
Microbial Diversity Characteristics of Areca Palm Rhizosphere Soil at Different Growth Stages

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Soil Samples Collection and Deoxyribonucleic Acid (DNA) Extraction
2.2. Diversity Analysis of Microbial Communities
3. Results and Discussion
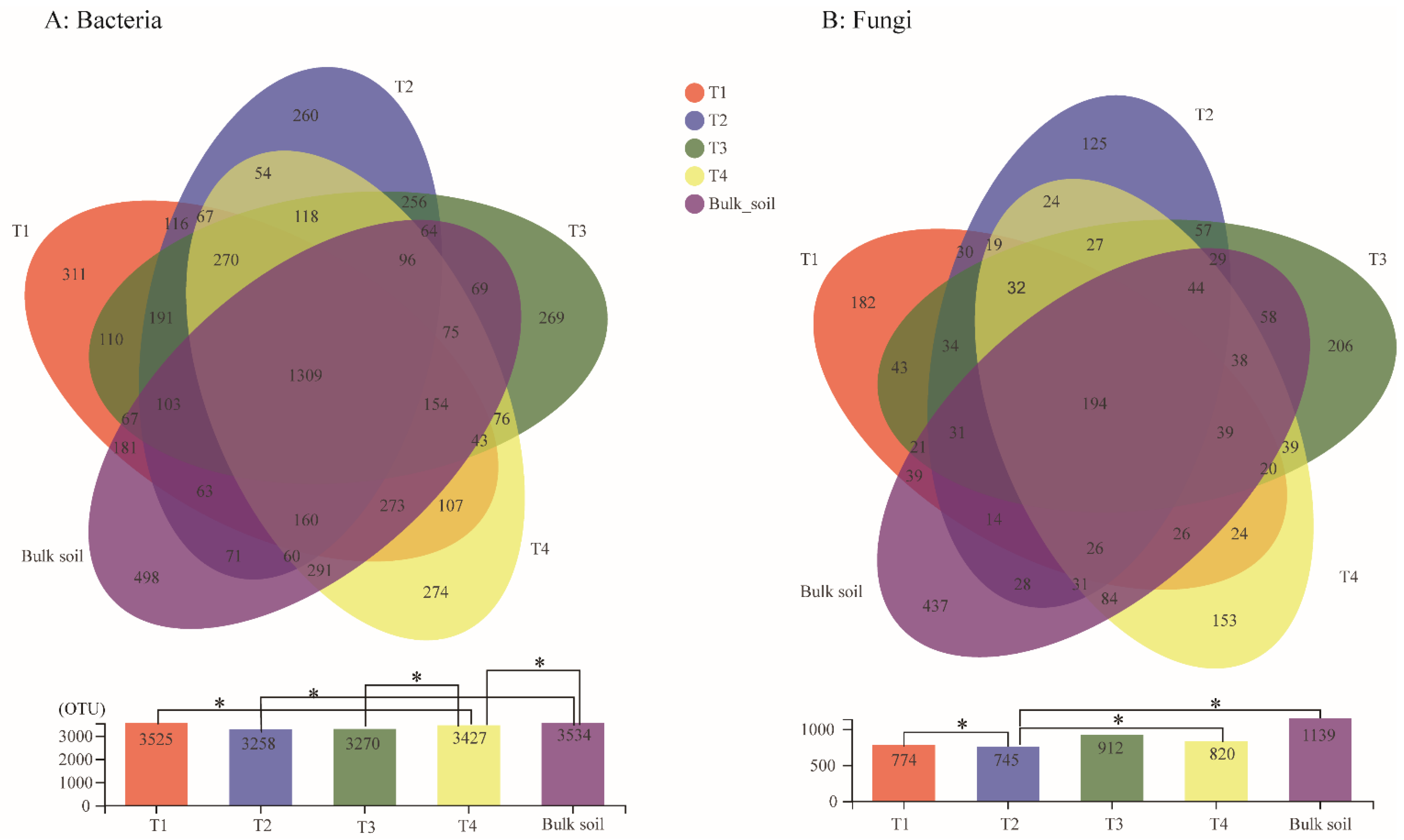
3.1. Bacterial and Fungal Diversity of the Areca Palm Rhizosphere
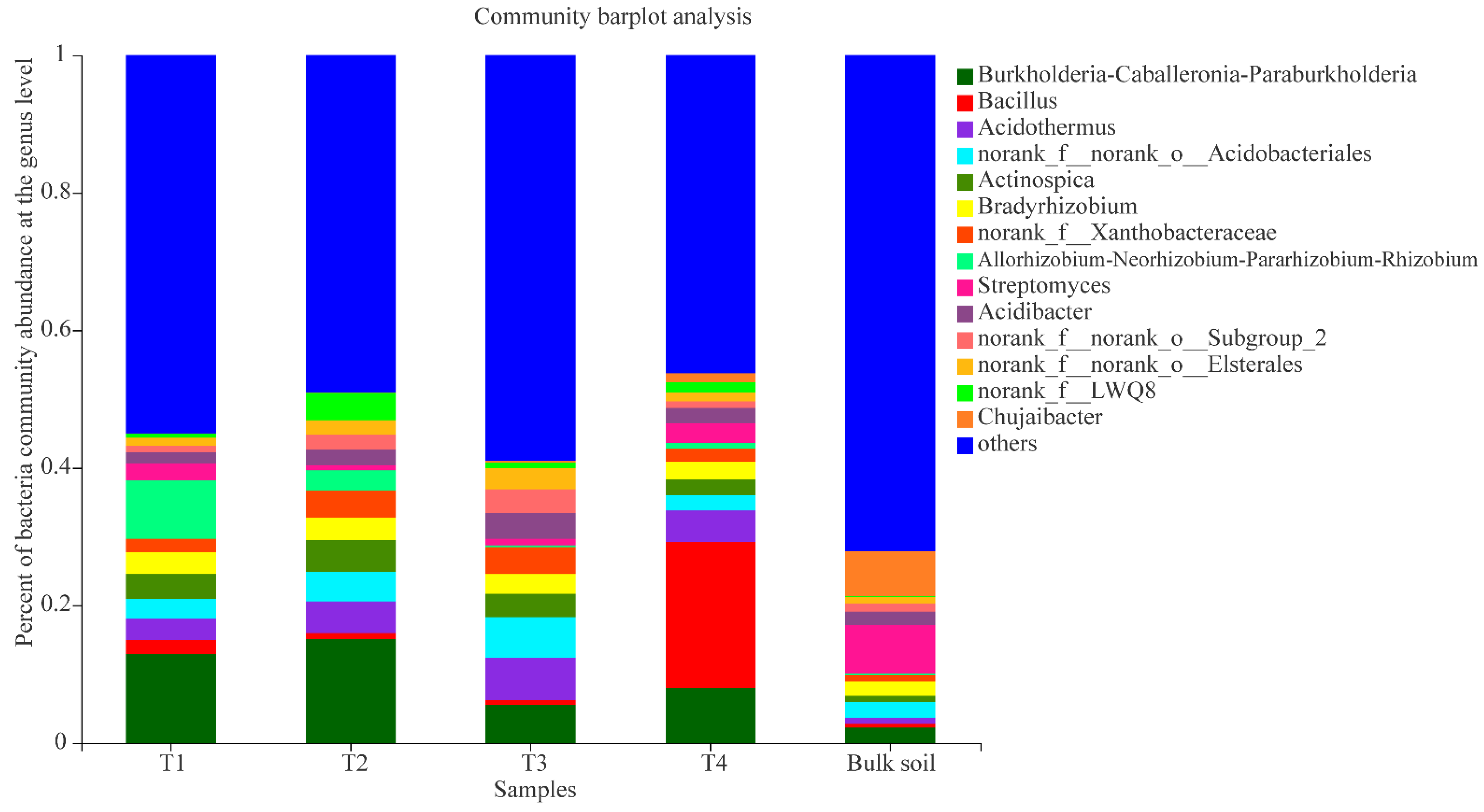
3.2. Bacterial Composition at Different Growth Stages
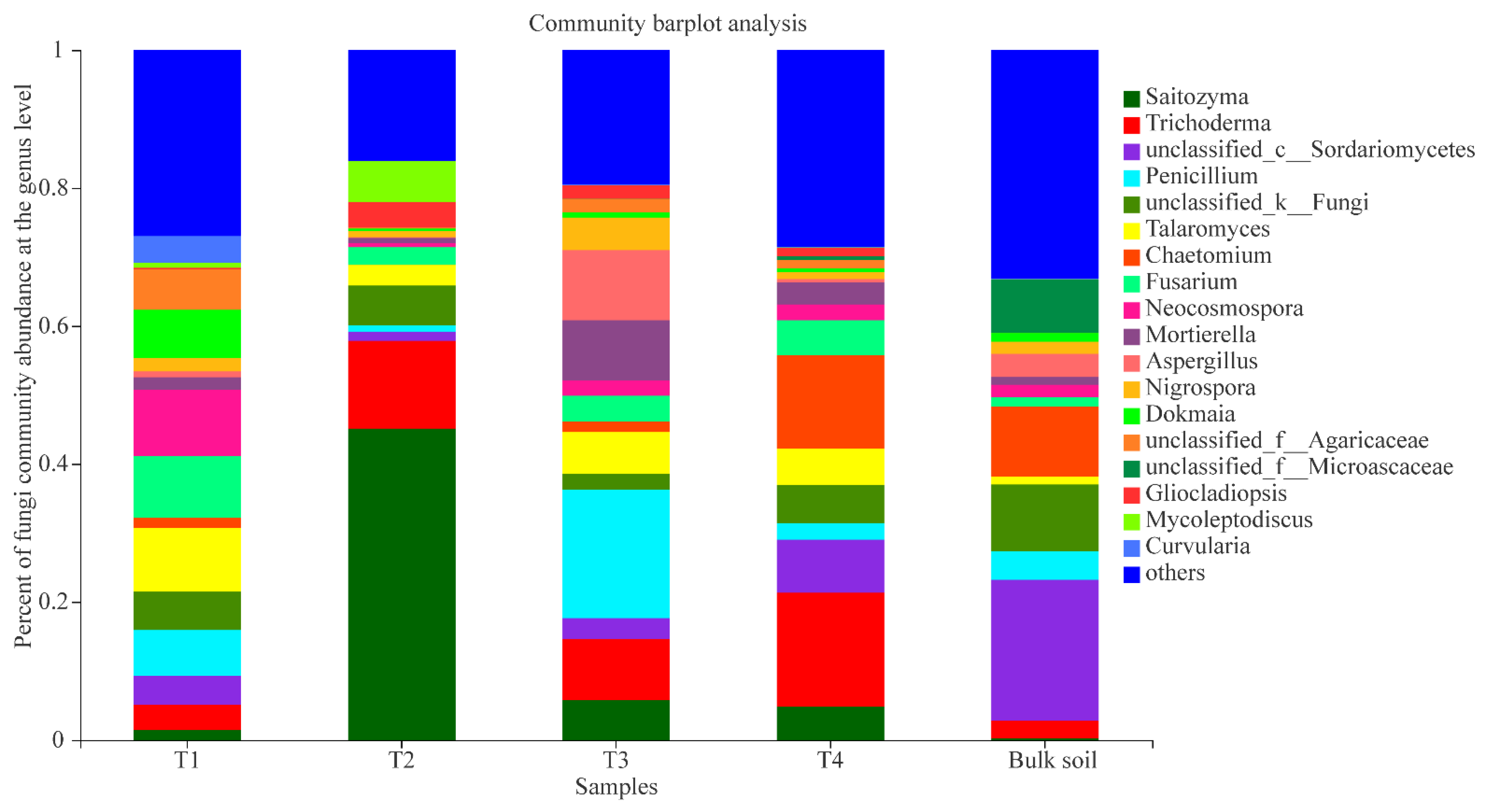
3.3. Fungal Composition at Different Growth Stages
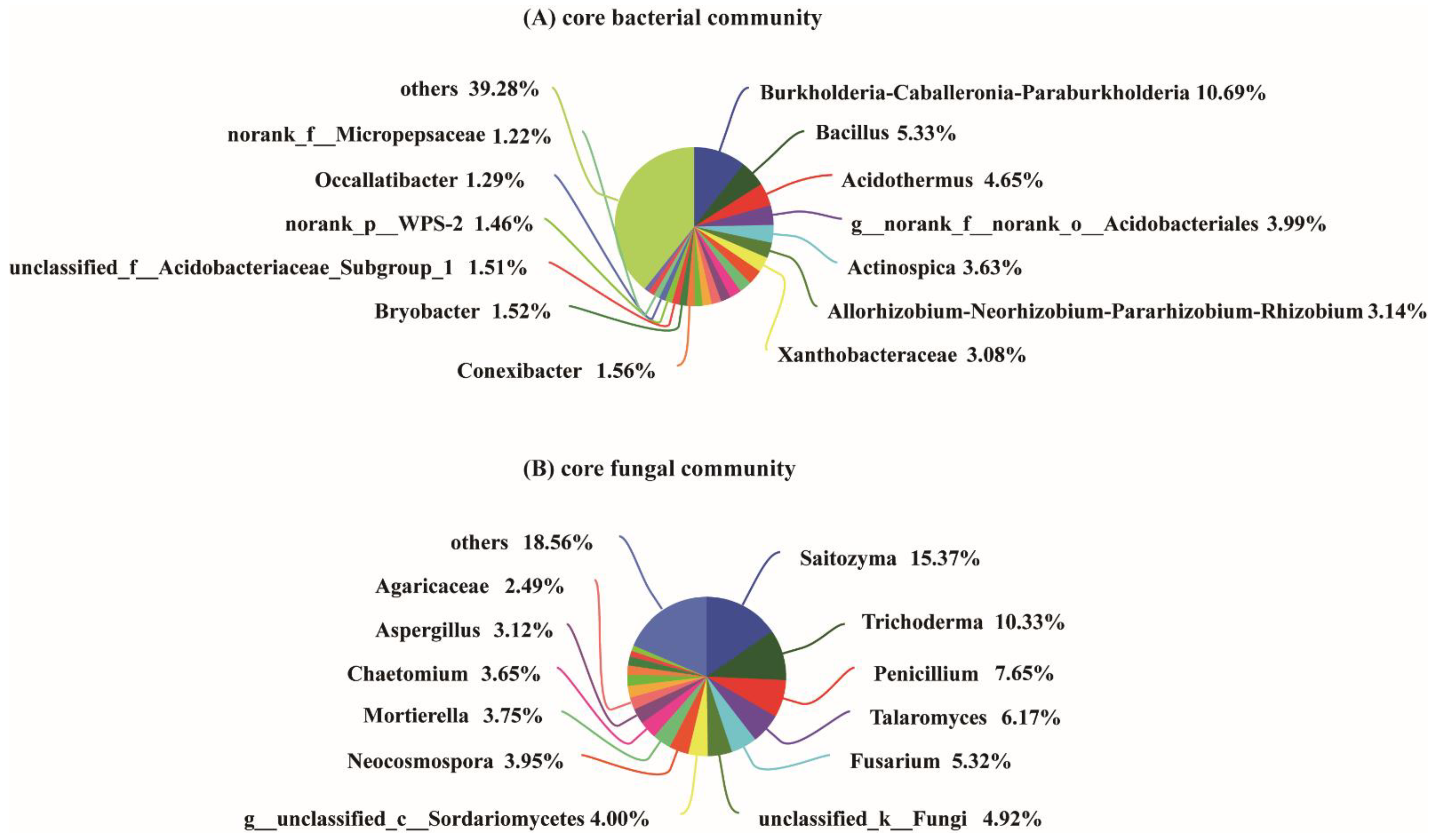
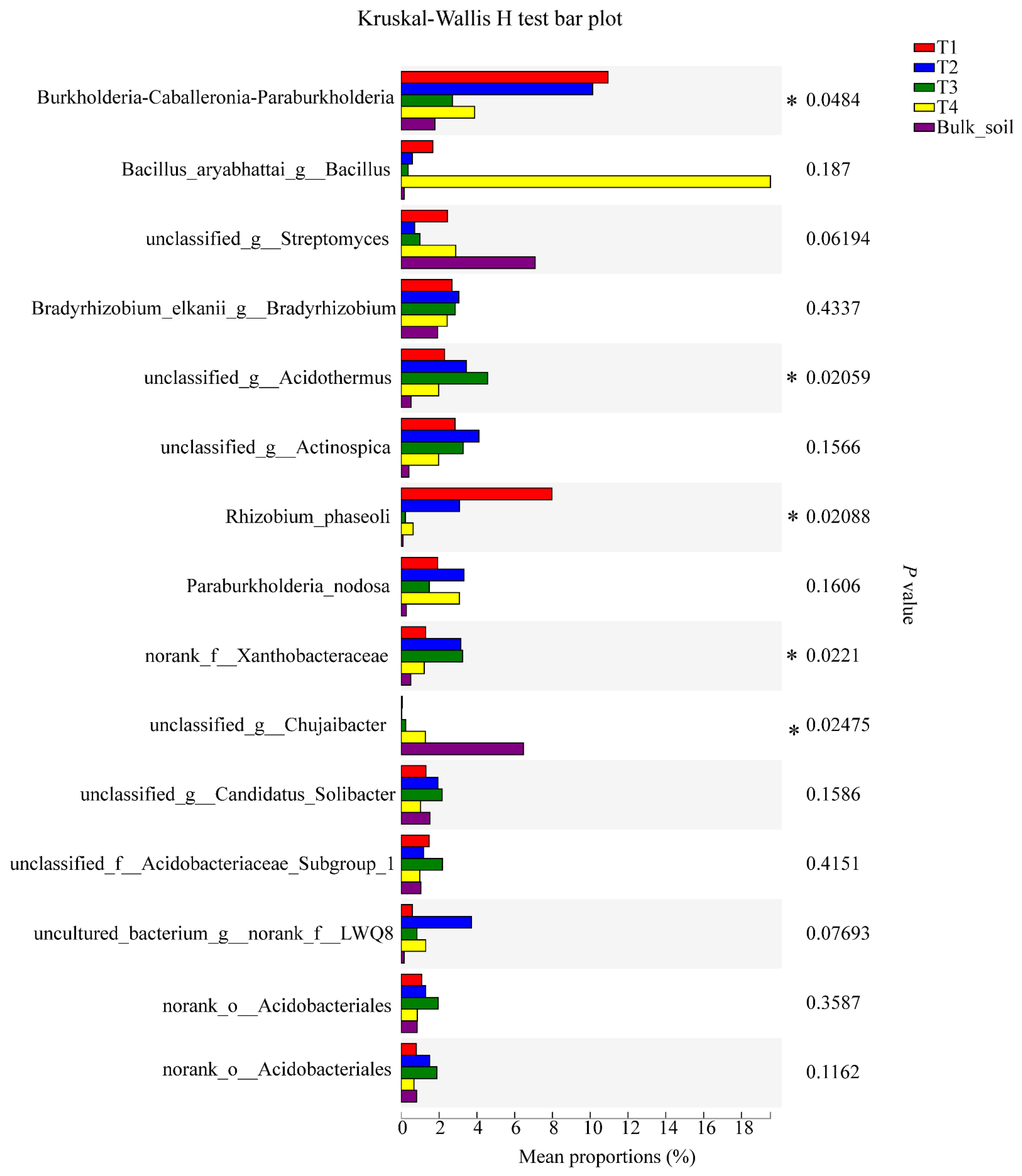
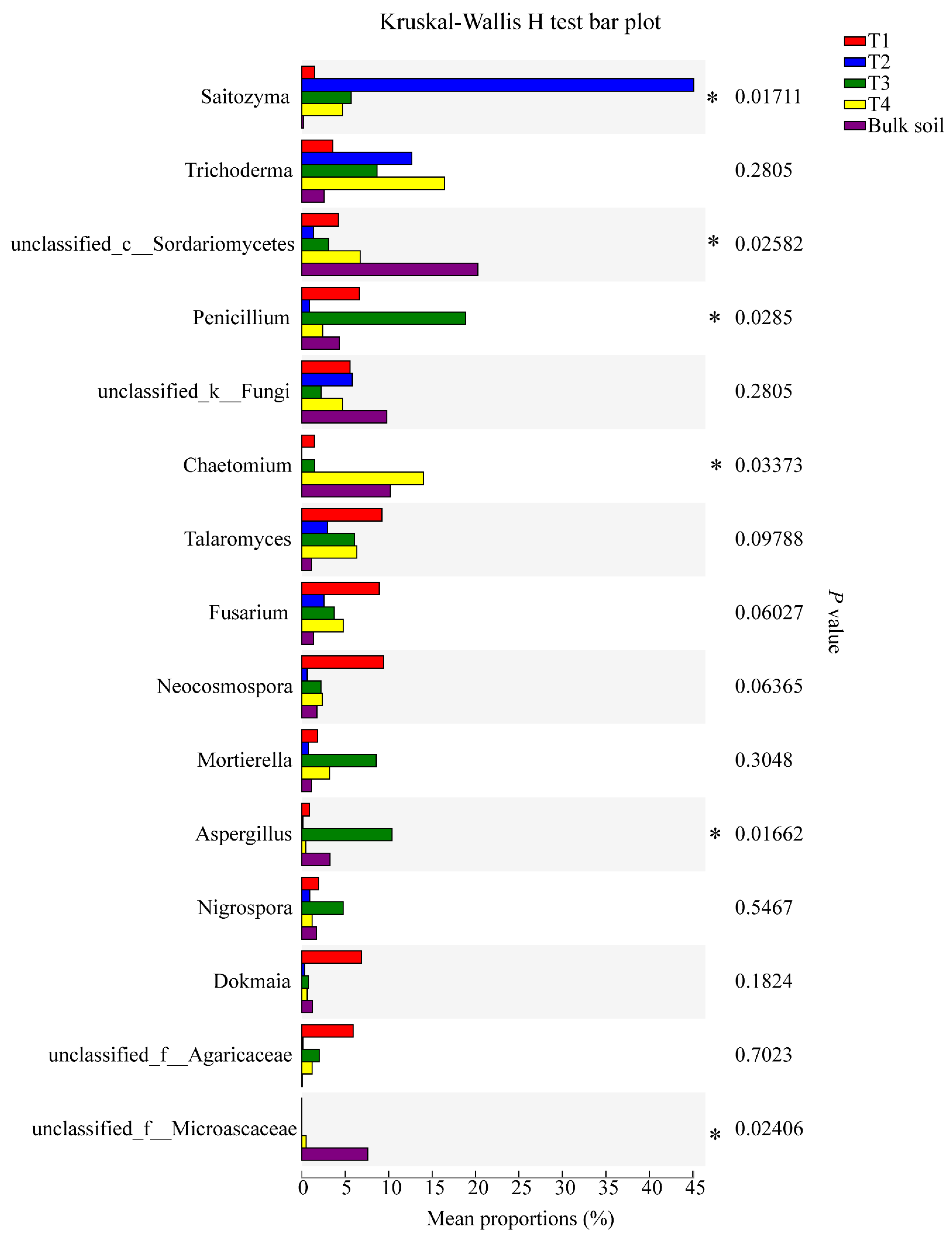
3.4. Microbial Genera with Significant Differences among the Samples
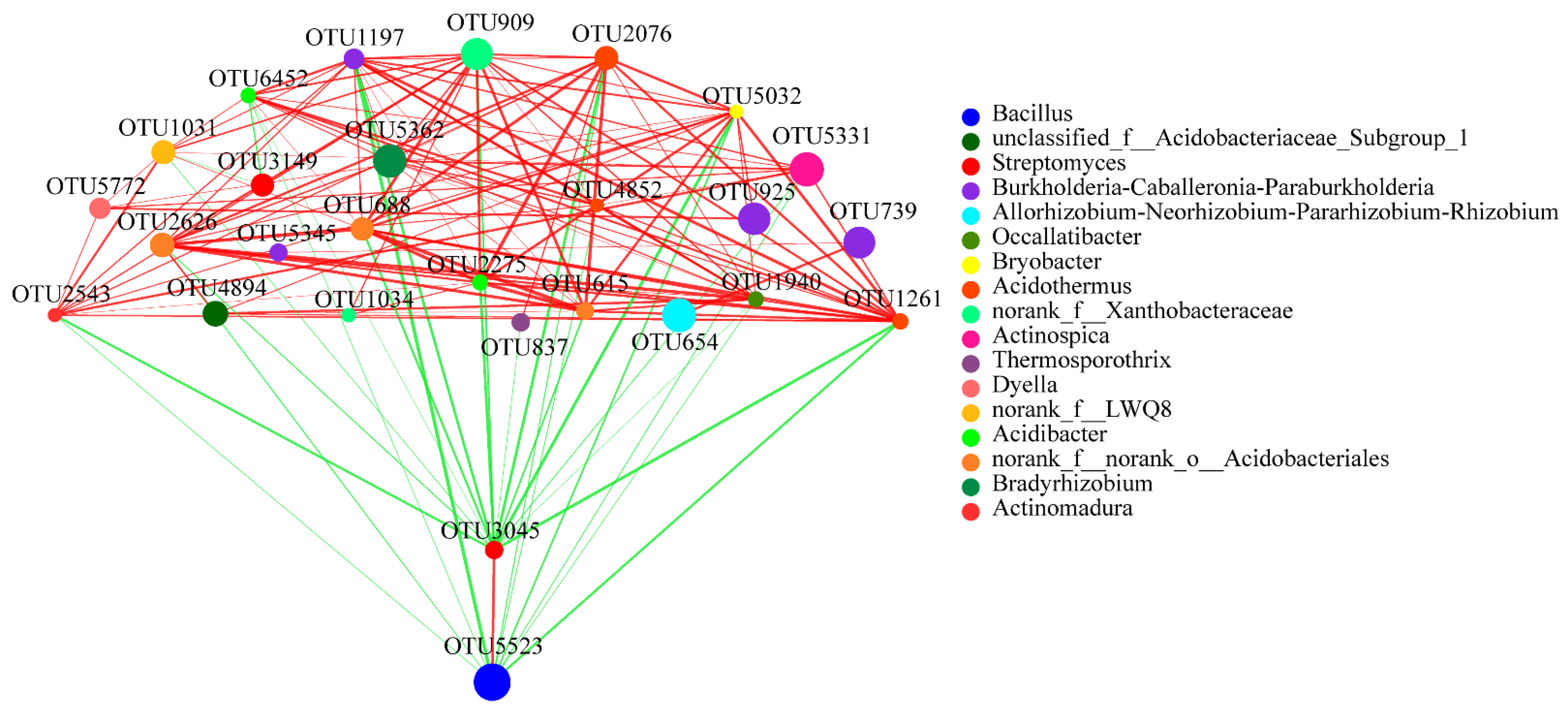
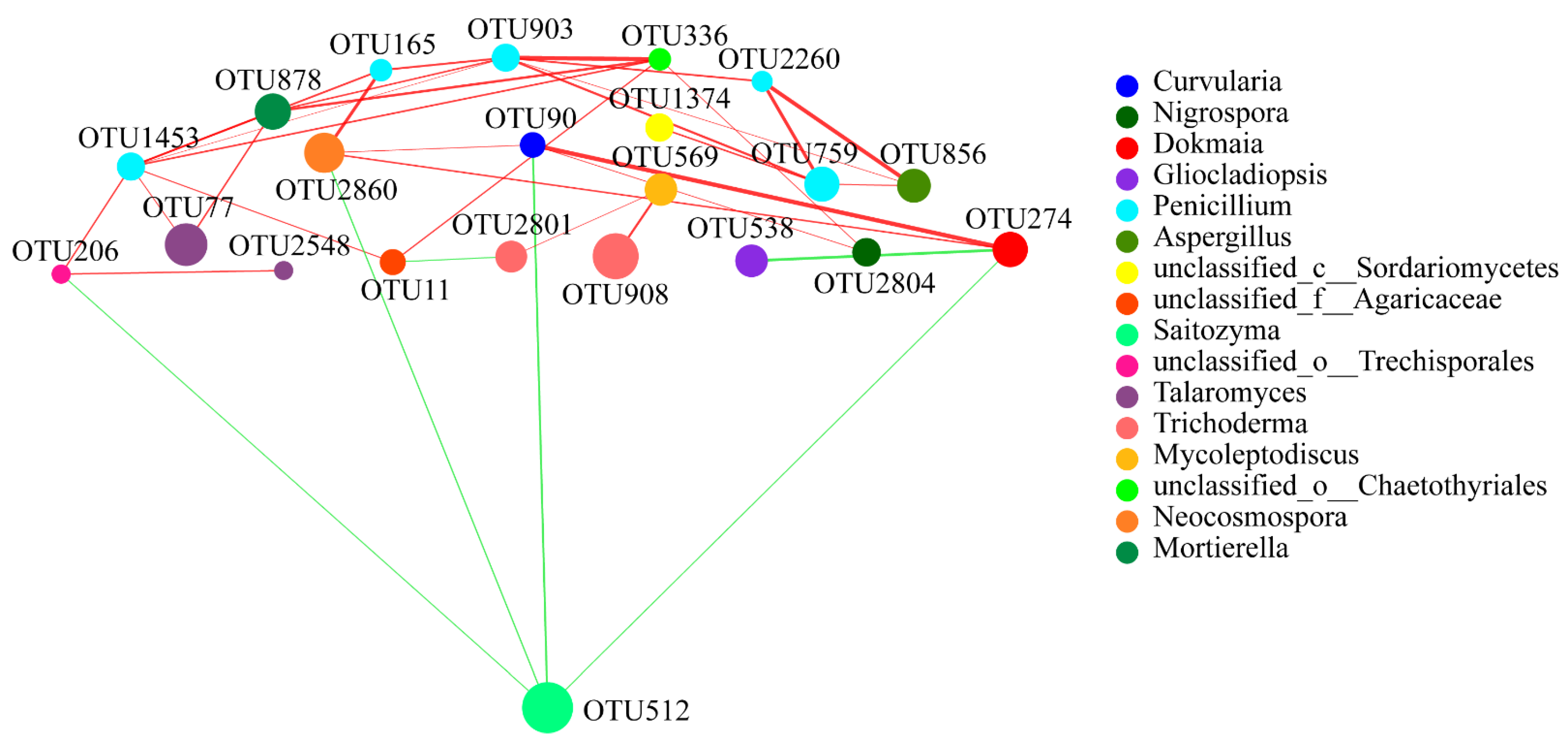
3.5. Bacterial and Fungal Network Analyses
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Henderson, A. (Ed.) The Palms of Southern Asia; Priceton University Press: Priceton, NJ, USA, 2009. [Google Scholar]
- Salehi, B.; Konovalov, D.A.; Fru, P.; Kapewangolo, P.; Peron, G.; Ksenija, M.S.; Cardoso, S.M.; Pereira, O.R.; Nigam, M.; Nicola, S.; et al. Areca catechu-From Farm to Food and Biomedical Applications. Phytother. Res. 2020, 34, 2140–2158. [Google Scholar] [CrossRef] [PubMed]
- Bharath, B.G.; Ananda, K.S.; Jayarajan, R.; Nagaraja, N.R. Studies on Genetic Relationships and Diversity in Arecanut (Areca catechu L.) Germplasm Utilizing RAPD Markers. J. Plant. Crop. 2015, 2015, 2. [Google Scholar]
- Yang, Y.; Huang, L.; Xu, C.; Qi, L.; Wu, Z.; Li, J.; Chen, H.; Wu, Y.; Fu, T.; Zhu, H.; et al. Chromosome-Scale Genome Assembly of Areca palm (Areca catechu). Mol. Ecol. Resour. 2021, 21, 2504–2519. [Google Scholar] [CrossRef]
- Manimekalai, R.; Nair, S.; Naganeeswaran, A.; Karun, A.; Malhotra, S.; Hubbali, V. Transcriptome Sequencing and De Novo Assembly in Arecanut, Areca catechu L. Elucidates The Secondary Metabolite Pathway Genes. Biotechnol. Rep. 2018, 17, 63–69. [Google Scholar] [CrossRef]
- Zhang, L.; Xiaojian, Y.; Zhang, J.; Wei, Y.; Huo, D.; Ma, C.; Chang, H.; Cai, K.; Shi, H. Comprehensive Microbiome and Metabolome Analyses Reveal the Physiological Mechanism of Chlorotic Areca Leaves. Tree Physiol. 2020, 41, 147–161. [Google Scholar] [CrossRef]
- Berg, G.; Smalla, K. Plant Species and Soil Type Cooperatively Shape the Structure and Function of Microbial Communities in The Rhizosphere. FEMS Microbiol. Ecol. 2009, 68, 1–13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Philippot, L.; Raaijmakers, J.; Lemanceau, P.; Putten, W. Going Back to The Roots: The Microbial Ecology of The Rhizosphere. Nat. Rev. Microbiol. 2013, 11, 789–799. [Google Scholar] [CrossRef] [PubMed]
- Rasche, F.; Hödl, V.; Poll, C.; Kandeler, E.; Gerzabek, M.H.; Elsas, J.; Sessitsch, A. Rhizosphere Bacteria Affected by Transgenic Potatoes with Antibacterial Activities Compared with The Effects of Soil, Wild-Type Potatoes, Vegetation Stage and Pathogen Exposure. FEMS Microbiol. Ecol. 2006, 56, 219–235. [Google Scholar] [CrossRef] [Green Version]
- Raaijmakers, J.; Paulitz, T.; Steinberg, C.; Alabouvette, C.; Moënne-Loccoz, Y. The Rhizosphere: A Playground and Battlefield for Soilborne Pathogens and Beneficial Microorganisms. Plant Soil 2009, 321, 341–361. [Google Scholar] [CrossRef] [Green Version]
- Inceoğlu, Ö.; Al-Soud, W.A.; Salles, J.; Semenov, A.; van Elsas, J. Comparative Analysis of Bacterial Communities in a Potato Field as Determined by Pyrosequencing. PLoS ONE 2011, 6, e23321. [Google Scholar] [CrossRef] [Green Version]
- Rajput, V.D.; Minkina, T.; Feizi, M.; Kumari, A.; Khan, M.; Mandzhieva, S.; Sushkova, S.; El-Ramady, H.; Verma, K.K.; Singh, A.; et al. Effects of silicon and silicon-based nanoparticles on rhizosphere microbiome, plant stress and growth. Biology 2021, 10, 791. [Google Scholar] [CrossRef]
- Chaparro, J.; Badri, D.; Vivanco, J. Rhizosphere Microbiome Assemblage is Affected by Plant Development. ISME J. 2013, 8, 790–803. [Google Scholar] [CrossRef] [Green Version]
- Rudrappa, T.; Czymmek, K.; Pare, P.; Bais, H. Root-Secreted Malic Acid Recruits Beneficial Soil Bacteria. Plant Physiol. 2008, 148, 1547–1556. [Google Scholar] [CrossRef] [Green Version]
- Berendsen, R.L.; Pieterse, C.M.; Bakker, P.A. The rhizosphere microbiome and plant health. Trends Plant Sci. 2012, 17, 478–486. [Google Scholar] [CrossRef] [PubMed]
- Giri, S.; Jeffrey, R.I.; Chen, C.; Zabriskie, T.M.; Krausz, K.W.; Gonzalez, F.J. A Metabolomic Approach to The Metabolism of The Areca Nut Alkaloids Arecoline and Arecaidine in The Mouse. Chem. Res. Toxicol. 2006, 19, 818–827. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lane, D. 16S/23S rRNA Sequencing. Nucleic Acid Techniques in Bacterial Systematics; Stackebrandt, E., Goodfellow, M., Eds.; John Wiley & Sons: Chichester, UK, 1990; pp. 115–147. [Google Scholar]
- Xiao, Y.; Xiong, T.; Peng, Z.; Liu, C.; Huang, T.; Yu, H.; Xie, M. Correlation Between Microbiota and Flavours in Fermentation of Chinese Sichuan Paocai. Food Res. Int. 2018, 114, 123–132. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Hu, A.; Zhou, J.; Zhang, W.; Li, P. Comparison of Bacterial Communities in Soil Samples with and without Tomato Bacterial Wilt Caused by Ralstonia solanacearum Species Complex. BMC Microbiol. 2020, 20, 89. [Google Scholar] [CrossRef]
- Qi, G.; Ma, G.; Chen, S.; Lin, C.; Zhao, X. Microbial Network and Soil Properties are Changed in Bacterial Wilt-Susceptible Soil. Appl. Environ. Microbiol. 2019, 85, e00162-19. [Google Scholar] [CrossRef] [Green Version]
- Wagner, M.R.; Lundberg, D.S.; del Rio, T.G.; Tringe, S.G.; Dangl, J.L.; Mitchell-Olds, T. Host genotype and age shape the leaf and root microbiomes of a wild perennial plant. Nat. Commun. 2016, 7, 12151. [Google Scholar] [CrossRef]
- Pfeiffer, S.; Mitter, B.; Oswald, A.; Schloter-Hai, B.; Schloter, M.; Declerck, S.; Sessitsch, A. Rhizosphere microbiomes of potato cultivated in the High Andes show stable and dynamic core microbiomes with different responses to plant development. FEMS Microbiol. Ecol. 2017, 93, fiw242. [Google Scholar] [CrossRef]
- de Ruiter, P.C.; Neutel, A.M.; Moore, J.C. Energetics, Patterns of Interaction Strengths, and Stability in Real Ecosystems. Science 1995, 269, 1257–1260. [Google Scholar] [CrossRef]
- Li, X.; Yang, W.; Gaedke, U.; de Ruiter, P.C. Energetic Constraints Imposed on Trophic Interaction Strengths Enhance Resilience in Empirical and Model Food Webs. J. Anim. Ecol. 2021, 90, 2065–2076. [Google Scholar] [CrossRef] [PubMed]
- Ongena, M.; Jacques, P. Bacillus lipopeptides: Versatile weapons for plant disease biocontrol. Trends Microbiol. 2008, 16, 115–125. [Google Scholar] [CrossRef] [PubMed]
- Tahir, H.A.; Gu, Q.; Wu, H.; Niu, Y.; Huo, R.; Gao, X. Bacillus volatiles adversely affect the physiology and ultra-structure of Ralstonia solanacearum and induce systemic resistance in tobacco against bacterial wilt. Sci. Rep. 2017, 7, 40481. [Google Scholar] [CrossRef]
- Asaka, O.; Shoda, M. Biocontrol of Rhizoctonia solani damping-off of tomato with Bacillus subtilis RB14. Appl. Environ. Microbiol. 1996, 62, 4081–4085. [Google Scholar] [CrossRef] [Green Version]
- Leclère, V.; Béchet, M.; Adam, A.; Guez, J.S.; Wathelet, B.; Ongena, M.; Thonart, P.; Gancel, F.; Chollet-Imbert, M.; Jacques, P. Mycosubtilin overproduction by Bacillus subtilis BBG100 enhances the organism’s antagonistic and biocontrol activities. Appl. Environ. Microbiol. 2005, 71, 4577–4584. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Romero, D.; de Vicente, A.; Rakotoaly, R.H.; Dufour, S.E.; Veening, J.W.; Arrebola, E.; Cazorla, F.M.; Kuipers, O.P.; Paquot, M.; Pérez-García, A. The iturin and fengycin families of lipopeptides are key factors in antagonism of Bacillus subtilis toward Podosphaera fusca. Mol. Plant-Microbe Interact. MPMI 2007, 20, 430–440. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shah, V.; Subramaniam, S. Bradyrhizobium japonicum USDA110: A representative model organism for studying the impact of pollutants on soil microbiota. Sci. Total. Environ. 2018, 624, 963–967. [Google Scholar] [CrossRef] [PubMed]
- Ma, L.J.; Geiser, D.M.; Proctor, R.H.; Rooney, A.P.; O’Donnell, K.; Trail, F.; Gardiner, D.M.; Manners, J.M.; Kazan, K. Fusarium Pathogenomics. Annu. Rev. Microbiol. 2013, 67, 399–416. [Google Scholar] [CrossRef] [PubMed] [Green Version]









Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ma, S.; Lin, Y.; Qin, Y.; Diao, X.; Li, P. Microbial Diversity Characteristics of Areca Palm Rhizosphere Soil at Different Growth Stages. Plants 2021, 10, 2706. https://doi.org/10.3390/plants10122706
Ma S, Lin Y, Qin Y, Diao X, Li P. Microbial Diversity Characteristics of Areca Palm Rhizosphere Soil at Different Growth Stages. Plants. 2021; 10(12):2706. https://doi.org/10.3390/plants10122706
Chicago/Turabian StyleMa, Siyuan, Yubin Lin, Yongqiang Qin, Xiaoping Diao, and Peng Li. 2021. "Microbial Diversity Characteristics of Areca Palm Rhizosphere Soil at Different Growth Stages" Plants 10, no. 12: 2706. https://doi.org/10.3390/plants10122706