Q.; Zhang, R. Identification of chemotaxis compounds in root exudates and their sensing chemoreceptors in plant-growth-promoting rhizobacteria Bacillus amyloliquefaciens SQR9. Mol. Plant-Microbe Interact. 2018, 31, 995–1005. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cherchali, A.; Boukhelata, N.; Kaci, Y.; Abrous-Belbachir, O.; Djebbar, R. Isolation and identification of a phosphate-solubilizing Paenibacillus polymyxa strain GOL 0202 from durum wheat (Triticum durum Desf.) rhizosphere and its effect on some seedlings morphophysiological parameters. Biocatal. Agric. Biotechnol. 2019, 19, 101087. [Google Scholar] [CrossRef]
- Seneviratne, G.; Weerasekara, M.L.M.A.W.; Seneviratne, K.A.C.N.; Zavahir, J.S.; Kecskés, M.L.; Kennedy, I.R. Importance of biofilm formation in plant growth promoting rhizobacterial action. In Plant Growth and Health Promoting Bacteria; Maheshwari, D.K., Ed.; Springer: Berlin/Heidelberg, Germany, 2011; pp. 81–95. [Google Scholar]
- Timmusk, S.; Grantcharova, N.; Wagner, E.G.H. Paenibacillus polymyxa invades plant roots and forms biofilms. Appl. Environ. Microbiol. 2005, 71, 7292–7300. [Google Scholar] [CrossRef] [Green Version]
- Hardoim, P.R.; van Overbeek, L.S.; van Elsas, J.D. Properties of bacterial endophytes and their proposed role in plant growth. Trends Microbiol. 2008, 16, 463–471. [Google Scholar] [CrossRef] [PubMed]
- Monteiro, R.A.; Balsanelli, E.; Wassem, R.; Marin, A.M.; Brusamarello-Santos, L.C.; Schmidt, M.A.; Tadra-Sfeir, M.Z.; Pankievicz, V.C.; Cruz, L.M.; Chubatsu, L.S.; et al. Herbaspirillum-plant interactions: Microscopical, histological and molecular aspects. Plant Soil 2012, 356, 175–196. [Google Scholar] [CrossRef]
- Ferreira, R.B.; Monteiro, S.; Freitas, R.; Santos, C.N.; Chen, Z.; Batista, L.M.; Duarte, J.; Borges, A.; Teixeira, A.R. The role of plant defence proteins in fungal pathogenesis. Mol. Plant Pathol. 2007, 8, 677–700. [Google Scholar] [CrossRef] [PubMed]
- Anjum, N.A.; Sharma, P.; Gill, S.S.; Hasanuzzaman, M.; Khan, E.A.; Kachhap, K.; Mohamed, A.A.; Thangavel, P.; Devi, G.D.; Vasudhevan, P.; et al. Catalase and ascorbate peroxidase—Representative H2O2-detoxifying heme enzymes in plants. Environ. Sci. Pollut. Res. Int. 2016, 23, 19002–19029. [Google Scholar] [CrossRef] [PubMed]
- Pii, Y.; Mimmo, T.; Tomasi, N.; Terzano, R.; Cesco, S.; Crecchio, C. Microbial interactions in the rhizosphere: Beneficial influences of plant growth-promoting rhizobacteria on nutrient acquisition process. A review. Biol. Fertil. Soils 2015, 51, 403–415. [Google Scholar] [CrossRef]
- Rodríguez-Navarro, A.; Rubio, F. High-affinity potassium and sodium transport systems in plants. J. Exp. Bot. 2006, 57, 1149–1160. [Google Scholar] [CrossRef] [Green Version]
- Sun, J.; Bankston, J.R.; Payandeh, J.; Hinds, T.R.; Zagotta, W.N.; Zheng, N. Crystal structure of the plant dual-affinity nitrate transporter NRT1.1. Nature 2014, 507, 73–77. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Courty, P.E.; Smith, P.; Koegel, S.; Redecker, D.; Wipf, D. Inorganic nitrogen uptake and transport in beneficial plant root-microbe interactions. Crit. Rev. Plant Sci. 2015, 34, 4–16. [Google Scholar] [CrossRef]
- Tegeder, M.; Masclaux-Daubresse, C. Source and sink mechanisms of nitrogen transport and use. New Phytol. 2018, 217, 35–53. [Google Scholar] [CrossRef] [Green Version]
- Quan, X.; Zeng, J.; Ye, L.; Chen, G.; Han, Z.; Shah, J.M.; Zhang, G. Transcriptome profiling analysis for two Tibetan wild barley genotypes in responses to low nitrogen. BMC Plant Bio. 2016, 16, 30. [Google Scholar] [CrossRef] [Green Version]
- Diaz, C.; Saliba-Colombani, V.; Loudet, O.; Belluomo, P.; Moreau, L.; Daniel-Vedele, F.; Morot-Gaudry, J.-F.; Masclaux-Daubresse, C. Leaf yellowing and anthocyanin accumulation are two genetically independent strategies in response to nitrogen limitation in Arabidopsis thaliana. Plant Cell Physiol. 2006, 47, 74–83. [Google Scholar] [CrossRef] [Green Version]
- Soubeyrand, E.; Basteau, C.; Hilbert, G.; van Leeuwen, C.; Delrot, S.; Gomès, E. Nitrogen supply affects anthocyanin biosynthetic and regulatory genes in grapevine cv. Cabernet-Sauvignon berries. Phytochemistry 2014, 103, 38–49. [Google Scholar] [CrossRef]
- Joshi, R.; Ramanarao, M.V.; Lee, S.; Kato, N.; Baisakh, N. Ectopic expression of ADP ribosylation factor 1 (SaARF1) from smooth cordgrass (Spartina alterniflora Loisel) confers drought and salt tolerance in transgenic rice and Arabidopsis. Plant Cell Tissue Organ Cult. 2014, 117, 17–30. [Google Scholar] [CrossRef]
- Lewis, D.R.; Negi, S.; Sukumar, P.; Muday, G.K. Ethylene inhibits lateral root development, increases IAA transport and expression of PIN3 and PIN7 auxin efflux carriers. Development 2011, 138, 3485–3495. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Swarup, R.; Perry, P.; Hagenbeek, D.; Van Der Straeten, D.; Beemster, G.T.S.; Sandberg, G.; Bhalerao, R.; Ljung, K.; Bennett, M.J. Ethylene upregulates auxin biosynthesis in Arabidopsis seedlings to enhance inhibition of root cell elongation. Plant Cell 2007, 19, 2186–2196. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blin, K.; Shaw, S.; Steinke, K.; Villebro, R.; Ziemert, N.; Lee, S.Y.; Medema, M.H.; Weber, T. antiSMASH 5.0: Updates to the secondary metabolite genome mining pipeline. Nucleic Acids Res. 2019, 47, W81–W87. [Google Scholar] [CrossRef] [Green Version]
- Tyc, O.; Song, C.; Dickschat, J.S.; Vos, M.; Garbeva, P. The ecological role of volatile and soluble secondary metabolites produced by soil bacteria. Trends Microbiol. 2017, 25, 280–292. [Google Scholar] [CrossRef]
- Sui, X.; Kiser, P.D.; Lintig, J.v.; Palczewski, K. Structural basis of carotenoid cleavage: From bacteria to mammals. Arch. Biochem. Biophys. 2013, 539, 203–213. [Google Scholar] [CrossRef] [Green Version]
- Bible, A.N.; Fletcher, S.J.; Pelletier, D.A.; Schadt, C.W.; Jawdy, S.S.; Weston, D.J.; Engle, N.L.; Tschaplinski, T.; Masyuko, R.; Polisetti, S.; et al. A carotenoid-deficient mutant in Pantoea sp. YR343, a bacteria isolated from the rhizosphere of Populus deltoides, is defective in root colonization. Front. Microbiol. 2016, 7, 491. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Qiu, S. Examining phylogenetic relationships of Erwinia and Pantoea species using whole genome sequence data. Antonie Van Leeuwenhoek 2015, 108, 1037–1046. [Google Scholar] [CrossRef]
- Bulgarelli, D.; Garrido-Oter, R.; Munch, P.C.; Weiman, A.; Droge, J.; Pan, Y.; McHardy, A.C.; Schulze-Lefert, P. Structure and function of the bacterial root microbiota in wild and domesticated barley. Cell Host Microbe 2015, 17, 392–403. [Google Scholar] [CrossRef] [Green Version]
- Bacon, C.W.; Palencia, E.R.; Hinton, D.M. Abiotic and biotic plant stress-tolerant and beneficial secondary metabolites produced by endophytic Bacillus species. In Plant Microbes Symbiosis: Applied Facets; Arora, N.K., Ed.; Springer: New Delhi, India, 2015; pp. 163–177. [Google Scholar]
- Mishra, J.; Fatima, T.; Arora, N.K. Role of secondary metabolites from plant growth-promoting rhizobacteria in combating salinity stress. In Plant Microbiome: Stress Response; Egamberdieva, D., Ahmad, P., Eds.; Springer: Singapore, 2018; pp. 127–163. [Google Scholar]
- Liu, H.; Liu, K.; Li, Y.; Wang, C.; Hou, Q.; Xu, W.; Fan, L.; Zhao, J.; Gou, J.; Du, B.; et al. Complete genome sequence of Paenibacillus polymyxa YC0136, a plant growth–promoting rhizobacterium isolated from tobacco rhizosphere. Genome Announc. 2017, 5, e01635-16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Korir, H.; Mungai, N.W.; Thuita, M.; Hamba, Y.; Masso, C. Co-inoculation effect of rhizobia and plant growth promoting rhizobacteria on common bean growth in a low phosphorus soil. Front. Plant Sci. 2017, 8, 141. [Google Scholar] [CrossRef] [Green Version]
- Yu, X.; Liu, X.; Zhu, T.-H.; Liu, G.-H.; Mao, C. Co-inoculation with phosphate-solubilzing and nitrogen-fixing bacteria on solubilization of rock phosphate and their effect on growth promotion and nutrient uptake by walnut. Eur. J. Soil Biol. 2012, 50, 112–117. [Google Scholar] [CrossRef]
- Razzaghi Komaresofla, B.; Alikhani, H.A.; Etesami, H.; Khoshkholgh-Sima, N.A. Improved growth and salinity tolerance of the halophyte Salicornia sp. by co–inoculation with endophytic and rhizosphere bacteria. Appl. Soil Ecol. 2019, 138, 160–170. [Google Scholar] [CrossRef]
- Chen, S.; Zhou, Y.; Chen, Y.; Gu, J. fastp: An ultra-fast all-in-one FASTQ preprocessor. Bioinformatics 2018, 34, i884–i890. [Google Scholar] [CrossRef] [PubMed]
- Patro, R.; Duggal, G.; Love, M.I.; Irizarry, R.A.; Kingsford, C. Salmon provides fast and bias-aware quantification of transcript expression. Nat. Methods 2017, 14, 417–419. [Google Scholar] [CrossRef] [Green Version]
- Li, T.; Mann, R.; Sawbridge, T.; Kaur, J.; Auer, D.; Spangenberg, G. Novel Xanthomonas species from the perennial ryegrass seed microbiome—Assessing the bioprotection activity of non-pathogenic relatives of pathogens. Front. Microbiol. 2020, 11, 1991. [Google Scholar] [CrossRef] [PubMed]
- Pimentel, H.; Bray, N.L.; Puente, S.; Melsted, P.; Pachter, L. Differential analysis of RNA-seq incorporating quantification uncertainty. Nat. Methods 2017, 14, 687–690. [Google Scholar] [CrossRef]
- Raudvere, U.; Kolberg, L.; Kuzmin, I.; Arak, T.; Adler, P.; Peterson, H.; Vilo, J. g:Profiler: A web server for functional enrichment analysis and conversions of gene lists (2019 update). Nucleic Acids Res. 2019, 47, W191–W198. [Google Scholar] [CrossRef] [Green Version]
- Hulsen, T.; de Vlieg, J.; Alkema, W. BioVenn—A web application for the comparison and visualization of biological lists using area-proportional Venn diagrams. BMC Genom. 2008, 9, 488. [Google Scholar] [CrossRef] [Green Version]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | Treatment | Medium | No. of Genes Passed the Abundance Filter | No. of Differentially Expressed Genes | |
|---|---|---|---|---|---|
| Bacteria | AR | Barley seedling | Nutrient Broth | 4009 | 1380 |
| S25 | 5013 | 2945 | |||
| S02 | 5266 | 2890 | |||
| Burk’s N-free | 5032 | 2524 | |||
| Plant | Barley seedling | AR | Nutrient Broth | 37,073 | 13,948 |
| S25 | 35,365 | 13,648 | |||
| S02 | 34,798 | 9129 | |||
| Burk’s N-free | 31,502 | 10,806 |
| Strain ID | Genome Size (bp) | No. of Genes |
|---|---|---|
| S02 (Paenibacillus sp.) | 6,060,529 | 5436 |
| S25 (Paenibacillus sp.) | 5,958,851 | 5306 |
| AR (Erwinia gerundensis) | 4,437,426 | 4091 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, T.; Mann, R.; Kaur, J.; Spangenberg, G.; Sawbridge, T. Transcriptome Analyses of Barley Roots Inoculated with Novel Paenibacillus sp. and Erwinia gerundensis Strains Reveal Beneficial Early-Stage Plant–Bacteria Interactions. Plants 2021, 10, 1802. https://doi.org/10.3390/plants10091802
Li T, Mann R, Kaur J, Spangenberg G, Sawbridge T. Transcriptome Analyses of Barley Roots Inoculated with Novel Paenibacillus sp. and Erwinia gerundensis Strains Reveal Beneficial Early-Stage Plant–Bacteria Interactions. Plants. 2021; 10(9):1802. https://doi.org/10.3390/plants10091802
Chicago/Turabian StyleLi, Tongda, Ross Mann, Jatinder Kaur, German Spangenberg, and Timothy Sawbridge. 2021. "Transcriptome Analyses of Barley Roots Inoculated with Novel Paenibacillus sp. and Erwinia gerundensis Strains Reveal Beneficial Early-Stage Plant–Bacteria Interactions" Plants 10, no. 9: 1802. https://doi.org/10.3390/plants10091802