
Molecular Challenges and Opportunities in Climate Change-Induced Kidney Diseases


Abstract
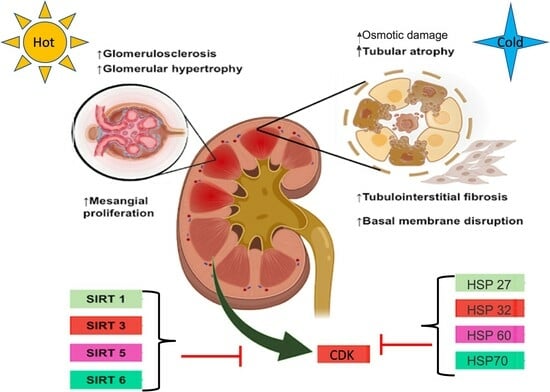
:
1. Introduction
2. Kidney Health in Extreme Temperature Conditions
3. Compensatory Mechanisms Activated under Extreme Temperatures Can Exert Further Kidney Injury
3.1. Mechanisms and Biomarkers Associated with Kidney Damage Due to High and Low Temperatures
3.1.1. Vasopressin, Fructose Uptake, Osmotic Damage, and Inflammation
3.1.2. Renin–Angiotensin–Aldosterone System
4. Heat Shock Proteins and Sirtuins as Renal Biomarkers of Protection against Extreme Temperatures
4.1. Heat Shock Proteins
4.1.1. Heat Shock Protein 27
4.1.2. Heat Shock Protein 70
4.1.3. Heat Shock Protein 90
4.1.4. Heat Shock Protein 60
4.1.5. Heat Shock Protein 32
4.2. Sirtuins
4.2.1. Sirtuins and RAAS
4.2.2. Sirtuins, Metabolism, and Adipose Tissue
5. Concluding Remarks and Future Perspectives
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Peters, A.; Schneider, A. Cardiovascular Risks of Climate Change. Nat. Rev. Cardiol. 2021, 18, 1–2. [Google Scholar] [CrossRef]
- Davídkovová, H.; Plavcová, E.; Kynčl, J.; Kyselý, J. Impacts of Hot and Cold Spells Differ for Acute and Chronic Ischaemic Heart Diseases. BMC Public Health 2014, 14, 480. [Google Scholar] [CrossRef]
- Gostimirovic, M.; Novakovic, R.; Rajkovic, J.; Djokic, V.; Terzic, D.; Putnik, S.; Gojkovic-Bukarica, L. The Influence of Climate Change on Human Cardiovascular Function. Arch. Environ. Occup. Health 2020, 75, 406–414. [Google Scholar] [CrossRef]
- Shor, E.; Roelfs, D. Climate Shock: Moving to Colder Climates and Immigrant Mortality. Soc. Sci. Med. 2019, 235, 112397. [Google Scholar] [CrossRef]
- Ponjoan, A.; Blanch, J.; Alves-Cabratosa, L.; Martí-Lluch, R.; Comas-Cufí, M.; Parramon, D.; del Mar Garcia-Gil, M.; Ramos, R.; Petersen, I. Effects of Extreme Temperatures on Cardiovascular Emergency Hospitalizations in a Mediterranean Region: A Self-Controlled Case Series Study. Environ. Health 2017, 16, 32. [Google Scholar] [CrossRef]
- Hurtado-Díaz, M.; Cruz, J.C.; Texcalac-Sangrador, J.L.; Félix-Arellano, E.E.; Gutiérrez-Ávila, I.; Briseño-Pérez, A.A.; Saavedra-Lara, N.; Tobías, A.; Riojas-Rodríguez, H. Short-Term Effects of Ambient Temperature on Non-External and Cardiovascular Mortality among Older Adults of Metropolitan Areas of Mexico. Int. J. Biometeorol. 2019, 63, 1641–1650. [Google Scholar] [CrossRef]
- Cicci, K.R.; Maltby, A.; Clemens, K.K.; Vicedo-Cabrera, A.M.; Gunz, A.C.; Lavigne, É.; Wilk, P. High Temperatures and Cardiovascular-Related Morbidity: A Scoping Review. Int. J. Environ. Res. Public Health 2022, 19, 11243. [Google Scholar] [CrossRef]
- Sasai, F.; Roncal-Jimenez, C.; Rogers, K.; Sato, Y.; Brown, J.M.; Glaser, J.; Garcia, G.; Sanchez-Lozada, L.G.; Rodriguez-Iturbe, B.; Dawson, J.B.; et al. Climate Change and Nephrology. Nephrol. Dial. Transplant. 2023, 38, 41–48. [Google Scholar] [CrossRef] [PubMed]
- Glaser, J.; Lemery, J.; Rajagopalan, B.; Diaz, H.F.; García-Trabanino, R.; Taduri, G.; Madero, M.; Amarasinghe, M.; Abraham, G.; Anutrakulchai, S.; et al. Climate Change and the Emergent Epidemic of CKD from Heat Stress in Rural Communities: The Case for Heat Stress Nephropathy. Clin. J. Am. Soc. Nephrol. 2016, 11, 1472–1483. [Google Scholar] [CrossRef] [PubMed]
- de Lorenzo, A.; Liaño, F. Altas temperaturas y nefrología: A propósito del cambio climático. Nefrología 2017, 37, 492–500. [Google Scholar] [CrossRef] [PubMed]
- Lee, W.-S.; Kim, W.-S.; Lim, Y.-H.; Hong, Y.-C. High Temperatures and Kidney Disease Morbidity: A Systematic Review and Meta-Analysis. J. Prev. Med. Public Health 2019, 52, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Correa-Rotter, R.; Wesseling, C.; Johnson, R.J. CKD of Unknown Origin in Central America: The Case for a Mesoamerican Nephropathy. Am. J. Kidney Dis. 2014, 63, 506–520. [Google Scholar] [CrossRef] [PubMed]
- Torres, C.; Aragón, A.; González, M.; López, I.; Jakobsson, K.; Elinder, C.-G.; Lundberg, I.; Wesseling, C. Decreased Kidney Function of Unknown Cause in Nicaragua: A Community-Based Survey. Am. J. Kidney Dis. 2010, 55, 485–496. [Google Scholar] [CrossRef] [PubMed]
- Orantes, C.M.; Herrera, R.; Almaguer, M.; Brizuela, E.G.; Hernández, C.E.; Bayarre, H.; Amaya, J.C.; Calero, D.J.; Orellana, P.; Colindres, R.M.; et al. Chronic Kidney Disease and Associated Risk Factors in the Bajo Lempa Region of El Salvador: Nefrolempa Study, 2009. MEDICC Rev. 2011, 13, 14–22. [Google Scholar] [CrossRef] [PubMed]
- Peraza, S.; Wesseling, C.; Aragon, A.; Leiva, R.; García-Trabanino, R.A.; Torres, C.; Jakobsson, K.; Elinder, C.G.; Hogstedt, C. Decreased Kidney Function Among Agricultural Workers in El Salvador. Am. J. Kidney Dis. 2012, 59, 531–540. [Google Scholar] [CrossRef] [PubMed]
- Wesseling, C.; Crowe, J.; Hogstedt, C.; Jakobsson, K.; Lucas, R.; Wegman, D. Mesoamerican Nephropathy: Report from the First International Research Workshop on Men; SALTRA/IRET-UNA, Universidad Nacional: Heredia, Costa Rica, 2013. [Google Scholar]
- John, O.; Gummudi, B.; Jha, A.; Gopalakrishnan, N.; Kalra, O.P.; Kaur, P.; Kher, V.; Kumar, V.; Machiraju, R.S.; Osborne, N.; et al. Chronic Kidney Disease of Unknown Etiology in India: What Do We Know and Where We Need to Go. Kidney Int. Rep. 2021, 6, 2743–2751. [Google Scholar] [CrossRef]
- Roncal-Jimenez, C.A.; García-Trabanino, R.; Wesseling, C.; Johnson, R.J. Mesoamerican Nephropathy or Global Warming Nephropathy? Blood Purif. 2016, 41, 135–138. [Google Scholar] [CrossRef]
- Crowe, J.; Rojas-Valverde, D.; Rojas-Garbanzo, M.; Gutiérrez-Vargas, R.; Ugalde-Ramírez, J.A.; Ledezma-Rojas, J.P.; Cabrera-Alpizar, W.; Salazar-Salazar, M.; Mauricio-La Torre, R.; Valera-Amador, L.; et al. Kidney Function in Rice Workers Exposed to Heat and Dehydration in Costa Rica. Int. J. Environ. Res. Public Health 2022, 19, 4962. [Google Scholar] [CrossRef]
- Hansson, E.; Glaser, J.; Jakobsson, K.; Weiss, I.; Wesseling, C.; Lucas, R.A.I.; Wei, J.L.K.; Ekström, U.; Wijkström, J.; Bodin, T.; et al. Pathophysiological Mechanisms by which Heat Stress Potentially Induces Kidney Inflammation and Chronic Kidney Disease in Sugarcane Workers. Nutrients 2020, 12, 1639. [Google Scholar] [CrossRef]
- Chang, J.C.; Yang, H.Y. Epidemiology of chronic kidney disease of undetermined aetiology in Taiwanese farmers: A cross-sectional study from Changhua Community-based Integrated Screening programme. Occup. Environ. Med. 2021, 78, 849–858. [Google Scholar] [CrossRef]
- Chang, C.J.; Yang, H.Y. Chronic Kidney Disease Among Agricultural Workers in Taiwan: A Nationwide Population-Based Study. Kidney Int. Rep. 2023, 8, 2677–2689. [Google Scholar] [CrossRef]
- Priyadarshani, W.V.D.; de Namor, A.F.D.; Silva, S.R.P. Rising of a global silent killer: Critical analysis of chronic kidney disease of uncertain aetiology (CKDu) worldwide and mitigation steps. Environ. Geochem. Health 2023, 45, 2647–2662. [Google Scholar] [CrossRef]
- Kupferman, J.; Ramírez-Rubio, O.; Amador, J.J.; López-Pilarte, D.; Wilker, E.H.; Laws, R.L.; Sennett, C.; Robles, N.V.; Lau, J.L.; Salinas, A.J.; et al. Acute Kidney Injury in Sugarcane Workers at Risk for Mesoamerican Nephropathy. Am. J. Kidney Dis. 2018, 72, 475–482. [Google Scholar] [CrossRef]
- Correa-Rotter, R.; García-Trabanino, R. Mesoamerican Nephropathy. Semin. Nephrol. 2019, 39, 263–271. [Google Scholar] [CrossRef] [PubMed]
- Fischer, R.S.B.; Vangala, C.; Truong, L.; Mandayam, S.; Chavarria, D.; Granera Llanes, O.M.; Fonseca Laguna, M.U.; Guerra Baez, A.; Garcia, F.; García-Trabanino, R.; et al. Early Detection of Acute Tubulointerstitial Nephritis in the Genesis of Mesoamerican Nephropathy. Kidney Int. 2018, 93, 681–690. [Google Scholar] [CrossRef]
- Wang, F.L.; Wang, W.Z.; Zhang, F.F.; Peng, S.Y.; Wang, H.Y.; Chen, R.; Wang, J.W.; Li, P.F.; Wang, Y.; Zhao, M.H.; et al. Heat exposure and hospitalizations for chronic kidney disease in China: A nationwide time series study in 261 major Chinese cities. Mil. Med. Res. 2023, 10, 41. [Google Scholar] [CrossRef]
- Chu, L.; Phung, D.; Crowley, S.; Dubrow, R. Relationships between short-term ambient temperature exposure and kidney disease hospitalizations in the warm season in Vietnam: A case-crossover study. Environ. Res. 2022, 209, 112776. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Varghese, B.M.; Hansen, A.; Borg, M.A.; Zhang, Y.; Driscoll, T.; Morgan, G.; Dear, K.; Gourley, M.; Capon, A.; et al. Hot weather as a risk factor for kidney disease outcomes: A systematic review and meta-analysis of epidemiological evidence. Sci. Total Environ. 2021, 801, 149806. [Google Scholar] [CrossRef]
- He, L.; Xue, B.; Wang, B.; Liu, C.; Gimeno Ruiz de Porras, D.; Delclos, G.L.; Hu, M.; Luo, B.; Zhang, K. Impact of high, low, and non-optimum temperatures on chronic kidney disease in a changing climate, 1990–2019: A global analysis. Environ. Res. 2022, 212 Pt A, 113172. [Google Scholar] [CrossRef]
- Kjellstrom, T.; Briggs, D.; Freyberg, C.; Lemke, B.; Otto, M.; Hyatt, O. Heat, Human Performance, and Occupational Health: A Key Issue for the Assessment of Global Climate Change Impacts. Annu. Rev. Public Health 2016, 37, 97–112. [Google Scholar] [CrossRef] [PubMed]
- Malig, B.J.; Wu, X.M.; Guirguis, K.; Gershunov, A.; Basu, R. Associations between Ambient Temperature and Hepatobiliary and Renal Hospitalizations in California, 1999 to 2009. Environ. Res. 2019, 177, 108566. [Google Scholar] [CrossRef]
- Robine, J.-M.; Cheung, S.L.K.; Le Roy, S.; Van Oyen, H.; Griffiths, C.; Michel, J.-P.; Herrmann, F.R. Death Toll Exceeded 70,000 in Europe during the Summer of 2003. Comptes Rendus Biol. 2008, 331, 171–178. [Google Scholar] [CrossRef] [PubMed]
- Gronlund, C.J.; Zanobetti, A.; Wellenius, G.A.; Schwartz, J.D.; O’Neill, M.S. Vulnerability to Renal, Heat and Respiratory Hospitalizations during Extreme Heat Among U.S. Elderly. Clim. Change 2016, 136, 631–645. [Google Scholar] [CrossRef] [PubMed]
- Hansen, A.L.; Bi, P.; Ryan, P.; Nitschke, M.; Pisaniello, D.; Tucker, G. The Effect of Heat Waves on Hospital Admissions for Renal Disease in a Temperate City of Australia. Int. J. Epidemiol. 2008, 37, 1359–1365. [Google Scholar] [CrossRef] [PubMed]
- Nitschke, M.; Tucker, G.R.; Hansen, A.L.; Williams, S.; Zhang, Y.; Bi, P. Impact of Two Recent Extreme Heat Episodes on Morbidity and Mortality in Adelaide, South Australia: A Case-Series Analysis. Environ. Health 2011, 10, 42. [Google Scholar] [CrossRef] [PubMed]
- Borg, M.; Bi, P.; Nitschke, M.; Williams, S.; McDonald, S. The Impact of Daily Temperature on Renal Disease Incidence: An Ecological Study. Environ. Health 2017, 16, 114. [Google Scholar] [CrossRef] [PubMed]
- Fletcher, B.A.; Lin, S.; Fitzgerald, E.F.; Hwang, S.-A. Association of Summer Temperatures with Hospital Admissions for Renal Diseases in New York State: A Case-Crossover Study. Am. J. Epidemiol. 2012, 175, 907–916. [Google Scholar] [CrossRef]
- Chen, T.; Sarnat, S.E.; Grundstein, A.J.; Winquist, A.; Chang, H.H. Time-Series Analysis of Heat Waves and Emergency Department Visits in Atlanta, 1993 to 2012. Environ. Health Perspect. 2017, 125, 057009. [Google Scholar] [CrossRef]
- Brikowski, T.H.; Lotan, Y.; Pearle, M.S. Climate-Related Increase in the Prevalence of Urolithiasis in the United States. Proc. Natl. Acad. Sci. USA 2008, 105, 9841–9846. [Google Scholar] [CrossRef]
- Elser, H.; Rowland, S.T.; Tartof, S.Y.; Parks, R.M.; Bruxvoort, K.; Morello-Frosch, R.; Robinson, S.C.; Pressman, A.R.; Wei, R.X.; Casey, J.A. Ambient Temperature and Risk of Urinary Tract Infection in California: A Time-Stratified Case-Crossover Study Using Electronic Health Records. Environ. Int. 2022, 165, 107303. [Google Scholar] [CrossRef]
- Brennan, M.; O’Keeffe, S.T.; Mulkerrin, E.C. Dehydration and Renal Failure in Older Persons during Heatwaves-Predictable, Hard to Identify but Preventable? Age Ageing 2019, 48, 615–618. [Google Scholar] [CrossRef] [PubMed]
- Kovats, R.; Hajat, S.; Wilkinson, P. Contrasting Patterns of Mortality and Hospital Admissions during Hot Weather and Heat Waves in Greater London, UK. Occup. Environ. Med. 2004, 61, 893–898. [Google Scholar] [CrossRef] [PubMed]
- Hifumi, T.; Kondo, Y.; Shimizu, K.; Miyake, Y. Heat Stroke. J. Intensive Care 2018, 6, 30. [Google Scholar] [CrossRef] [PubMed]
- Ingimarsson, J.P.; Krambeck, A.E.; Pais, V.M. Diagnosis and Management of Nephrolithiasis. Surg. Clin. N. Am. 2016, 96, 517–532. [Google Scholar] [CrossRef] [PubMed]
- Tasian, G.E.; Pulido, J.E.; Gasparrini, A.; Saigal, C.S.; Horton, B.P.; Landis, J.R.; Madison, R.; Keren, R.; Urologic Diseases in America Project. Daily Mean Temperature and Clinical Kidney Stone Presentation in Five U.S. Metropolitan Areas: A Time-Series Analysis. Environ. Health Perspect. 2014, 122, 1081–1087. [Google Scholar] [CrossRef] [PubMed]
- Johnson, R.J.; Rodriguez-Iturbe, B.; Roncal-Jimenez, C.; Lanaspa, M.A.; Ishimoto, T.; Nakagawa, T.; Correa-Rotter, R.; Wesseling, C.; Bankir, L.; Sanchez-Lozada, L.G. Hyperosmolarity Drives Hypertension and CKD—Water and Salt Revisited. Nat. Rev. Nephrol. 2014, 10, 415–420. [Google Scholar] [CrossRef] [PubMed]
- Roncal-Jimenez, C.A.; Milagres, T.; Andres-Hernando, A.; Kuwabara, M.; Jensen, T.; Song, Z.; Bjornstad, P.; Garcia, G.E.; Sato, Y.; Sanchez-Lozada, L.G.; et al. Effects of Exogenous Desmopressin on a Model of Heat Stress Nephropathy in Mice. Am. J. Physiol. Renal Physiol. 2017, 312, F418–F426. [Google Scholar] [CrossRef]
- Cirillo, P.; Gersch, M.S.; Mu, W.; Scherer, P.M.; Kim, K.M.; Gesualdo, L.; Henderson, G.N.; Johnson, R.J.; Sautin, Y.Y. Ketohexokinase-Dependent Metabolism of Fructose Induces Proinflammatory Mediators in Proximal Tubular Cells. J. Am. Soc. Nephrol. 2009, 20, 545–553. [Google Scholar] [CrossRef]
- Gutierrez, J.A.; Liu, W.; Perez, S.; Xing, G.; Sonnenberg, G.; Kou, K.; Blatnik, M.; Allen, R.; Weng, Y.; Vera, N.B.; et al. Pharmacologic Inhibition of Ketohexokinase Prevents Fructose-Induced Metabolic Dysfunction. Mol. Metab. 2021, 48, 101196. [Google Scholar] [CrossRef]
- Burg, M.B. Molecular Basis of Osmotic Regulation. Am. J. Physiol. Renal Physiol. 1995, 268, F983–F996. [Google Scholar] [CrossRef]
- Heung, M.; Chawla, L.S. Acute Kidney Injury: Gateway to Chronic Kidney Disease. Nephron Clin. Pract. 2014, 127, 30–34. [Google Scholar] [CrossRef] [PubMed]
- Jefferies, J.L.; Devarajan, P. Early Detection of Acute Kidney Injury after Pediatric Cardiac Surgery. Prog. Pediatr. Cardiol. 2016, 41, 9–16. [Google Scholar] [CrossRef] [PubMed]
- Wei, Q.; Xiao, X.; Fogle, P.; Dong, Z. Changes in Metabolic Profiles during Acute Kidney Injury and Recovery Following Ischemia/Reperfusion. PLoS ONE 2014, 9, e106647. [Google Scholar] [CrossRef] [PubMed]
- Jimenez, C.A.R.; Ishimoto, T.; Lanaspa, M.A.; Rivard, C.J.; Nakagawa, T.; Ejaz, A.A.; Cicerchi, C.; Inaba, S.; Le, M.; Miyazaki, M.; et al. Fructokinase Activity Mediates Dehydration-Induced Renal Injury. Kidney Int. 2014, 86, 294–302. [Google Scholar] [CrossRef]
- Song, Z.; Roncal-Jimenez, C.A.; Lanaspa-Garcia, M.A.; Oppelt, S.A.; Kuwabara, M.; Jensen, T.; Milagres, T.; Andres-Hernando, A.; Ishimoto, T.; Garcia, G.E.; et al. Role of Fructose and Fructokinase in Acute Dehydration-Induced Vasopressin Gene Expression and Secretion in Mice. J. Neurophysiol. 2017, 117, 646–654. [Google Scholar] [CrossRef] [PubMed]
- Khosla, U.M.; Zharikov, S.; Finch, J.L.; Nakagawa, T.; Roncal, C.; Mu, W.; Krotova, K.; Block, E.R.; Prabhakar, S.; Johnson, R.J. Hyperuricemia Induces Endothelial Dysfunction. Kidney Int. 2005, 67, 1739–1742. [Google Scholar] [CrossRef]
- Alelign, T.; Petros, B. Kidney Stone Disease: An Update on Current Concepts. Adv. Urol. 2018, 2018, e3068365. [Google Scholar] [CrossRef]
- Johnson, R.J.; Sánchez-Lozada, L.G.; Newman, L.S.; Lanaspa, M.A.; Diaz, H.F.; Lemery, J.; Rodriguez-Iturbe, B.; Tolan, D.R.; Butler-Dawson, J.; Sato, Y.; et al. Climate Change and the Kidney. Ann. Nutr. Metab. 2019, 74, 38–44. [Google Scholar] [CrossRef]
- Nakayama, T.; Kosugi, T.; Gersch, M.; Connor, T.; Sanchez-Lozada, L.G.; Lanaspa, M.A.; Roncal, C.; Perez-Pozo, S.E.; Johnson, R.J.; Nakagawa, T. Dietary Fructose Causes Tubulointerstitial Injury in the Normal Rat Kidney. Am. J. Physiol. Renal Physiol. 2010, 298, F712–F720. [Google Scholar] [CrossRef]
- Makris, K.; Spanou, L. Acute Kidney Injury: Definition, Pathophysiology and Clinical Phenotypes. Clin. Biochem. Rev. 2016, 37, 85–98. [Google Scholar]
- Akcay, A.; Nguyen, Q.; Edelstein, C.L. Mediators of Inflammation in Acute Kidney Injury. Mediat. Inflamm. 2009, 2009, 137072. [Google Scholar] [CrossRef] [PubMed]
- Rabb, H.; Griffin, M.D.; McKay, D.B.; Swaminathan, S.; Pickkers, P.; Rosner, M.H.; Kellum, J.A.; Ronco, C.; Acute Dialysis Quality Initiative Consensus XIII Work Group. Inflammation in AKI: Current Understanding, Key Questions, and Knowledge Gaps. J. Am. Soc. Nephrol. 2016, 27, 371–379. [Google Scholar] [CrossRef] [PubMed]
- Pavlakou, P.; Liakopoulos, V.; Eleftheriadis, T.; Mitsis, M.; Dounousi, E. Oxidative Stress and Acute Kidney Injury in Critical Illness: Pathophysiologic Mechanisms—Biomarkers—Interventions, and Future Perspectives. Oxid. Med. Cell. Longev. 2017, 2017, e6193694. [Google Scholar] [CrossRef]
- Lanaspa, M.A.; Ishimoto, T.; Cicerchi, C.; Tamura, Y.; Roncal-Jimenez, C.A.; Chen, W.; Tanabe, K.; Andres-Hernando, A.; Orlicky, D.J.; Finol, E.; et al. Endogenous Fructose Production and Fructokinase Activation Mediate Renal Injury in Diabetic Nephropathy. J. Am. Soc. Nephrol. 2014, 25, 2526–2538. [Google Scholar] [CrossRef] [PubMed]
- Luo, Y.; Zheng, S.G. Hall of Fame among Pro-Inflammatory Cytokines: Interleukin-6 Gene and Its Transcriptional Regulation Mechanisms. Front. Immunol. 2016, 7, 604. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; Li, L.; Lewington, S.; Guo, Y.; Sherliker, P.; Bian, Z.; Collins, R.; Peto, R.; Liu, Y.; Yang, R.; et al. Outdoor Temperature, Blood Pressure, and Cardiovascular Disease Mortality among 23 000 Individuals with Diagnosed Cardiovascular Diseases from China. Eur. Heart J. 2015, 36, 1178–1185. [Google Scholar] [CrossRef]
- Hanazawa, T.; Asayama, K.; Watabe, D.; Hosaka, M.; Satoh, M.; Yasui, D.; Obara, T.; Inoue, R.; Metoki, H.; Kikuya, M.; et al. Seasonal Variation in Self-Measured Home Blood Pressure among Patients on Antihypertensive Medications: HOMED-BP Study. Hypertens. Res. 2017, 40, 284–290. [Google Scholar] [CrossRef]
- Wyse, C.A.; Celis Morales, C.A.; Ward, J.; Lyall, D.; Smith, D.J.; Mackay, D.; Curtis, A.M.; Bailey, M.E.S.; Biello, S.; Gill, J.M.R.; et al. Population-Level Seasonality in Cardiovascular Mortality, Blood Pressure, BMI and Inflammatory Cells in UK Biobank. Ann. Med. 2018, 50, 410–419. [Google Scholar] [CrossRef]
- Huang, C.-C.; Chen, Y.-H.; Hung, C.-S.; Lee, J.-K.; Hsu, T.-P.; Wu, H.-W.; Chuang, P.-Y.; Chen, M.-F.; Ho, Y.-L. Assessment of the Relationship Between Ambient Temperature and Home Blood Pressure in Patients From a Web-Based Synchronous Telehealth Care Program: Retrospective Study. J. Med. Internet Res. 2019, 21, e12369. [Google Scholar] [CrossRef]
- Hiramatsu, K.; Yamada, T.; Katakura, M. Acute Effects of Cold on Blood Pressure, Renin-Angiotensinaldosterone System, Catecholamines and Adrenal Steroids in Man. Clin. Exp. Pharmacol. Physiol. 1984, 11, 171–179. [Google Scholar] [CrossRef]
- Marino, F.; Sockler, J.M.; Fry, J.M. Thermoregulatory, Metabolic and Sympathoadrenal Responses to Repeated Brief Exposure to Cold. Scand. J. Clin. Lab. Investig. 1998, 58, 537–546. [Google Scholar] [CrossRef]
- Cummings, M.F.; Steele, P.M.; Mahar, L.J.; Frewin, D.B.; Russell, W.J. The Role of Adrenal Medullary Catecholamine Release in the Response to a Cold Pressor Test. Cardiovasc. Res. 1983, 17, 189–191. [Google Scholar] [CrossRef]
- Lenders, J.W.; Peters, J.H.; Pieters, G.F.; Willemsen, J.J.; Thien, T. Hemodynamic Reactivity to Sympathoadrenal Stimulation in Adrenalectomized Women. J. Clin. Endocrinol. Metab. 1988, 67, 139–143. [Google Scholar] [CrossRef] [PubMed]
- Gasic, S.; Ratheiser, K.; Wagner, O.; Nowotny, P.; Vierhapper, H.; Waldhäusl, W. Alterations in Angiotensin II Release and Vascular Reactivity in Hypertensive Men: A Pilot Study. Am. J. Hypertens. 1999, 12, 1055–1062. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Zhang, S.; Wang, C.; Wang, B.; Guo, P. Effects of Moderate Strength Cold Air Exposure on Blood Pressure and Biochemical Indicators among Cardiovascular and Cerebrovascular Patients. Int. J. Environ. Res. Public Health 2014, 11, 2472–2487. [Google Scholar] [CrossRef] [PubMed]
- Šrámek, P.; Šimečková, M.; Janský, L.; Šavlíková, J.; Vybíral, S. Human Physiological Responses to Immersion into Water of Different Temperatures. Eur. J. Appl. Physiol. 2000, 81, 436–442. [Google Scholar] [CrossRef] [PubMed]
- Polonia, J.J.; Monteiro, A.; Esteves, A.; Cunha, M.E.; Santos, M.L.; Coutinho, J.; Coelho, J.L.; Brandao, F.A.; Cerqueira-Gomes, M. Influence of Sublingual Captopril on Plasma Catecholamine Levels during Hypertensive Emergencies and Cold Immersion. Am. J. Med. 1988, 84, 148–151. [Google Scholar] [CrossRef] [PubMed]
- Bailey, R.E.; Bartos, D.; Barton, F.; Castro, A.; Dobson, R.L.; Grettie, D.P.; Kramer, R.; Macfarlane, D.; Sato, K. Activation of aldosterone and renin secretion by thermal stress. Experientia 1972, 28, 159–160. [Google Scholar] [CrossRef] [PubMed]
- Kosunen, K.J.; Pakarinen, A.J.; Kuoppasalmi, K.; Adlercreutz, H. Plasma renin activity, angiotensin II, and aldosterone during intense heat stress. J. Appl. Physiol. 1976, 41, 323–327. [Google Scholar] [CrossRef]
- Berlyne, G.M.; Finberg, J.P.; Yoran, C. The effect of β-adrenoceptor blockade on body temperature and plasma renin activity in heat-exposed man. Br. J. Clin. Pharmacol. 1974, 1, 307–312. [Google Scholar] [CrossRef]
- Nielsen, B.; Hales, J.R.; Strange, S.; Christensen, N.J.; Warberg, J.; Saltin, B. Human circulatory and thermoregulatory adaptations with heat acclimation and exercis.e in a hot, dry environment. J. Physiol. 1993, 460, 467–485. [Google Scholar] [CrossRef]
- Finberg, J.P.; Katz, M.; Gazit, H.; Berlyne, G.M. Plasma renin activity after acute heat exposure in nonacclimatized and naturally acclimatized man. J. Appl. Physiol. 1974, 36, 519–523. [Google Scholar] [CrossRef]
- Nielsen, B.; Strange, S.; Christensen, N.J.; Warberg, J.; Saltin, B. Acute and adaptive responses in humans to exercise in a warm, humid environment. Pflug. Arch. Eur. J. Physiol. 1997, 434, 49–56. [Google Scholar] [CrossRef]
- Finberg, J.P.; Berlyne, G.M. Modification of renin and aldosterone response to heat by acclimatization in man. J. Appl. Physiol. Respir. Environ. Exerc. Physiol. 1977, 42, 554–558. [Google Scholar] [CrossRef]
- Armstrong, L.E.; Francesconi, R.P.; Kraemer, W.J.; Leva, N.; De Luca, J.P.; Hubbard, R.W. Plasma cortisol, renin, and aldosterone during an intense heat acclimation program. Int. J. Sports Med. 1989, 10, 38–42. [Google Scholar] [CrossRef] [PubMed]
- Francesconi, R.P.; Sawka, M.N.; Pandolf, K.B. Hypohydration and heat acclimation: Plasma renin and aldosterone during exercise. J. Appl. Physiol. Respir. Environ. Exerc. Physiol. 1983, 55, 1790–1794. [Google Scholar] [CrossRef] [PubMed]
- Guerrero-Beltrán, C.E.; Mijares-Rojas, I.A.; Salgado-Garza, G.; Garay-Gutiérrez, N.F.; Carrión-Chavarría, B. Peptidic Vaccines: The New Cure for Heart Diseases? Pharmacol. Res. 2021, 164, 105372. [Google Scholar] [CrossRef] [PubMed]
- Garay-Gutiérrez, N.F.; Hernandez-Fuentes, C.P.; García-Rivas, G.; Lavandero, S.; Guerrero-Beltrán, C.E. Vaccines against Components of the Renin–Angiotensin System. Heart Fail. Rev. 2021, 26, 711–726. [Google Scholar] [CrossRef] [PubMed]
- Chapman, C.L.; Johnson, B.D.; Parker, M.D.; Hostler, D.; Pryor, R.R.; Schlader, Z. Kidney physiology and pathophysiology during heat stress and the modification by exercise, dehydration, heat acclimation and aging. Temperature 2020, 8, 108–159. [Google Scholar] [CrossRef] [PubMed]
- Song, R.; Li, Q.; Hu, J.; Yi, H.; Mao, Z.; Zhou, F. A mouse model of exertional heatstroke-related acute kidney injury. Ann. Transl. Med. 2022, 10, 276. [Google Scholar] [CrossRef]
- Dubey, A.; Prajapati, K.S.; Swamy, M.; Pachauri, V. Heat Shock Proteins: A Therapeutic Target Worth to Consider. Vet. World. 2015, 8, 46–51. [Google Scholar] [CrossRef]
- Krishnan-Sivadoss, I.; Mijares-Rojas, I.A.; Villarreal-Leal, R.A.; Torre-Amione, G.; Knowlton, A.A.; Guerrero-Beltrán, C.E. Heat Shock Protein 60 and Cardiovascular Diseases: An Intricate Love-Hate Story. Med. Res. Rev. 2021, 41, 29–71. [Google Scholar] [CrossRef] [PubMed]
- Vila-Casahonda, R.G.; Lozano-Aponte, J.; Guerrero-Beltrán, C.E. HSP60-Derived Peptide as an LPS/TLR4 Modulator: An in Silico Approach. Front. Cardiovasc. Med. 2022, 9, 731376. [Google Scholar] [CrossRef] [PubMed]
- Chen, B.; Feder, M.E.; Kang, L. Evolution of Heat-Shock Protein Expression Underlying Adaptive Responses to Environmental Stress. Mol. Ecol. 2018, 27, 3040–3054. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.-H.; Kang, L. Differences in Egg Thermotolerance between Tropical and Temperate Populations of the Migratory Locust Locusta migratoria (Orthoptera: Acridiidae). J. Insect. Physiol. 2005, 51, 1277–1285. [Google Scholar] [CrossRef] [PubMed]
- Dunbar, H.E.; Wilson, A.C.C.; Ferguson, N.R.; Moran, N.A. Aphid Thermal Tolerance Is Governed by a Point Mutation in Bacterial Symbionts. PLoS Biol. 2007, 5, e96. [Google Scholar] [CrossRef]
- Velichko, A.K.; Markova, E.N.; Petrova, N.V.; Razin, S.V.; Kantidze, O.L. Mechanisms of Heat Shock Response in Mammals. Cell. Mol. Life Sci. 2013, 70, 4229–4241. [Google Scholar] [CrossRef]
- Williams, C.M.; McCue, M.D.; Sunny, N.E.; Szejner-Sigal, A.; Morgan, T.J.; Allison, D.B.; Hahn, D.A. Cold Adaptation Increases Rates of Nutrient Flow and Metabolic Plasticity during Cold Exposure in Drosophila Melanogaster. Proc. R. Soc. B Biol. Sci. 2016, 283, 20161317. [Google Scholar] [CrossRef]
- Nayak Rao, S. The Role of Heat Shock Proteins in Kidney Disease. J. Transl. Intern. Med. 2016, 4, 114–117. [Google Scholar] [CrossRef]
- Matsumoto, T.; Urushido, M.; Ide, H.; Ishihara, M.; Hamada-Ode, K.; Shimamura, Y.; Ogata, K.; Inoue, K.; Taniguchi, Y.; Taguchi, T.; et al. Small Heat Shock Protein Beta-1 (HSPB1) Is Upregulated and Regulates Autophagy and Apoptosis of Renal Tubular Cells in Acute Kidney Injury. PLoS ONE 2015, 10, e0126229. [Google Scholar] [CrossRef]
- Sanchez-Niño, M.D.; Sanz, A.B.; Sanchez-Lopez, E.; Ruiz-Ortega, M.; Benito-Martin, A.; Saleem, M.A.; Mathieson, P.W.; Mezzano, S.; Egido, J.; Ortiz, A. HSP27/HSPB1 as an Adaptive Podocyte Antiapoptotic Protein Activated by High Glucose and Angiotensin II. Lab. Investig. 2012, 92, 32–45. [Google Scholar] [CrossRef]
- Tsagalis, G.C.; Nikolopoulou, N.; Sotsiou, F.; Hadjiconstantinou, V. The Expression of Heat Shock Proteins 27 and 70 in Lupus Nephritis. Hosp. Chron. 2006, 1, 125–129. [Google Scholar] [CrossRef]
- Kim, M.; Park, S.W.; Kim, M.; Chen, S.W.C.; Gerthoffer, W.T.; D’Agati, V.D.; Lee, H.T. Selective Renal Overexpression of Human Heat Shock Protein 27 Reduces Renal Ischemia-Reperfusion Injury in Mice. Am. J. Physiol. Renal Physiol. 2010, 299, F347–F358. [Google Scholar] [CrossRef]
- O’Neill, S.; Ingman, T.G.; Wigmore, S.J.; Harrison, E.M.; Bellamy, C.O. Differential Expression of Heat Shock Proteins in Healthy and Diseased Human Renal Allografts. Ann. Transplant. 2013, 18, 550–557. [Google Scholar] [CrossRef]
- Smoyer, W.E.; Ransom, R.F. Hsp27 Regulates Podocyte Cytoskeletal Changes in an In Vitro Model of Podocyte Process Retraction. FASEB J. 2002, 16, 315–326. [Google Scholar] [CrossRef] [PubMed]
- Fitri, L.E.; Rosmarwati, E.; Rizky, Y.; Budiarti, N.; Samsu, N.; Mintaroem, K. Strong Renal Expression of Heat Shock Protein 70, High Mobility Group Box 1, Inducible Nitric Oxide Synthase, and Nitrotyrosine in Mice Model of Severe Malaria. Rev. Soc. Bras. Med. Trop. 2017, 50, 489–498. [Google Scholar] [CrossRef] [PubMed]
- Ferns, G.; Shams, S.; Shafi, S. Heat Shock Protein 27: Its Potential Role in Vascular Disease. Int. J. Exp. Pathol. 2006, 87, 253–274. [Google Scholar] [CrossRef] [PubMed]
- Hernádez-Pando, R.; Pedraza-Chaverri, J.; Orozco-Estévez, H.; Silva-Serna, P.; Moreno, I.; Rondán-Zárate, A.; Elinos, M.; Correa-Rotter, R.; Larriva-Sahd, J. Histological and Subcellular Distribution of 65 and 70 kD Heat Shock Proteins in Experimental Nephrotoxic Injury. Exp. Toxicol. Pathol. 1995, 47, 501–508. [Google Scholar] [CrossRef] [PubMed]
- Mayer, M.P.; Bukau, B. Hsp70 Chaperones: Cellular Functions and Molecular Mechanism. Cell. Mol. Life Sci. 2005, 62, 670–684. [Google Scholar] [CrossRef] [PubMed]
- Hassan, F.-U.; Nawaz, A.; Rehman, M.S.; Ali, M.A.; Dilshad, S.M.R.; Yang, C. Prospects of HSP70 as a Genetic Marker for Thermo-Tolerance and Immuno-Modulation in Animals under Climate Change Scenario. Anim. Nutr. 2019, 5, 340–350. [Google Scholar] [CrossRef]
- Truman, A.W.; Kristjansdottir, K.; Wolfgeher, D.; Hasin, N.; Polier, S.; Zhang, H.; Perrett, S.; Prodromou, C.; Jones, G.W.; Kron, S.J. CDK-Dependent Hsp70 Phosphorylation Controls G1 Cyclin Abundance and Cell-Cycle Progression. Cell 2012, 151, 1308–1318. [Google Scholar] [CrossRef]
- Meyer, K.D.; Patil, D.P.; Zhou, J.; Zinoviev, A.; Skabkin, M.A.; Elemento, O.; Pestova, T.V.; Qian, S.-B.; Jaffrey, S.R. 5’ UTR m(6)A Promotes Cap-Independent Translation. Cell 2015, 163, 999–1010. [Google Scholar] [CrossRef]
- Ruete, M.C.; Carrizo, L.C.; Vallés, P.G. Na+/K+-ATPase Stabilization by Hsp70 in the Outer Stripe of the Outer Medulla in Rats during Recovery from a Low-Protein Diet. Cell Stress Chaperon. 2008, 13, 157–167. [Google Scholar] [CrossRef]
- Lebherz-Eichinger, D.; Ankersmit, H.J.; Hacker, S.; Hetz, H.; Kimberger, O.; Schmidt, E.M.; Reiter, T.; Hörl, W.H.; Haas, M.; Krenn, C.G.; et al. HSP27 and HSP70 Serum and Urine Levels in Patients Suffering from Chronic Kidney Disease. Clin. Chim. Acta 2012, 413, 282–286. [Google Scholar] [CrossRef]
- Yang, B.; Hosgood, S.A.; Da, Z.; Harper, S.J.F.; Waller, H.L.; Kay, M.D.; Furness, P.N.; Nicholson, M.L. Biomarkers Assessing Warm Ischemic Injury Using an Isolated Porcine Kidney Hemoreperfusion Model. Exp. Biol. Med. 2012, 237, 1462–1473. [Google Scholar] [CrossRef]
- Wang, Z.; Gall, J.M.; Bonegio, R.G.B.; Havasi, A.; Hunt, C.R.; Sherman, M.Y.; Schwartz, J.H.; Borkan, S.C. Induction of Heat Shock Protein 70 Inhibits Ischemic Renal Injury. Kidney Int. 2011, 79, 861–870. [Google Scholar] [CrossRef]
- Mao, H.; Li, Z.; Zhou, Y.; Li, Z.; Zhuang, S.; An, X.; Zhang, B.; Chen, W.; Nie, J.; Wang, Z.; et al. HSP72 Attenuates Renal Tubular Cell Apoptosis and Interstitial Fibrosis in Obstructive Nephropathy. Am. J. Physiol. Renal Physiol. 2008, 295, F202–F214. [Google Scholar] [CrossRef]
- Zhipeng, W.; Li, L.; Qibing, M.; Linna, L.; Yuhua, R.; Rong, Z. Increased Expression of Heat Shock Protein (HSP)72 in a Human Proximal Tubular Cell Line (HK-2) with Gentamicin-Induced Injury. J. Toxicol. Sci. 2006, 31, 61–70. [Google Scholar] [CrossRef]
- Noh, H.; Kim, H.J.; Yu, M.R.; Kim, W.-Y.; Kim, J.; Ryu, J.H.; Kwon, S.H.; Jeon, J.S.; Han, D.C.; Ziyadeh, F. Heat Shock Protein 90 Inhibitor Attenuates Renal Fibrosis through Degradation of Transforming Growth Factor-β Type II Receptor. Lab. Investig. 2012, 92, 1583–1596. [Google Scholar] [CrossRef]
- Shang, Y.; Xu, X.; Duan, X.; Guo, J.; Wang, Y.; Ren, F.; He, D.; Chang, Z. Hsp70 and Hsp90 Oppositely Regulate TGF-β Signaling through CHIP/Stub1. Biochem. Biophys. Res. Commun. 2014, 446, 387–392. [Google Scholar] [CrossRef]
- Krivoruchko, A.; Storey, K.B. Regulation of the Heat Shock Response under Anoxia in the Turtle, Trachemys Scripta Elegans. J. Comp. Physiol. B 2010, 180, 403–414. [Google Scholar] [CrossRef]
- Benoit, J.B.; Lopez-Martinez, G.; Phillips, Z.P.; Patrick, K.R.; Denlinger, D.L. Heat Shock Proteins Contribute to Mosquito Dehydration Tolerance. J. Insect. Physiol. 2010, 56, 151–156. [Google Scholar] [CrossRef]
- Chen, B.; Yang, B.; Zhu, J.; Wu, J.; Sha, J.; Sun, J.; Bao, E.; Zhang, X. Hsp90 Relieves Heat Stress-Induced Damage in Mouse Kidneys: Involvement of Antiapoptotic PKM2-AKT and Autophagic HIF-1α Signaling. Int. J. Mol. Sci. 2020, 21, 1646. [Google Scholar] [CrossRef]
- Manissorn, J.; Singhto, N.; Thongboonkerd, V. Characterizations of HSP90-Interacting Complex in Renal Cells Using Tandem Affinity Purification and Its Potential Role in Kidney Stone Formation. Proteomics 2018, 18, e1800004. [Google Scholar] [CrossRef]
- Eiam-Ong, S.; Sinphitukkul, K.; Manotham, K.; Eiam-Ong, S. Rapid Nongenomic Action of Aldosterone on Protein Expressions of Hsp90( α and β ) and Pc-Src in Rat Kidney. Biomed. Res. Int. 2013, 2013, 346480. [Google Scholar] [CrossRef]
- O’Neill, S.; Humphries, D.; Tse, G.; Marson, L.P.; Dhaliwal, K.; Hughes, J.; Ross, J.A.; Wigmore, S.J.; Harrison, E.M. Heat Shock Protein 90 Inhibition Abrogates TLR4-Mediated NF-κB Activity and Reduces Renal Ischemia-Reperfusion Injury. Sci. Rep. 2015, 5, 12958. [Google Scholar] [CrossRef]
- Schreiner, A.D.; Brzezinski, W.A. Acute Kidney Injury, an Id Reaction and HSP90. Am. J. Med. Sci. 2015, 350, 157–158. [Google Scholar] [CrossRef]
- Ray, S.C.; Mason, J.; O’Connor, P.M. Ischemic Renal Injury: Can Renal Anatomy and Associated Vascular Congestion Explain Why the Medulla and Not the Cortex Is Where the Trouble Starts? Semin. Nephrol. 2019, 39, 520–529. [Google Scholar] [CrossRef]
- Pfaller, W. Structure Function Correlation on Rat Kidney. Quantitative Correlation of Structure and Function in the Normal and Injured Rat Kidney. Adv. Anat. Embryol. Cell Biol. 1982, 70, 1–106. [Google Scholar] [PubMed]
- Scholz, H.; Boivin, F.J.; Schmidt-Ott, K.M.; Bachmann, S.; Eckardt, K.-U.; Scholl, U.I.; Persson, P.B. Kidney Physiology and Susceptibility to Acute Kidney Injury: Implications for Renoprotection. Nat. Rev. Nephrol. 2021, 17, 335–349. [Google Scholar] [CrossRef] [PubMed]
- Wojtaszek, P.A.; Heasley, L.E.; Berl, T. In Vivo Regulation of MAP Kinases in Ratus Norvegicus Renal Papilla by Water Loading and Restriction. J. Clin. Investig. 1998, 102, 1874–1881. [Google Scholar] [CrossRef]
- Tang, S.; Zhou, S.; Yin, B.; Xu, J.; Di, L.; Zhang, J.; Bao, E. Heat Stress-Induced Renal Damage in Poultry and the Protective Effects of HSP60 and HSP47. Cell Stress Chaperon. 2018, 23, 1033–1040. [Google Scholar] [CrossRef] [PubMed]
- Itoh, H.; Komatsuda, A.; Ohtani, H.; Wakui, H.; Imai, H.; Sawada, K.-I.; Otaka, M.; Ogura, M.; Suzuki, A.; Hamada, F. Mammalian HSP60 Is Quickly Sorted into the Mitochondria under Conditions of Dehydration. Eur. J. Biochem. 2002, 269, 5931–5938. [Google Scholar] [CrossRef] [PubMed]
- Goo, J.S.; Kim, Y.N.; Choi, K.M.; Hwang, I.S.; Kim, J.E.; Lee, Y.J.; Kwak, M.H.; Shim, S.B.; Jee, S.W.; Lim, C.J.; et al. Proteomic Analysis of Kidneys from Selenoprotein M Transgenic Rats in Response to Increased Bioability of Selenium. Clin. Proteom. 2013, 10, 10. [Google Scholar] [CrossRef]
- El-Gamasy, M.A.; El-Sadek, A.E.; Fakhreldin, A.R.; Kamel, A.; Elbehery, E.G. Heat Shock Protein 60 as a Biomarker for Acute Kidney Injury Secondary to Septic Shock in Pediatric Patients, Egyptian Multicenter Experience. Saudi J. Kidney Dis. Transplant. 2018, 29, 852–862. [Google Scholar] [CrossRef]
- Lever, J.M.; Boddu, R.; George, J.F.; Agarwal, A. Heme Oxygenase-1 in Kidney Health and Disease. Antioxid. Redox Signal. 2016, 25, 165–183. [Google Scholar] [CrossRef]
- Bolisetty, S.; Zarjou, A.; Agarwal, A. Heme Oxygenase 1 as a Therapeutic Target in Acute Kidney Injury. Am. J. Kidney Dis. 2017, 69, 531–545. [Google Scholar] [CrossRef]
- Nath, K.A. Heme Oxygenase-1 and Acute Kidney Injury. Curr. Opin. Nephrol. Hypertens. 2014, 23, 17–24. [Google Scholar] [CrossRef]
- Sakai, K.; Nozaki, Y.; Murao, Y.; Yano, T.; Ri, J.; Niki, K.; Kinoshita, K.; Funauchi, M.; Matsumura, I. Protective Effect and Mechanism of IL-10 on Renal Ischemia-Reperfusion Injury. Lab. Investig. 2019, 99, 671–683. [Google Scholar] [CrossRef]
- Konrad, F.M.; Zwergel, C.; Ngamsri, K.-C.; Reutershan, J. Anti-Inflammatory Effects of Heme Oxygenase-1 Depend on Adenosine A2A- and A2B-Receptor Signaling in Acute Pulmonary Inflammation. Front. Immunol. 2017, 8, 1874. [Google Scholar] [CrossRef]
- Bolisetty, S.; Traylor, A.; Zarjou, A.; Johnson, M.S.; Benavides, G.A.; Ricart, K.; Boddu, R.; Moore, R.D.; Landar, A.; Barnes, S.; et al. Mitochondria-Targeted Heme Oxygenase-1 Decreases Oxidative Stress in Renal Epithelial Cells. Am. J. Physiol. Renal Physiol. 2013, 305, F255–F264. [Google Scholar] [CrossRef] [PubMed]
- da Silva, J.L.; Zand, B.A.; Yang, L.M.; Sabaawy, H.E.; Lianos, E.; Abraham, N.G. Heme Oxygenase Isoform-Specific Expression and Distribution in the Rat Kidney. Kidney Int. 2001, 59, 1448–1457. [Google Scholar] [CrossRef] [PubMed]
- Choi, B.-M.; Pae, H.-O.; Jeong, Y.-R.; Kim, Y.-M.; Chung, H.-T. Critical Role of Heme Oxygenase-1 in Foxp3-Mediated Immune Suppression. Biochem. Biophys. Res. Commun. 2005, 327, 1066–1071. [Google Scholar] [CrossRef] [PubMed]
- Morimoto, K.; Ohta, K.; Yachie, A.; Yang, Y.; Shimizu, M.; Goto, C.; Toma, T.; Kasahara, Y.; Yokoyama, H.; Miyata, T.; et al. Cytoprotective Role of Heme Oxygenase (HO)-1 in Human Kidney with Various Renal Diseases. Kidney Int. 2001, 60, 1858–1866. [Google Scholar] [CrossRef] [PubMed]
- Kie, J.-H.; Kapturczak, M.H.; Traylor, A.; Agarwal, A.; Hill-Kapturczak, N. Heme Oxygenase-1 Deficiency Promotes Epithelial-Mesenchymal Transition and Renal Fibrosis. J. Am. Soc. Nephrol. 2008, 19, 1681–1691. [Google Scholar] [CrossRef]
- Chen, X.; Wei, S.-Y.; Li, J.-S.; Zhang, Q.-F.; Wang, Y.-X.; Zhao, S.-L.; Yu, J.; Wang, C.; Qin, Y.; Wei, Q.-J.; et al. Overexpression of Heme Oxygenase-1 Prevents Renal Interstitial Inflammation and Fibrosis Induced by Unilateral Ureter Obstruction. PLoS ONE 2016, 11, e0147084. [Google Scholar] [CrossRef]
- Vidyasagar, A.; Wilson, N.A.; Djamali, A. Heat Shock Protein 27 (HSP27): Biomarker of Disease and Therapeutic Target. Fibrogenesis Tissue Repair 2012, 5, 7. [Google Scholar] [CrossRef]
- Wang, Z.; Jin, H.; Li, C.; Hou, Y.; Mei, Q.; Fan, D. Heat Shock Protein 72 Protects Kidney Proximal Tubule Cells from Injury Induced by Triptolide by Means of Activation of the MEK/ERK Pathway. Int. J Toxicol. 2009, 28, 177–189. [Google Scholar] [CrossRef]
- Kupis, W.; Pałyga, J.; Tomal, E.; Niewiadomska, E. The Role of Sirtuins in Cellular Homeostasis. J. Physiol. Biochem. 2016, 72, 371–380. [Google Scholar] [CrossRef]
- Morigi, M.; Perico, L.; Benigni, A. Sirtuins in Renal Health and Disease. J. Am. Soc. Nephrol. 2018, 29, 1799–1809. [Google Scholar] [CrossRef]
- Vasquez, M.C.; Martinez, D.A.; Tomanek, L. Multiple Stressor Responses Are Regulated by Sirtuins in Mytilus Congeners. Comp. Biochem. Physiol. A Mol. Integr. Physiol. 2020, 246, 110719. [Google Scholar] [CrossRef]
- Zhang, D.; Li, S.; Cruz, P.; Kone, B.C. Sirtuin 1 Functionally and Physically Interacts with Disruptor of Telomeric Silencing-1 to Regulate α-ENaC Transcription in Collecting Duct. J. Biol. Chem. 2009, 284, 20917–20926. [Google Scholar] [CrossRef]
- Lin, Q.; Geng, Y.; Lin, S.; Tian, Z. Sirtuin1 (SIRT1) Regulates Tumor Necrosis Factor-Alpha (TNF-α-Induced) Aquaporin-2 (AQP2) Expression in Renal Medullary Collecting Duct Cells Through Inhibiting the NF-κB Pathway. Med. Sci. Monit. Basic Res. 2016, 22, 165–174. [Google Scholar] [CrossRef]
- Khositseth, S.; Bruce, L.J.; Walsh, S.B.; Bawazir, W.M.; Ogle, G.D.; Unwin, R.J.; Thong, M.-K.; Sinha, R.; Choo, K.E.; Chartapisak, W.; et al. Tropical Distal Renal Tubular Acidosis: Clinical and Epidemiological Studies in 78 Patients. QJM Int. J. Med. 2012, 105, 861–877. [Google Scholar] [CrossRef]
- Nimmannit, S.; Malasit, P.; Susaengrat, W.; Ong-Aj-Yooth, S.; Vasuvattakul, S.; Pidetcha, P.; Shayakul, C.; Nilwarangkur, S. Prevalence of Endemic Distal Renal Tubular Acidosis and Renal Stone in the Northeast of Thailand. Nephron 1996, 72, 604–610. [Google Scholar] [CrossRef]
- Noriega, L.G.; Melo, Z.; Rajaram, R.D.; Mercado, A.; Tovar, A.R.; Velazquez-Villegas, L.A.; Castañeda-Bueno, M.; Reyes-López, Y.; Ryu, D.; Rojas-Vega, L.; et al. SIRT7 Modulates the Stability and Activity of the Renal K-Cl Cotransporter KCC4 through Deacetylation. EMBO Rep. 2021, 22, e50766. [Google Scholar] [CrossRef]
- Feng, X.; Su, H.; He, X.; Chen, J.-X.; Zeng, H. SIRT3 Deficiency Sensitizes Angiotensin-II-Induced Renal Fibrosis. Cells 2020, 9, 2510. [Google Scholar] [CrossRef]
- Guo, J.; Wang, Z.; Wu, J.; Liu, M.; Li, M.; Sun, Y.; Huang, W.; Li, Y.; Zhang, Y.; Tang, W.; et al. Endothelial SIRT6 Is Vital to Prevent Hypertension and Associated Cardiorenal Injury Through Targeting Nkx3.2-GATA5 Signaling. Circ. Res. 2019, 124, 1448–1461. [Google Scholar] [CrossRef] [PubMed]
- Xi, J.; Jing, J.; Zhang, Y.; Liang, C.; Hao, Z.; Zhang, L.; Chen, Y. SIRT3 Inhibited the Formation of Calcium Oxalate-Induced Kidney Stones through Regulating NRF2/HO-1 Signaling Pathway. J. Cell. Biochem. 2019, 120, 8259–8271. [Google Scholar] [CrossRef] [PubMed]
- Xi, J.; Chen, Y.; Jing, J.; Zhang, Y.; Liang, C.; Hao, Z.; Zhang, L. Sirtuin 3 Suppresses the Formation of Renal Calcium Oxalate Crystals through Promoting M2 Polarization of Macrophages. J. Cell. Physiol. 2019, 234, 11463–11473. [Google Scholar] [CrossRef] [PubMed]
- Bai, C.Q.; Ouyang, J.; Su, C.H.; Cui, Q.Q.; Liu, D.; Gao, Z.H.; Chen, S.Y.; Zhao, Y.Y. Association of hyperuricemia-induced renal damage with sirtuin 1 and endothelial nitric oxide synthase in rats. Zhonghua Yi Xue Za Zhi 2021, 101, 429–434. [Google Scholar] [CrossRef] [PubMed]
- van Marken Lichtenbelt, W.D.; Vanhommerig, J.W.; Smulders, N.M.; Drossaerts, J.M.A.F.L.; Kemerink, G.J.; Bouvy, N.D.; Schrauwen, P.; Teule, G.J.J. Cold-Activated Brown Adipose Tissue in Healthy Men. N. Engl. J. Med. 2009, 360, 1500–1508. [Google Scholar] [CrossRef] [PubMed]
- Hanssen, M.J.W.; Hoeks, J.; Brans, B.; van der Lans, A.A.J.J.; Schaart, G.; van den Driessche, J.J.; Jörgensen, J.A.; Boekschoten, M.V.; Hesselink, M.K.C.; Havekes, B.; et al. Short-Term Cold Acclimation Improves Insulin Sensitivity in Patients with Type 2 Diabetes Mellitus. Nat. Med. 2015, 21, 863–865. [Google Scholar] [CrossRef]
- Hoeke, G.; Kooijman, S.; Boon, M.R.; Rensen, P.C.N.; Berbée, J.F.P. Role of Brown Fat in Lipoprotein Metabolism and Atherosclerosis. Circ. Res. 2016, 118, 173–182. [Google Scholar] [CrossRef]
- Lee, P.; Bova, R.; Schofield, L.; Bryant, W.; Dieckmann, W.; Slattery, A.; Govendir, M.A.; Emmett, L.; Greenfield, J.R. Brown Adipose Tissue Exhibits a Glucose-Responsive Thermogenic Biorhythm in Humans. Cell Metab. 2016, 23, 602–609. [Google Scholar] [CrossRef] [PubMed]
- Blauw, L.L.; Aziz, N.A.; Tannemaat, M.R.; Blauw, C.A.; de Craen, A.J.; Pijl, H.; Rensen, P.C.N. Diabetes Incidence and Glucose Intolerance Prevalence Increase with Higher Outdoor Temperature. BMJ Open Diabetes Res. Care 2017, 5, e000317. [Google Scholar] [CrossRef] [PubMed]
- Symonds, M.E.; Farhat, G.; Aldiss, P.; Pope, M.; Budge, H. Brown Adipose Tissue and Glucose Homeostasis—The Link between Climate Change and the Global Rise in Obesity and Diabetes. Adipocyte 2019, 8, 46–50. [Google Scholar] [CrossRef]
- Lee, P.; Smith, S.; Linderman, J.; Courville, A.B.; Brychta, R.J.; Dieckmann, W.; Werner, C.D.; Chen, K.Y.; Celi, F.S. Temperature-Acclimated Brown Adipose Tissue Modulates Insulin Sensitivity in Humans. Diabetes 2014, 63, 3686–3698. [Google Scholar] [CrossRef]
- Jia, P.; Wu, X.; Pan, T.; Xu, S.; Hu, J.; Ding, X. Uncoupling Protein 1 Inhibits Mitochondrial Reactive Oxygen Species Generation and Alleviates Acute Kidney Injury. EBioMedicine 2019, 49, 331–340. [Google Scholar] [CrossRef]
- Cai, Y.-Y.; Zhang, H.-B.; Fan, C.-X.; Zeng, Y.-M.; Zou, S.-Z.; Wu, C.-Y.; Wang, L.; Fang, S.; Li, P.; Xue, Y.-M.; et al. Renoprotective Effects of Brown Adipose Tissue Activation in Diabetic Mice. J. Diabetes 2019, 11, 958–970. [Google Scholar] [CrossRef]
- Kralisch, S.; Hoffmann, A.; Klöting, N.; Frille, A.; Kuhn, H.; Nowicki, M.; Paeschke, S.; Bachmann, A.; Blüher, M.; Zhang, M.-Z.; et al. The Brown Fat-Secreted Adipokine Neuregulin 4 Is Decreased in Human and Murine Chronic Kidney Disease. Eur. J. Endocrinol. 2019, 181, 151–159. [Google Scholar] [CrossRef] [PubMed]
- Sebaa, R.; Johnson, J.; Pileggi, C.; Norgren, M.; Xuan, J.; Sai, Y.; Tong, Q.; Krystkowiak, I.; Bondy-Chorney, E.; Davey, N.E.; et al. SIRT3 Controls Brown Fat Thermogenesis by Deacetylation Regulation of Pathways Upstream of UCP1. Mol. Metab. 2019, 25, 35–49. [Google Scholar] [CrossRef] [PubMed]
- Yao, L.; Cui, X.; Chen, Q.; Yang, X.; Fang, F.; Zhang, J.; Liu, G.; Jin, W.; Chang, Y. Cold-Inducible SIRT6 Regulates Thermogenesis of Brown and Beige Fat. Cell Rep. 2017, 20, 641–654. [Google Scholar] [CrossRef] [PubMed]
- Wang, F.; Tong, Q. SIRT2 Suppresses Adipocyte Differentiation by Deacetylating FOXO1 and Enhancing FOXO1’s Repressive Interaction with PPARγ. Mol. Biol. Cell. 2009, 20, 801–808. [Google Scholar] [CrossRef] [PubMed]
- Shuai, L.; Zhang, L.-N.; Li, B.-H.; Tang, C.-L.; Wu, L.-Y.; Li, J.; Li, J.-Y. SIRT5 Regulates Brown Adipocyte Differentiation and Browning of Subcutaneous White Adipose Tissue. Diabetes 2019, 68, 1449–1461. [Google Scholar] [CrossRef]
- Molinari, F.; Feraco, A.; Mirabilii, S.; Saladini, S.; Sansone, L.; Vernucci, E.; Tomaselli, G.; Marzolla, V.; Rotili, D.; Russo, M.A.; et al. SIRT5 Inhibition Induces Brown Fat-Like Phenotype in 3T3-L1 Preadipocytes. Cells 2021, 10, 1126. [Google Scholar] [CrossRef]
- Hong, Q.; Zhang, L.; Das, B.; Li, Z.; Liu, B.; Cai, G.; Chen, X.; Chuang, P.Y.; He, J.C.; Lee, K. Increased Podocyte Sirtuin-1 Function Attenuates Diabetic Kidney Injury. Kidney Int. 2018, 93, 1330–1343. [Google Scholar] [CrossRef]
- Zhong, Y.; Lee, K.; He, J.C. SIRT1 Is a Potential Drug Target for Treatment of Diabetic Kidney Disease. Front. Endocrinol. 2018, 9, 624. [Google Scholar] [CrossRef]
- Zhou, D.; Zhou, M.; Wang, Z.; Fu, Y.; Jia, M.; Wang, X.; Liu, M.; Zhang, Y.; Sun, Y.; Lu, Y.; et al. PGRN Acts as a Novel Regulator of Mitochondrial Homeostasis by Facilitating Mitophagy and Mitochondrial Biogenesis to Prevent Podocyte Injury in Diabetic Nephropathy. Cell Death Dis. 2019, 10, 524. [Google Scholar] [CrossRef]
- Sun, H.-J.; Xiong, S.-P.; Cao, X.; Cao, L.; Zhu, M.-Y.; Wu, Z.-Y.; Bian, J.-S. Polysulfide-Mediated Sulfhydration of SIRT1 Prevents Diabetic Nephropathy by Suppressing Phosphorylation and Acetylation of P65 NF-κB and STAT3. Redox Biol. 2021, 38, 101813. [Google Scholar] [CrossRef]
- Wang, X.-L.; Wu, L.-Y.; Zhao, L.; Sun, L.-N.; Liu, H.-Y.; Liu, G.; Guan, G.-J. SIRT1 Activator Ameliorates the Renal Tubular Injury Induced by Hyperglycemia in Vivo and in Vitro via Inhibiting Apoptosis. Biomed. Pharmacother. 2016, 83, 41–50. [Google Scholar] [CrossRef]
- Huang, K.; Gao, X.; Wei, W. The Crosstalk between Sirt1 and Keap1/Nrf2/ARE Anti-Oxidative Pathway Forms a Positive Feedback Loop to Inhibit FN and TGF-Β1 Expressions in Rat Glomerular Mesangial Cells. Exp. Cell Res. 2017, 361, 63–72. [Google Scholar] [CrossRef]
- Kida, Y.; Zullo, J.A.; Goligorsky, M.S. Endothelial Sirtuin 1 Inactivation Enhances Capillary Rarefaction and Fibrosis Following Kidney Injury through Notch Activation. Biochem. Biophys. Res. Commun. 2016, 478, 1074–1079. [Google Scholar] [CrossRef]
- Muraoka, H.; Hasegawa, K.; Sakamaki, Y.; Minakuchi, H.; Kawaguchi, T.; Yasuda, I.; Kanda, T.; Tokuyama, H.; Wakino, S.; Itoh, H. Role of Nampt-Sirt6 Axis in Renal Proximal Tubules in Extracellular Matrix Deposition in Diabetic Nephropathy. Cell Rep. 2019, 27, 199–212. [Google Scholar] [CrossRef] [PubMed]
- Esmaeili, S.; Motamedrad, M.; Hemmati, M.; Mehrpour, O.; Khorashadizadeh, M. Prevention of Kidney Cell Damage in Hyperglycaemia Condition by Adiponectin. Cell Biochem. Funct. 2019, 37, 148–152. [Google Scholar] [CrossRef] [PubMed]
- Wei, S.; Gao, Y.; Dai, X.; Fu, W.; Cai, S.; Fang, H.; Zeng, Z.; Chen, Z. SIRT1-Mediated HMGB1 Deacetylation Suppresses Sepsis-Associated Acute Kidney Injury. Am. J. Physiol. Renal Physiol. 2019, 316, F20–F31. [Google Scholar] [CrossRef] [PubMed]
- Gao, Q.; Zhu, H. The Overexpression of Sirtuin1 (SIRT1) Alleviated Lipopolysaccharide (LPS)-Induced Acute Kidney Injury (AKI) via Inhibiting the Activation of Nucleotide-Binding Oligomerization Domain-Like Receptors (NLR) Family Pyrin Domain Containing 3 (NLRP3) Inflammasome. Med. Sci. Monit. 2019, 25, 2718–2726. [Google Scholar] [CrossRef] [PubMed]
- Ryu, D.R.; Yu, M.R.; Kong, K.H.; Kim, H.; Kwon, S.H.; Jeon, J.S.; Han, D.C.; Noh, H. Sirt1-Hypoxia-Inducible Factor-1α Interaction Is a Key Mediator of Tubulointerstitial Damage in the Aged Kidney. Aging Cell 2019, 18, e12904. [Google Scholar] [CrossRef] [PubMed]
- Li, P.; Liu, Y.; Qin, X.; Chen, K.; Wang, R.; Yuan, L.; Chen, X.; Hao, C.; Huang, X. SIRT1 Attenuates Renal Fibrosis by Repressing HIF-2α. Cell Death Discov. 2021, 7, 59. [Google Scholar] [CrossRef] [PubMed]
- Chou, X.; Ding, F.; Zhang, X.; Ding, X.; Gao, H.; Wu, Q. Sirtuin-1 Ameliorates Cadmium-Induced Endoplasmic Reticulum Stress and Pyroptosis through XBP-1s Deacetylation in Human Renal Tubular Epithelial Cells. Arch. Toxicol. 2019, 93, 965–986. [Google Scholar] [CrossRef] [PubMed]
- Huang, K.; Li, R.; Wei, W. Sirt1 Activation Prevents Anti-Thy 1.1 Mesangial Proliferative Glomerulonephritis in the Rat through the Nrf2/ARE Pathway. Eur. J. Pharmacol. 2018, 832, 138–144. [Google Scholar] [CrossRef] [PubMed]
- Gong, L.; He, J.; Sun, X.; Li, L.; Zhang, X.; Gan, H. Activation of Sirtuin1 Protects against Ischemia/Reperfusion-Induced Acute Kidney Injury. Biomed. Pharmacother. 2020, 125, 110021. [Google Scholar] [CrossRef] [PubMed]
- Fujino, T.; Yokokawa, R.; Oshima, T.; Hayakawa, M. SIRT1 Knockdown Up-Regulates P53 and P21/Cip1 Expression in Renal Adenocarcinoma Cells but Not in Normal Renal-Derived Cells in a Deacetylase-Independent Manner. J. Toxicol. Sci. 2018, 43, 711–715. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Zhu, Y.; Sheng, Y.; Xiao, J.; Xiao, Y.; Cheng, N.; Chai, Y.; Wu, X.; Zhang, S.; Xiang, T. SIRT1 Downregulated FGB Expression to Inhibit RCC Tumorigenesis by Destabilizing STAT3. Exp. Cell Res. 2019, 382, 111466. [Google Scholar] [CrossRef] [PubMed]
- He, F.-F.; You, R.-Y.; Ye, C.; Lei, C.-T.; Tang, H.; Su, H.; Zhang, C. Inhibition of SIRT2 Alleviates Fibroblast Activation and Renal Tubulointerstitial Fibrosis via MDM2. Cell. Physiol. Biochem. 2018, 46, 451–460. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Mu, Y.; Zhou, X.; Ji, H.; Gao, X.; Cai, W.W.; Guan, Q.; Xu, T. SIRT2-Mediated FOXO3a Deacetylation Drives Its Nuclear Translocation Triggering FasL-Induced Cell Apoptosis during Renal Ischemia Reperfusion. Apoptosis 2017, 22, 519–530. [Google Scholar] [CrossRef] [PubMed]
- Jung, Y.J.; Park, W.; Kang, K.P.; Kim, W. SIRT2 Is Involved in Cisplatin-Induced Acute Kidney Injury through Regulation of Mitogen-Activated Protein Kinase Phosphatase-1. Nephrol. Dial. Transplant. 2020, 35, 1145–1156. [Google Scholar] [CrossRef] [PubMed]
- Srivastava, S.P.; Li, J.; Kitada, M.; Fujita, H.; Yamada, Y.; Goodwin, J.E.; Kanasaki, K.; Koya, D. SIRT3 Deficiency Leads to Induction of Abnormal Glycolysis in Diabetic Kidney with Fibrosis. Cell Death Dis. 2018, 9, 997. [Google Scholar] [CrossRef]
- Jiao, X.; Li, Y.; Zhang, T.; Liu, M.; Chi, Y. Role of Sirtuin3 in High Glucose-Induced Apoptosis in Renal Tubular Epithelial Cells. Biochem. Biophys. Res. Commun. 2016, 480, 387–393. [Google Scholar] [CrossRef]
- Ogura, Y.; Kitada, M.; Monno, I.; Kanasaki, K.; Watanabe, A.; Koya, D. Renal Mitochondrial Oxidative Stress Is Enhanced by the Reduction of Sirt3 Activity, in Zucker Diabetic Fatty Rats. Redox Rep. 2018, 23, 153–159. [Google Scholar] [CrossRef]
- Li, M.; Li, C.-M.; Ye, Z.-C.; Huang, J.; Li, Y.; Lai, W.; Peng, H.; Lou, T.-Q. Sirt3 Modulates Fatty Acid Oxidation and Attenuates Cisplatin-Induced AKI in Mice. J. Cell. Mol. Med. 2020, 24, 5109–5121. [Google Scholar] [CrossRef] [PubMed]
- Lin, J.-R.; Zheng, Y.-J.; Zhang, Z.-B.; Shen, W.-L.; Li, X.-D.; Wei, T.; Ruan, C.-C.; Chen, X.-H.; Zhu, D.-L.; Gao, P.-J. Suppression of Endothelial-to-Mesenchymal Transition by SIRT (Sirtuin) 3 Alleviated the Development of Hypertensive Renal Injury. Hypertension 2018, 72, 350–360. [Google Scholar] [CrossRef] [PubMed]
- He, P.; Li, Z.; Yue, Z.; Gao, H.; Feng, G.; Wang, P.; Huang, Y.; Luo, W.; Hong, H.; Liang, L.; et al. SIRT3 Prevents Angiotensin II-Induced Renal Tubular Epithelial-Mesenchymal Transition by Ameliorating Oxidative Stress and Mitochondrial Dysfunction. Mol. Cell. Endocrinol. 2018, 460, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Li, N.; Zhang, J.; Yan, X.; Zhang, C.; Liu, H.; Shan, X.; Li, J.; Yang, Y.; Huang, C.; Zhang, P.; et al. SIRT3-KLF15 Signaling Ameliorates Kidney Injury Induced by Hypertension. Oncotarget 2017, 8, 39592–39604. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Xu, J.; Li, X.; Liu, Z.; Han, Y.; Xu, X.; Li, X.; Tang, Y.; Liu, Y.; Yu, T.; et al. Sirt3 Modulate Renal Ischemia-Reperfusion Injury through Enhancing Mitochondrial Fusion and Activating the ERK-OPA1 Signaling Pathway. J. Cell. Physiol. 2019, 234, 23495–23506. [Google Scholar] [CrossRef]
- Liu, H.; Li, S.; Liu, X.; Chen, Y.; Deng, H. SIRT3 Overexpression Inhibits Growth of Kidney Tumor Cells and Enhances Mitochondrial Biogenesis. J. Proteome Res. 2018, 17, 3143–3152. [Google Scholar] [CrossRef] [PubMed]
- Shi, J.-X.; Wang, Q.-J.; Li, H.; Huang, Q. SIRT4 Overexpression Protects against Diabetic Nephropathy by Inhibiting Podocyte Apoptosis. Exp. Ther. Med. 2017, 13, 342–348. [Google Scholar] [CrossRef]
- Wei, R.; He, D.; Zhang, X. Role of SIRT2 in Regulation of Stemness of Cancer Stem-Like Cells in Renal Cell Carcinoma. Cell. Physiol. Biochem. 2018, 49, 2348–2357. [Google Scholar] [CrossRef]
- Li, W.; Yang, Y.; Li, Y.; Zhao, Y.; Jiang, H. Sirt5 Attenuates Cisplatin-Induced Acute Kidney Injury through Regulation of Nrf2/HO-1 and Bcl-2. Biomed. Res. Int. 2019, 2019, e4745132. [Google Scholar] [CrossRef]
- Yoon, S.P.; Kim, J. Poly(ADP-Ribose) Polymerase 1 Contributes to Oxidative Stress through Downregulation of Sirtuin 3 during Cisplatin Nephrotoxicity. Anat. Cell Biol. 2016, 49, 165–176. [Google Scholar] [CrossRef] [PubMed]
- Chiba, T.; Peasley, K.D.; Cargill, K.R.; Maringer, K.V.; Bharathi, S.S.; Mukherjee, E.; Zhang, Y.; Holtz, A.; Basisty, N.; Yagobian, S.D.; et al. Sirtuin 5 Regulates Proximal Tubule Fatty Acid Oxidation to Protect against AKI. J. Am. Soc. Nephrol. 2019, 30, 2384–2398. [Google Scholar] [CrossRef]
- Ma, Y.; Qi, Y.; Wang, L.; Zheng, Z.; Zhang, Y.; Zheng, J. SIRT5-Mediated SDHA Desuccinylation Promotes Clear Cell Renal Cell Carcinoma Tumorigenesis. Free Radic. Biol. Med. 2019, 134, 458–467. [Google Scholar] [CrossRef] [PubMed]
- Choi, J.; Koh, E.; Lee, Y.S.; Lee, H.-W.; Kang, H.G.; Yoon, Y.E.; Han, W.K.; Choi, K.H.; Kim, K.-S. Mitochondrial Sirt3 Supports Cell Proliferation by Regulating Glutamine-Dependent Oxidation in Renal Cell Carcinoma. Biochem. Biophys. Res. Commun. 2016, 474, 547–553. [Google Scholar] [CrossRef] [PubMed]
- Liu, M.; Liang, K.; Zhen, J.; Zhou, M.; Wang, X.; Wang, Z.; Wei, X.; Zhang, Y.; Sun, Y.; Zhou, Z.; et al. Sirt6 Deficiency Exacerbates Podocyte Injury and Proteinuria through Targeting Notch Signaling. Nat. Commun. 2017, 8, 413. [Google Scholar] [CrossRef]
- Yang, Q.; Hu, J.; Yang, Y.; Chen, Z.; Feng, J.; Zhu, Z.; Wang, H.; Yang, D.; Liang, W.; Ding, G. Sirt6 Deficiency Aggravates Angiotensin II-Induced Cholesterol Accumulation and Injury in Podocytes. Theranostics 2020, 10, 7465–7479. [Google Scholar] [CrossRef]
- Zhang, Y.; Wang, L.; Meng, L.; Cao, G.; Wu, Y. Sirtuin 6 Overexpression Relieves Sepsis-Induced Acute Kidney Injury by Promoting Autophagy. Cell Cycle 2019, 18, 425–436. [Google Scholar] [CrossRef]
- Cai, J.; Liu, Z.; Huang, X.; Shu, S.; Hu, X.; Zheng, M.; Tang, C.; Liu, Y.; Chen, G.; Sun, L.; et al. The Deacetylase Sirtuin 6 Protects against Kidney Fibrosis by Epigenetically Blocking β-Catenin Target Gene Expression. Kidney Int. 2020, 97, 106–118. [Google Scholar] [CrossRef] [PubMed]
- Fan, Y.; Yang, Q.; Yang, Y.; Gao, Z.; Ma, Y.; Zhang, L.; Liang, W.; Ding, G. Sirt6 Suppresses High Glucose-Induced Mitochondrial Dysfunction and Apoptosis in Podocytes through AMPK Activation. Int. J. Biol. Sci. 2019, 15, 701–713. [Google Scholar] [CrossRef]
- Ji, L.; Chen, Y.; Wang, H.; Zhang, W.; He, L.; Wu, J.; Liu, Y. Overexpression of Sirt6 Promotes M2 Macrophage Transformation, Alleviating Renal Injury in Diabetic Nephropathy. Int. J. Oncol. 2019, 55, 103–115. [Google Scholar] [CrossRef]
- Li, Z.; Xu, K.; Zhang, N.; Amador, G.; Wang, Y.; Zhao, S.; Li, L.; Qiu, Y.; Wang, Z. Overexpressed SIRT6 Attenuates Cisplatin-Induced Acute Kidney Injury by Inhibiting ERK1/2 Signaling. Kidney Int. 2018, 93, 881–892. [Google Scholar] [CrossRef]
- Wang, X.; Lin, B.; Nie, L.; Li, P. microRNA-20b Contributes to High Glucose-Induced Podocyte Apoptosis by Targeting SIRT7. Mol. Med. Rep. 2017, 16, 5667–5674. [Google Scholar] [CrossRef] [PubMed]
- Miyasato, Y.; Yoshizawa, T.; Sato, Y.; Nakagawa, T.; Miyasato, Y.; Kakizoe, Y.; Kuwabara, T.; Adachi, M.; Ianni, A.; Braun, T.; et al. Sirtuin 7 Deficiency Ameliorates Cisplatin-Induced Acute Kidney Injury Through Regulation of the Inflammatory Response. Sci. Rep. 2018, 8, 5927. [Google Scholar] [CrossRef] [PubMed]
- Clearfield, M.; Davis, G.; Weis, J.; Gayer, G.; Shubrook, J.H. Cardiovascular Disease as a Result of the Interactions Between Obesity, Climate Change, and Inflammation: The COCCI Syndemic. J. Am. Osteopath. Assoc. 2018, 118, 719–729. [Google Scholar] [CrossRef] [PubMed]
- Borg, M.; Nitschke, M.; Williams, S.; McDonald, S.; Nairn, J.; Bi, P. Using the Excess Heat Factor to Indicate Heatwave-Related Urinary Disease: A Case Study in Adelaide, South Australia. Int. J. Biometeorol. 2019, 63, 435–447. [Google Scholar] [CrossRef]
- Bobb, J.F.; Obermeyer, Z.; Wang, Y.; Dominici, F. Cause-Specific Risk of Hospital Admission Related to Extreme Heat in Older Adults. JAMA 2014, 312, 2659–2667. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
| HSP | Nephron Location | Heat Stress Pathophysiological Pathways Involved | Main Renal Protective Functions | References |
|---|---|---|---|---|
| HSP27 | PCT and medullary (distal) CT | AKI due to dehydration or drug toxicity (cyclophilin). | ↓ TNF-α injury, ↓ Bad, Bax, and caspase activation, ↓ osmotic damage, ↑ podocyte barrier function, ↑ glutathione bioavailability, ↓ ROS, ↑ cytoskeleton maintenance | [101,102,103,104,107,108,148] |
| HSP32 | Mesangium, cortical PCT | Heavy metal intoxication, AKI due to dehydration or ischemia, or oxidative stress. | ↓ IL-1, IL-6, and IL-8, ↓ ROS bioavailability, ↓ TGF beta | [137,138,139,140,141,142,143,144,145,146] |
| HSP60 | Corticomedullary PCT and Henle’s loop | AKI due to dehydration, hypoperfusion, osmotic damage, or heavy metal toxicity. | ↓ Tubular necrosis, cytoskeleton repairing, and mitochondrial protein re-folding | [109,129,131,133,134,135,136] |
| HSP70 | PCT, medullary CT, Henle’s loop, podocyte slits | AKI due to dehydration and ischemia. | ↓ Bax and caspase 3, ↑ preservation of Na+-K+-ATPase and electrolyte transport maintenance, ↓ NF-kB p65 translocation, ↓ TGF-β, ↓ COL1 deposition, ↑ ERK | [99,100,111,114,115,116,118,149] |
| HSP90 | Bowman’s space, Mesangium, medullary Henle’s loop | AKI due to dehydration and ischemia or nephrolithiasis. | ↑ Kidney function due to ↑ PKM2-Akt, ↓caspase activity, ↑ autophagy by ↑ HIF-1α-BNIP3/BNIP3L, ↓ β-actin, vimentin, and calpain-1 oxalate binding, ↓TLR4 and TNF-α | [120,121,122,123,125,127,128,129] |
| Class | SIRT | Main Activity | Nephropathy | Molecular Mechanisms | Reference |
|---|---|---|---|---|---|
| I | 1 | Deacylase | DKD | ↑ Mitochondrial function and mitophagy, ↑ (PGRN-PGC1α/PPARɣ) ↓ Inflammation, ↓ (NF-kB, STAT3) ↓ Apoptosis, ↓ (caspase 3, cleaved PARP), ↑ mTOR ↓ Fibrosis (Keap1/Nrf2, ARE), ↓ (Notch, Timp-1, FN, and TGF-β) | [178,179,180,181,182,183,184,185,186] |
| Sepsis-related AKI | ↓ Inflammation, ↓(HMGB-1, NLRP3 inflammasome, IL18, and IL-1β) | [187,188] | |||
| Tubulointerstitial fibrosis | ↓ Fibrosis, ↓ (HIF-1α, HIF-2α, TGF-β, and ECM genes) | [189,190] | |||
| Toxic AKI (heavy metals) | ↓ Pyroptosis, ↓ (XBP-1/IRE-1α/NLRP3 inflammasome) | [191] | |||
| Glomerulosclerosis | ↓ Mesangial proliferation and fibrosis, ↓ (COL4, α-SMA, and TGF-β) ↓ Oxidative stress, ↑ (Nrf2/ARE, SOD1, and HO-1) | [183,192] | |||
| Ischemia-reperfusion | ↑ Autophagy, ↑ (LC3B-II, beclin1) | [193] | |||
| Renal cell carcinoma | ↓ Tumorigenesis, ↓ STAT3/FGB ↑ Tumorigenesis, ↓ (p53, p21/Cip1) | [194,195] | |||
| 2 | Deacylase | Tubulointerstitial fibrosis | ↑ Fibrosis, ↑ TGF-β1/MDM2 | [196] | |
| Ischemia-reperfusion | ↑ Apoptosis, ↑ FOXO3α/FasL | [197] | |||
| AKI [cisplatin] | ↑ Apoptosis and necroptosis, ↑ (MKP-1, JNK) | [198] | |||
| 3 | Deacylase | DKD | ↓ Fibrosis, ↓ (HIF-1α, TGF-β/Smad3/4, and aberrant glycolysis) ↓ Apoptosis, ↑ Akt/FoxO ↓ Oxidative stress, ↑ (IDH2, SOD2) | [199,200,201] | |
| Toxic AKI (cisplatin) | ↓ Oxidative stress, ↑ (Nrf2/SOD2, GPx, and catalase) ↑ FAO, ↑ LKB1/AMPK | [202] | |||
| Nephrolithiasis | ↓ Apoptosis, ↓Bax ↑ Crystal-cell adherence, ↑ NRF2/HO-1 | [160,161] | |||
| Hypertensive nephropathy | ↓ Endothelial-to-mesenchymal transition ↑ FOXO3α/catalase ↓ Fibrosis, ↓ (KLF15, FN, and COL4) | [196,203,204,205] | |||
| Ischemia-reperfusion | ↑ Mitochondrial fusion, ↑ ERK/OPA1 ↓ Apoptosis, ↑ Bcl-2/c-IAP, ↓ (cytochrome c, caspase 9) ↓ Inflammation, ↓ (MCP1, IL-6, and TNF-α) ↓ Oxidative stress markers: GSSG, MDA, and LPO, ↑ (SOD, GPX) | [206] | |||
| Renal cell carcinoma | Warburg effect reversal ↑ Mitochondriogenesis, ↑ TFAM | [207] | |||
| DKD | ↓ Inflammation, ↓ (IL-1β, IL-6, and TNF-α) ↓ Apoptosis, ↑ Bcl-2, ↓ (NOX1, Bax, and p38) | [208] | |||
| II | 4 | Ribosyl-transferase | Renal cell carcinoma | Potential role in chemoresistance and metastasis | [209] |
| Toxic AKI (cisplatin) | ↓ Apoptosis, ↑ Bcl-2, ↓ (caspase 3, cleaved PARP, and cytochrome c) ↓ Oxidative stress, ↑ Nrf2/HO-1 ↓ Mitochondrial fission, ↓ Drp1 | [210,211] | |||
| III | 5 | Deacylase | Ischemia-reperfusion | Mitochondrial vs. peroxisomal FAO ratio dysregulation | [212] |
| Renal cell carcinoma | ↑ Mitochondrial O2 consumption, ↑ GDH ↑ Tumorigenesis, ↓ SDH5 | [213,214] | |||
| Various podocytopathies | ↓ Inflammation, ↓ (IL-1β, IL-6, TNF-α, and Notch) ↑ Autophagy, ↑ LC3B-II | [215] | |||
| IV | 6 | Ribosyl-transferase Deacylase | Hypertensive nephropathy | ↓ Endothelial cell permeability, ↓ (MMPs, WT1, and GATA5), ↑ ZO-1 ↓ Endothelial senescence, ↓b-galactosidase ↑ Autophagy, ↑ LC3B-II ↑ Cholesterol efflux, ↑ ABCG1 ↓ Fibrosis, ↓ Angiopoietins/TGF-β | [158,159,216] |
| Sepsis-related AKI | ↓ Inflammation: IL-6 and TNF-α ↑ Autophagy, ↑ LC3B-II | [217] | |||
| Tubulointerstitial fibrosis | ↓ Fibrosis, ↓ (Wnt/β -catenin/TGF-β, GSK3β) | [218] | |||
| DKD | ↑ Mitochondrial function, ↑ AMPK ↓ Inflammation, ↑ M2 macrophage transformation | [219,220] | |||
| Toxic AKI (cisplatin) | ↓ Inflammation and apoptosis, ↓ (NF-kB, ERK1/2) | [221] | |||
| DKD | ↓ Apoptosis, ↓ Caspase 3 | [222] | |||
| 7 | Deacylase | Toxic AKI (cisplatin) | ↑ Inflammation: NF-kB, TNF-α | [223] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Luna-Cerón, E.; Pherez-Farah, A.; Krishnan-Sivadoss, I.; Guerrero-Beltrán, C.E. Molecular Challenges and Opportunities in Climate Change-Induced Kidney Diseases. Biomolecules 2024, 14, 251. https://doi.org/10.3390/biom14030251
Luna-Cerón E, Pherez-Farah A, Krishnan-Sivadoss I, Guerrero-Beltrán CE. Molecular Challenges and Opportunities in Climate Change-Induced Kidney Diseases. Biomolecules. 2024; 14(3):251. https://doi.org/10.3390/biom14030251
Chicago/Turabian StyleLuna-Cerón, Eder, Alfredo Pherez-Farah, Indumathi Krishnan-Sivadoss, and Carlos Enrique Guerrero-Beltrán. 2024. "Molecular Challenges and Opportunities in Climate Change-Induced Kidney Diseases" Biomolecules 14, no. 3: 251. https://doi.org/10.3390/biom14030251