
LILRB4 Checkpoint for Immunotherapy: Structure, Mechanism and Disease Targets
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
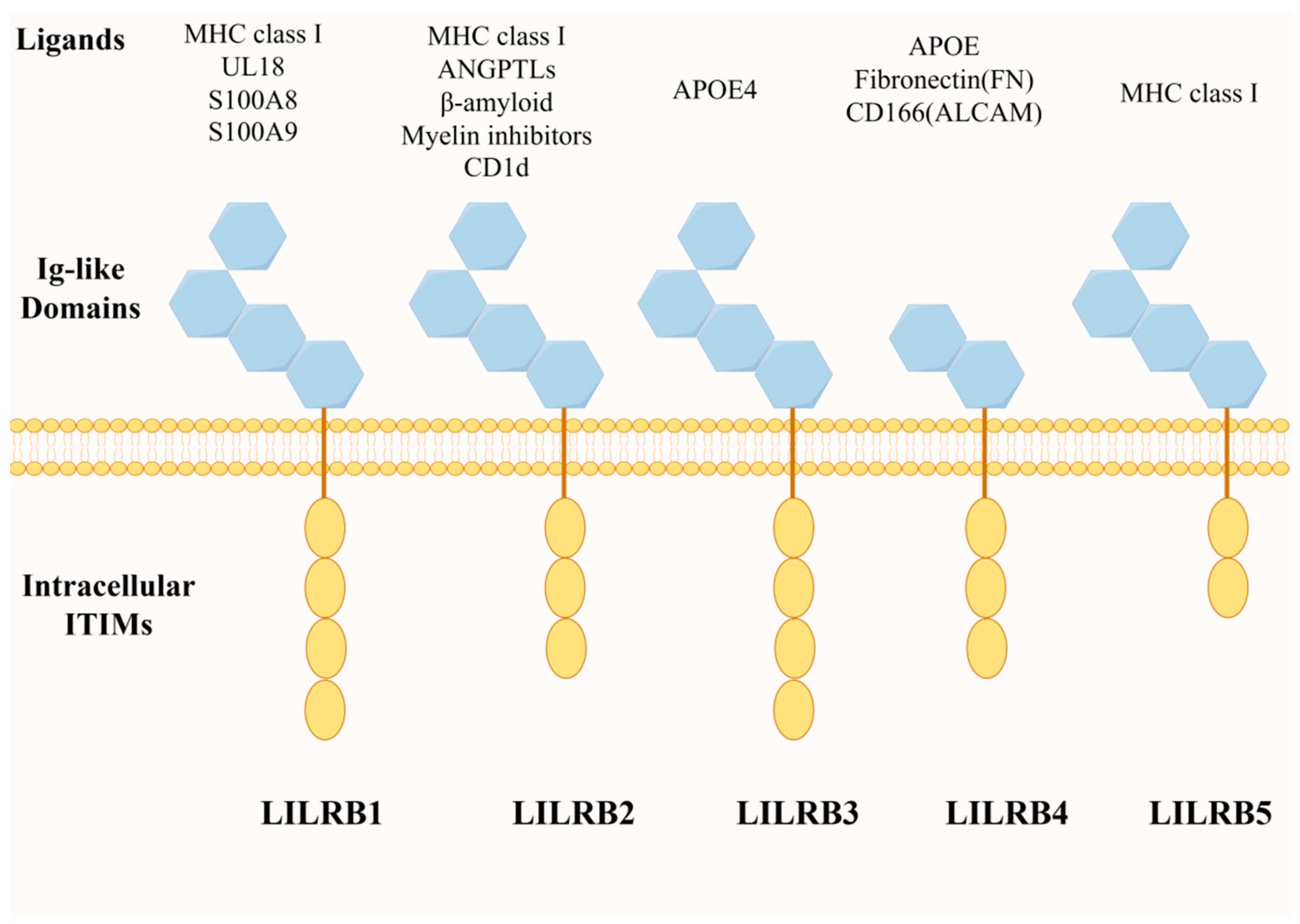
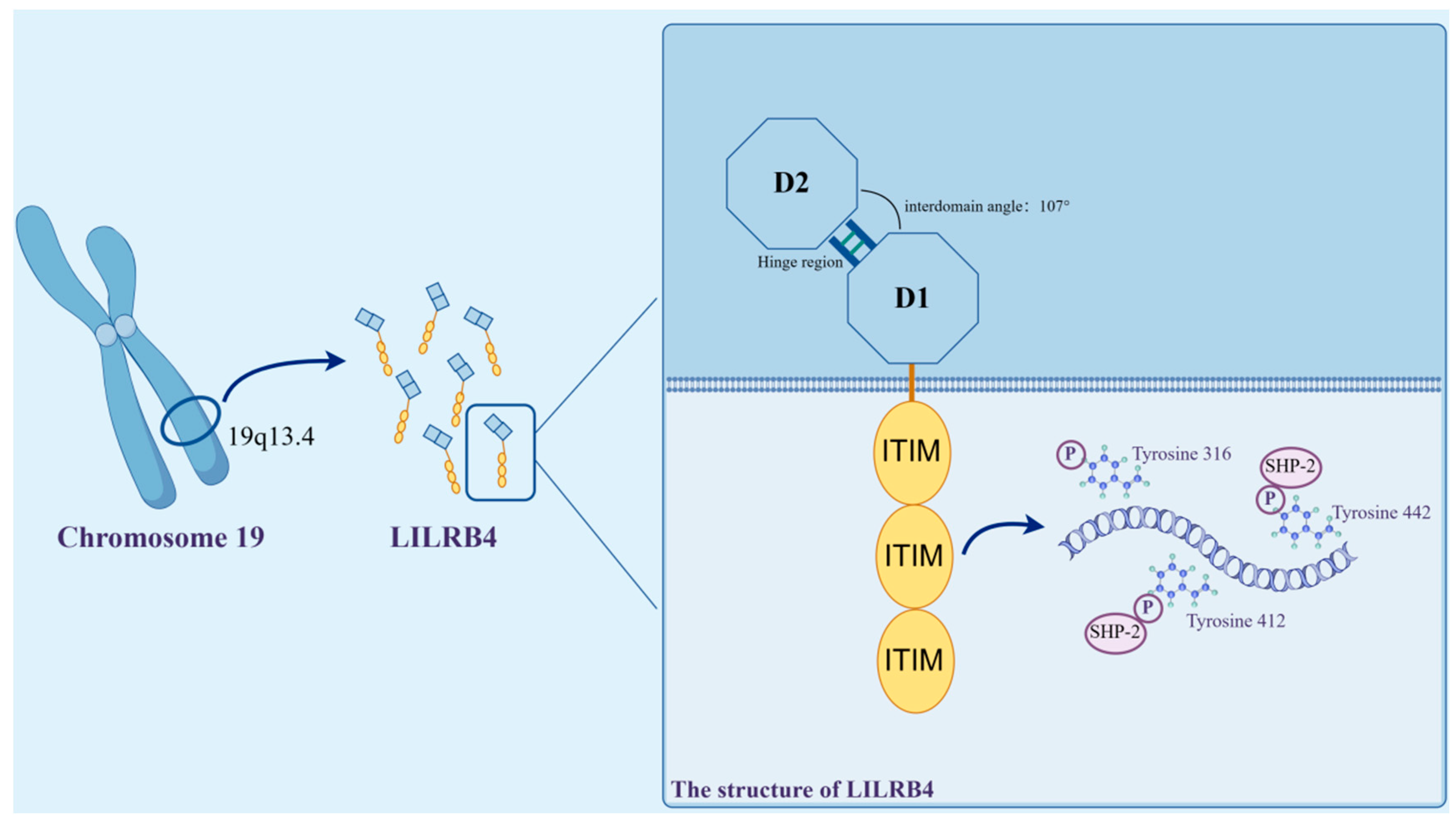
2. Structure and Expression of LILRB4 and Its Ligand Molecules
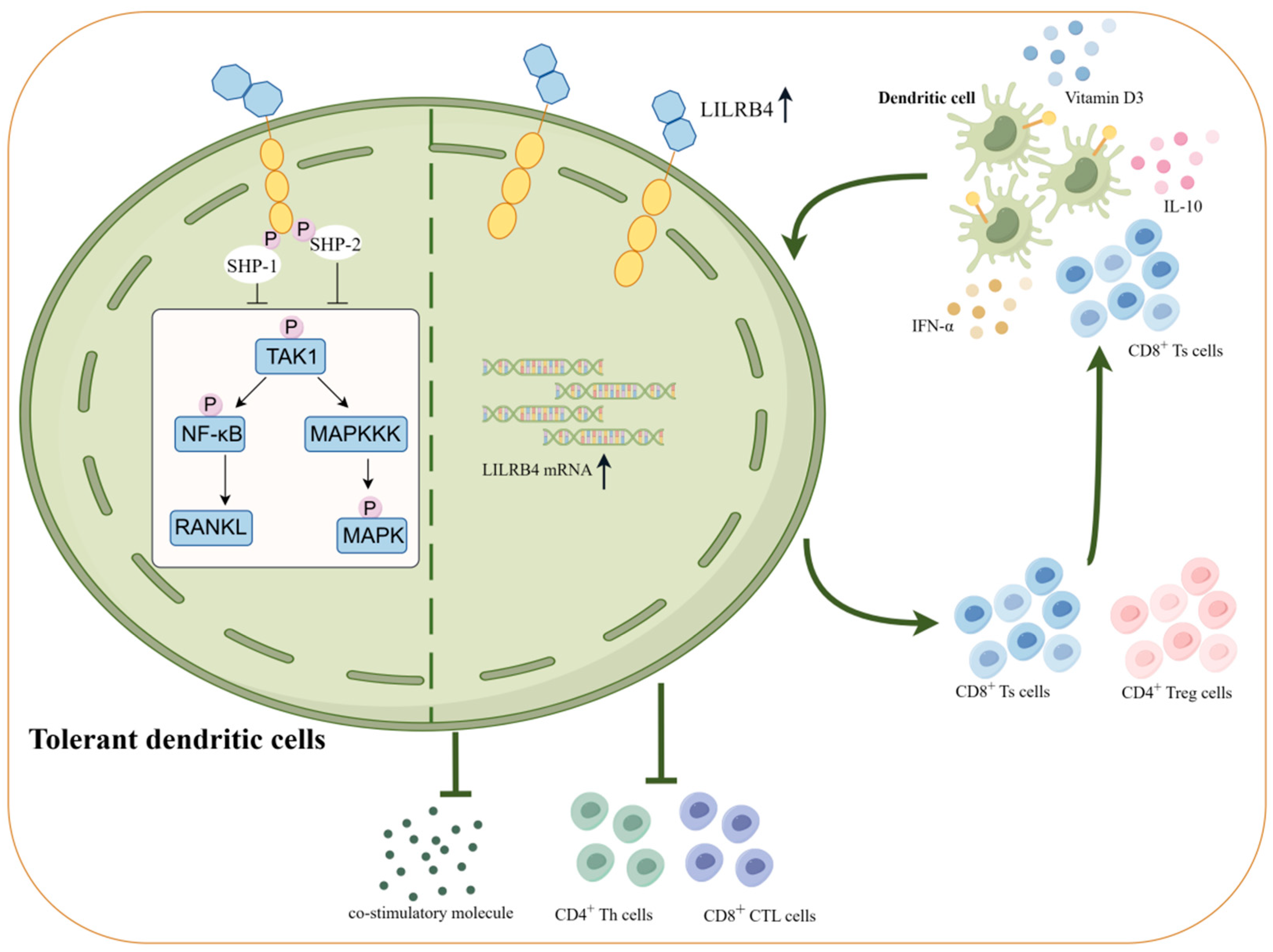
3. LILRB4 and Immune Tolerance
4. Role of LILRB4 in the Immune Response
4.1. High Expression of LILRB4 Decreases Cytokine Expression and Induces Inhibitory Cell Differentiation
4.2. LILRB4 Modulates Intracellular Signaling Pathways
4.3. LILRB4 Plays a Unique Role in Epigenetic Modification
5. Regulatory Role of LILRB4 Molecules in Disease
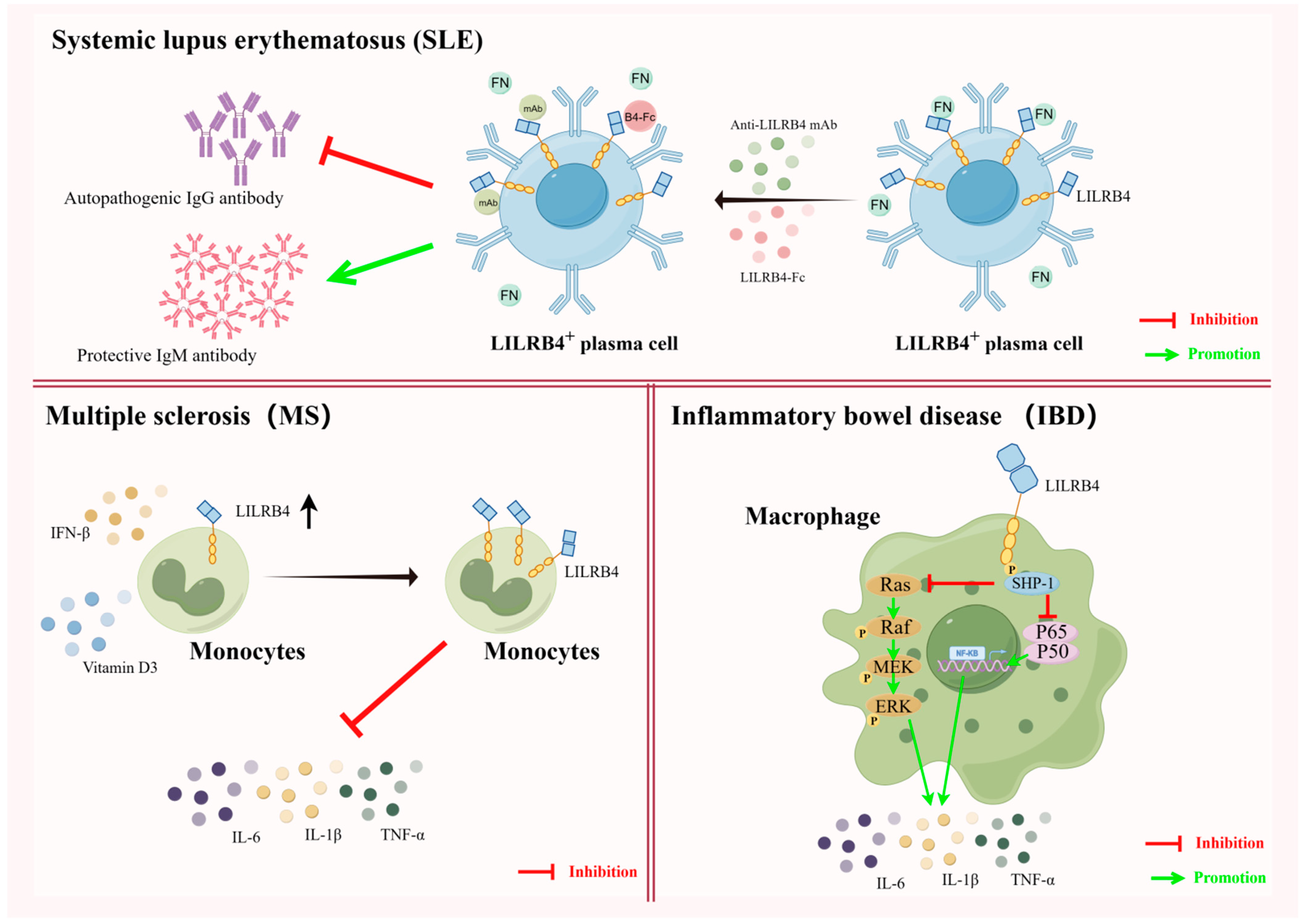
5.1. LILRB4 and Autoimmune Tolerance
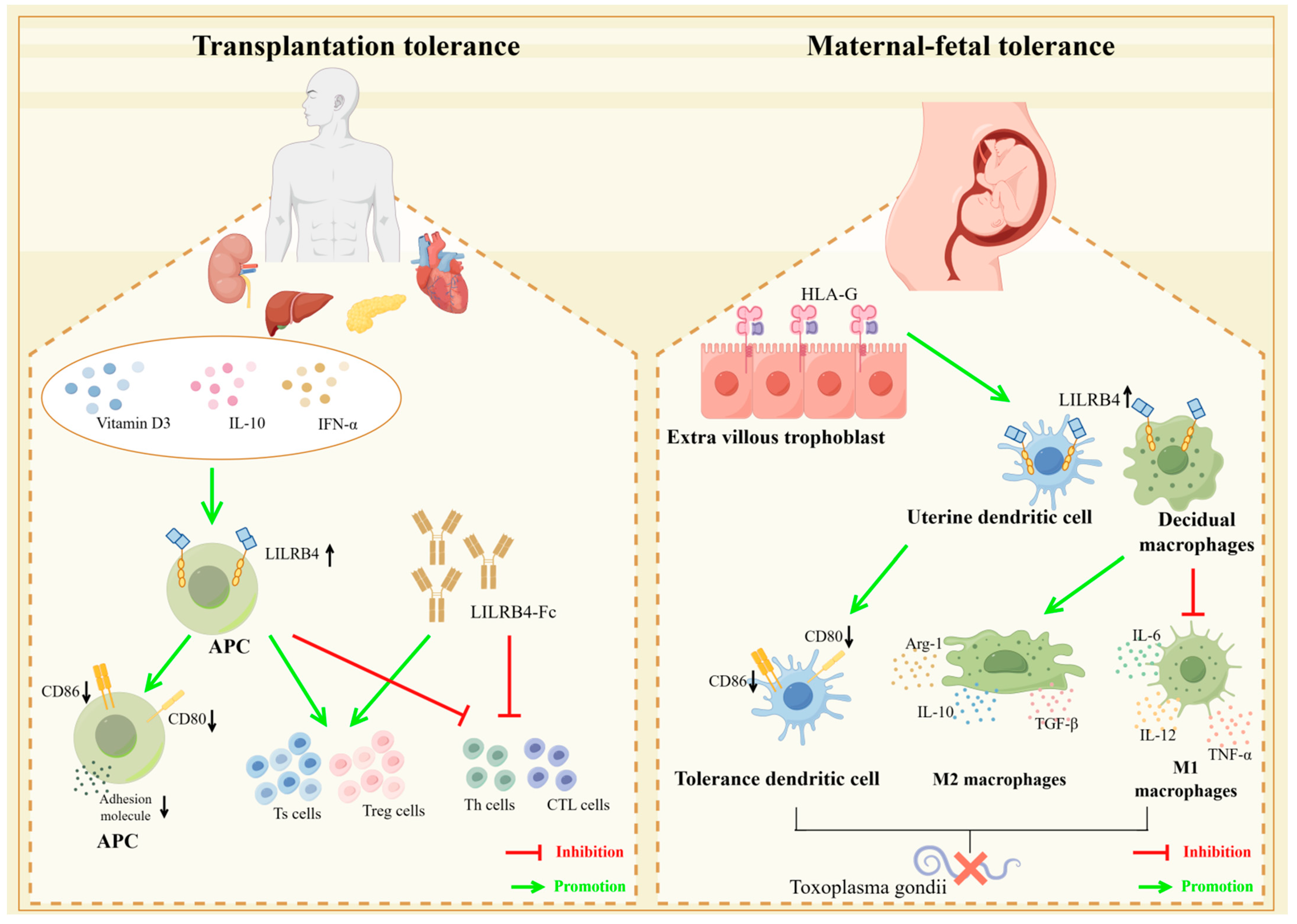
5.2. LILRB4 and Transplantation Immune Tolerance
5.3. LILRB4 and Maternal–Fetal Immune Tolerance
5.4. Studies on the Clinical Application of Blocking LILRB4 Receptors in Tumor Immunotherapy
5.5. Studies on the Association of LILRB4 with Other Diseases
6. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Bluestone, J.A.; Auchincloss, H.; Nepom, G.T.; Rotrosen, D.; St Clair, E.W.; Turka, L.A. The Immune Tolerance Network at 10 years: Tolerance research at the bedside. Nat. Rev. Immunol. 2010, 10, 797–803. [Google Scholar] [CrossRef] [PubMed]
- Yuan, J.; Bagley, J.; Iacomini, J. Hyperlipidemia Promotes Anti-Donor Th17 Responses That Accelerate Allograft Rejection. Am. J. Transplant. 2015, 15, 2336–2345. [Google Scholar] [CrossRef] [PubMed]
- Pearson, R.M.; Casey, L.M.; Hughes, K.R.; Miller, S.D.; Shea, L.D. In vivo reprogramming of immune cells: Technologies for induction of antigen-specific tolerance. Adv. Drug Deliv. Rev. 2017, 114, 240–255. [Google Scholar] [CrossRef] [PubMed]
- Galluzzi, L.; Humeau, J.; Buqué, A.; Zitvogel, L.; Kroemer, G. Immunostimulation with chemotherapy in the era of immune checkpoint inhibitors. Nat. Rev. Clin. Oncol. 2020, 17, 725–741. [Google Scholar] [CrossRef]
- Postow, M.A.; Sidlow, R.; Hellmann, M.D. Immune-Related Adverse Events Associated with Immune Checkpoint Blockade. N. Engl. J. Med. 2018, 378, 158–168. [Google Scholar] [CrossRef]
- Darvin, P.; Toor, S.M.; Sasidharan Nair, V.; Elkord, E. Immune checkpoint inhibitors: Recent progress and potential biomarkers. Exp. Mol. Med. 2018, 50, 1–11. [Google Scholar] [CrossRef]
- Matthews, H.K.; Bertoli, C.; de Bruin, R.A.M. Cell cycle control in cancer. Nat. Rev. Mol. Cell Biol. 2022, 23, 74–88. [Google Scholar] [CrossRef]
- Gutting, T.; Burgermeister, E.; Härtel, N.; Ebert, M.P. Checkpoints and beyond-Immunotherapy in colorectal cancer. Semin. Cancer Biol. 2019, 55, 78–89. [Google Scholar] [CrossRef] [PubMed]
- Turner, J.A.; Stephen-Victor, E.; Wang, S.; Rivas, M.N.; Abdel-Gadir, A.; Harb, H.; Cui, Y.; Fanny, M.; Charbonnier, L.M.; Fong, J.J.H.; et al. Regulatory T Cell-Derived TGF-β1 Controls Multiple Checkpoints Governing Allergy and Autoimmunity. Immunity 2020, 53, 1202–1214.e1206. [Google Scholar] [CrossRef]
- Jiang, J.; Huang, H.; Chen, R.; Lin, Y.; Ling, Q. Immunotherapy for hepatocellular carcinoma recurrence after liver transplantation, can we harness the power of immune checkpoint inhibitors? Front. Immunol. 2023, 14, 1092401. [Google Scholar] [CrossRef]
- Sharpe, A.H.; Pauken, K.E. The diverse functions of the PD1 inhibitory pathway. Nat. Rev. Immunol. 2018, 18, 153–167. [Google Scholar] [CrossRef]
- Rowshanravan, B.; Halliday, N.; Sansom, D.M. CTLA-4: A moving target in immunotherapy. Blood 2018, 131, 58–67. [Google Scholar] [CrossRef]
- Wu, G.; Xu, Y.; Schultz, R.D.; Chen, H.; Xie, J.; Deng, M.; Liu, X.; Gui, X.; John, S.; Lu, Z.; et al. LILRB3 supports acute myeloid leukemia development and regulates T-cell antitumor immune responses through the TRAF2-cFLIP-NF-κB signaling axis. Nat. Cancer 2021, 2, 1170–1184. [Google Scholar] [CrossRef]
- Chen, H.M.; van der Touw, W.; Wang, Y.S.; Kang, K.; Mai, S.; Zhang, J.; Alsina-Beauchamp, D.; Duty, J.A.; Mungamuri, S.K.; Zhang, B.; et al. Blocking immunoinhibitory receptor LILRB2 reprograms tumor-associated myeloid cells and promotes antitumor immunity. J. Clin. Investig. 2018, 128, 5647–5662. [Google Scholar] [CrossRef]
- van der Touw, W.; Chen, H.M.; Pan, P.Y.; Chen, S.H. LILRB receptor-mediated regulation of myeloid cell maturation and function. Cancer Immunol. Immunother. 2017, 66, 1079–1087. [Google Scholar] [CrossRef] [PubMed]
- Redondo-García, S.; Barritt, C.; Papagregoriou, C.; Yeboah, M.; Frendeus, B.; Cragg, M.S.; Roghanian, A. Human leukocyte immunoglobulin-like receptors in health and disease. Front. Immunol. 2023, 14, 1282874. [Google Scholar] [CrossRef]
- Deng, M.; Gui, X.; Kim, J.; Xie, L.; Chen, W.; Li, Z.; He, L.; Chen, Y.; Chen, H.; Luo, W.; et al. LILRB4 signalling in leukaemia cells mediates T cell suppression and tumour infiltration. Nature 2018, 562, 605–609. [Google Scholar] [CrossRef] [PubMed]
- Sugahara-Tobinai, A.; Inui, M.; Metoki, T.; Watanabe, Y.; Onuma, R.; Takai, T.; Kumaki, S. Augmented ILT3/LILRB4 Expression of Peripheral Blood Antibody Secreting Cells in the Acute Phase of Kawasaki Disease. Pediatr. Infect. Dis. J. 2019, 38, 431–438. [Google Scholar] [CrossRef] [PubMed]
- Inui, M.; Sugahara-Tobinai, A.; Fujii, H.; Itoh-Nakadai, A.; Fukuyama, H.; Kurosaki, T.; Ishii, T.; Harigae, H.; Takai, T. Tolerogenic immunoreceptor ILT3/LILRB4 paradoxically marks pathogenic auto-antibody-producing plasmablasts and plasma cells in non-treated SLE. Int. Immunol. 2016, 28, 597–604. [Google Scholar] [CrossRef] [PubMed]
- Jensen, M.A.; Patterson, K.C.; Kumar, A.A.; Kumabe, M.; Franek, B.S.; Niewold, T.B. Functional genetic polymorphisms in ILT3 are associated with decreased surface expression on dendritic cells and increased serum cytokines in lupus patients. Ann. Rheum. Dis. 2013, 72, 596–601. [Google Scholar] [CrossRef]
- Farré, D.; Martínez-Vicente, P.; Engel, P.; Angulo, A. Immunoglobulin superfamily members encoded by viruses and their multiple roles in immune evasion. Eur. J. Immunol. 2017, 47, 780–796. [Google Scholar] [CrossRef]
- Hirayasu, K.; Arase, H. Functional and genetic diversity of leukocyte immunoglobulin-like receptor and implication for disease associations. J. Hum. Genet. 2015, 60, 703–708. [Google Scholar] [CrossRef]
- De Louche, C.D.; Roghanian, A. Human inhibitory leukocyte Ig-like receptors: From immunotolerance to immunotherapy. JCI Insight 2022, 7, e151553. [Google Scholar] [CrossRef]
- Colonna, M.; Navarro, F.; Bellón, T.; Llano, M.; García, P.; Samaridis, J.; Angman, L.; Cella, M.; López-Botet, M. A common inhibitory receptor for major histocompatibility complex class I molecules on human lymphoid and myelomonocytic cells. J. Exp. Med. 1997, 186, 1809–1818. [Google Scholar] [CrossRef]
- Nakajima, H.; Samaridis, J.; Angman, L.; Colonna, M. Human myeloid cells express an activating ILT receptor (ILT1) that associates with Fc receptor gamma-chain. J. Immunol. 1999, 162, 5–8. [Google Scholar] [CrossRef] [PubMed]
- Xu, D.; Qu, C.K. Protein tyrosine phosphatases in the JAK/STAT pathway. Front. Biosci. 2008, 13, 4925–4932. [Google Scholar] [CrossRef]
- Arm, J.P.; Nwankwo, C.; Austen, K.F. Molecular identification of a novel family of human Ig superfamily members that possess immunoreceptor tyrosine-based inhibition motifs and homology to the mouse gp49B1 inhibitory receptor. J. Immunol. 1997, 159, 2342–2349. [Google Scholar] [CrossRef] [PubMed]
- Gleissner, C.A.; Dengler, T.J. Induction of ILT expression on nonprofessional antigen presenting cells: Clinical applications. Hum. Immunol. 2009, 70, 357–359. [Google Scholar] [CrossRef] [PubMed]
- Coxon, C.H.; Geer, M.J.; Senis, Y.A. ITIM receptors: More than just inhibitors of platelet activation. Blood 2017, 129, 3407–3418. [Google Scholar] [CrossRef]
- Li, Z.; Deng, M.; Huang, F.; Jin, C.; Sun, S.; Chen, H.; Liu, X.; He, L.; Sadek, A.H.; Zhang, C.C. LILRB4 ITIMs mediate the T cell suppression and infiltration of acute myeloid leukemia cells. Cell Mol. Immunol. 2020, 17, 272–282. [Google Scholar] [CrossRef]
- Park, M.; Liu, R.W.; An, H.; Geczy, C.L.; Thomas, P.S.; Tedla, N. A dual positive and negative regulation of monocyte activation by leukocyte Ig-like receptor B4 depends on the position of the tyrosine residues in its ITIMs. Innate Immun. 2017, 23, 381–391. [Google Scholar] [CrossRef]
- Cella, M.; Döhring, C.; Samaridis, J.; Dessing, M.; Brockhaus, M.; Lanzavecchia, A.; Colonna, M. A novel inhibitory receptor (ILT3) expressed on monocytes, macrophages, and dendritic cells involved in antigen processing. J. Exp. Med. 1997, 185, 1743–1751. [Google Scholar] [CrossRef]
- Cheng, H.; Mohammed, F.; Nam, G.; Chen, Y.; Qi, J.; Garner, L.I.; Allen, R.L.; Yan, J.; Willcox, B.E.; Gao, G.F. Crystal structure of leukocyte Ig-like receptor LILRB4 (ILT3/LIR-5/CD85k): A myeloid inhibitory receptor involved in immune tolerance. J. Biol. Chem. 2011, 286, 18013–18025. [Google Scholar] [CrossRef]
- Castells, M.C.; Klickstein, L.B.; Hassani, K.; Cumplido, J.A.; Lacouture, M.E.; Austen, K.F.; Katz, H.R. gp49B1-alpha(v)beta3 interaction inhibits antigen-induced mast cell activation. Nat. Immunol. 2001, 2, 436–442. [Google Scholar] [CrossRef] [PubMed]
- Xu, Z.; Chang, C.C.; Li, M.; Zhang, Q.Y.; Vasilescu, E.M.; D’Agati, V.; Floratos, A.; Vlad, G.; Suciu-Foca, N. ILT3.Fc-CD166 Interaction Induces Inactivation of p70 S6 Kinase and Inhibits Tumor Cell Growth. J. Immunol. 2018, 200, 1207–1219. [Google Scholar] [CrossRef] [PubMed]
- Itoi, S.; Takahashi, N.; Saito, H.; Miyata, Y.; Su, M.T.; Kezuka, D.; Itagaki, F.; Endo, S.; Fujii, H.; Harigae, H.; et al. Myeloid immune checkpoint ILT3/LILRB4/gp49B can co-tether fibronectin with integrin on macrophages. Int. Immunol. 2022, 34, 435–444. [Google Scholar] [CrossRef]
- Su, M.T.; Inui, M.; Wong, Y.L.; Takahashi, M.; Sugahara-Tobinai, A.; Ono, K.; Miyamoto, S.; Murakami, K.; Itoh-Nakadai, A.; Kezuka, D.; et al. Blockade of checkpoint ILT3/LILRB4/gp49B binding to fibronectin ameliorates autoimmune disease in BXSB/Yaa mice. Int. Immunol. 2021, 33, 447–458. [Google Scholar] [CrossRef]
- Zhan, S.; Zheng, J.; Zhang, H.; Zhao, M.; Liu, X.; Jiang, Y.; Yang, C.; Ren, L.; Liu, Z.; Hu, X. LILRB4 Decrease on uDCs Exacerbate Abnormal Pregnancy Outcomes Following Toxoplasma gondii Infection. Front. Microbiol. 2018, 9, 588. [Google Scholar] [CrossRef] [PubMed]
- Singh, L.; Muise, E.S.; Bhattacharya, A.; Grein, J.; Javaid, S.; Stivers, P.; Zhang, J.; Qu, Y.; Joyce-Shaikh, B.; Loboda, A.; et al. ILT3 (LILRB4) Promotes the Immunosuppressive Function of Tumor-Educated Human Monocytic Myeloid-Derived Suppressor Cells. Mol. Cancer Res. 2021, 19, 702–716. [Google Scholar] [CrossRef]
- Su, M.T.; Kumata, S.; Endo, S.; Okada, Y.; Takai, T. LILRB4 promotes tumor metastasis by regulating MDSCs and inhibiting miR-1 family miRNAs. Oncoimmunology 2022, 11, 2060907. [Google Scholar] [CrossRef]
- Lee, H.N.; Manangeeswaran, M.; Lewkowicz, A.P.; Engel, K.; Chowdhury, M.; Garige, M.; Eckhaus, M.A.; Sourbier, C.; Ireland, D.D.; Verthelyi, D. NK cells require immune checkpoint receptor LILRB4/gp49B to control neurotropic Zika virus infections in mice. JCI Insight 2022, 7, e151420. [Google Scholar] [CrossRef]
- Kretzschmar, F.; Piecha, R.; Jahn, J.; Potru, P.S.; Spittau, B. Characterization of the Leucocyte Immunoglobulin-like Receptor B4 (Lilrb4) Expression in Microglia. Biology 2021, 10, 1300. [Google Scholar] [CrossRef]
- Theofilopoulos, A.N.; Kono, D.H.; Baccala, R. The multiple pathways to autoimmunity. Nat. Immunol. 2017, 18, 716–724. [Google Scholar] [CrossRef] [PubMed]
- Mora, T.; Walczak, A.M. Towards a quantitative theory of tolerance. Trends Immunol. 2023, 44, 512–518. [Google Scholar] [CrossRef]
- Nemazee, D. Mechanisms of central tolerance for B cells. Nat. Rev. Immunol. 2017, 17, 281–294. [Google Scholar] [CrossRef]
- Schwartz, R.H. Historical overview of immunological tolerance. Cold Spring Harb. Perspect. Biol. 2012, 4, a006908. [Google Scholar] [CrossRef] [PubMed]
- Schietinger, A.; Greenberg, P.D. Tolerance and exhaustion: Defining mechanisms of T cell dysfunction. Trends Immunol. 2014, 35, 51–60. [Google Scholar] [CrossRef]
- Anderson, M.S.; Su, M.A. AIRE expands: New roles in immune tolerance and beyond. Nat. Rev. Immunol. 2016, 16, 247–258. [Google Scholar] [CrossRef] [PubMed]
- Palmer, E. Negative selection--clearing out the bad apples from the T-cell repertoire. Nat. Rev. Immunol. 2003, 3, 383–391. [Google Scholar] [CrossRef]
- Talmage, D.W.; Pearlman, D.S. The antibody response: A model based on antagonistic actions of antigen. J. Theor. Biol. 1963, 5, 321–339. [Google Scholar] [CrossRef]
- Hähnlein, J.S.; Nadafi, R.; Jong, T.A.; Semmelink, J.F.; Remmerswaal, E.B.M.; Safy, M.; Lienden, K.P.V.; Maas, M.; Gerlag, D.M.; Tak, P.P.; et al. Human Lymph Node Stromal Cells Have the Machinery to Regulate Peripheral Tolerance during Health and Rheumatoid Arthritis. Int. J. Mol. Sci. 2020, 21, 5713. [Google Scholar] [CrossRef] [PubMed]
- Alpdogan, O.; van den Brink, M.R. Immune tolerance and transplantation. Semin. Oncol. 2012, 39, 629–642. [Google Scholar] [CrossRef]
- Honey, K.; Cobbold, S.P.; Waldmann, H. Dominant tolerance and linked suppression induced by therapeutic antibodies do not depend on Fas-FasL interactions. Transplantation 2000, 69, 1683–1689. [Google Scholar] [CrossRef]
- Bluestone, J.A.; Anderson, M. Tolerance in the Age of Immunotherapy. N. Engl. J. Med. 2020, 383, 1156–1166. [Google Scholar] [CrossRef] [PubMed]
- Pan, P.Y.; Ozao, J.; Zhou, Z.; Chen, S.H. Advancements in immune tolerance. Adv. Drug Deliv. Rev. 2008, 60, 91–105. [Google Scholar] [CrossRef]
- Fitch, Z.W.; Kang, L.; Li, J.; Knechtle, S.J.; Turek, J.W.; Kirk, A.D.; Markert, M.L.; Kwun, J. Introducing thymus for promoting transplantation tolerance. J. Allergy Clin. Immunol. 2022, 150, 549–556. [Google Scholar] [CrossRef]
- Sánchez-Fueyo, A.; Whitehouse, G.; Grageda, N.; Cramp, M.E.; Lim, T.Y.; Romano, M.; Thirkell, S.; Lowe, K.; Fry, L.; Heward, J.; et al. Applicability, safety, and biological activity of regulatory T cell therapy in liver transplantation. Am. J. Transplant. 2020, 20, 1125–1136. [Google Scholar] [CrossRef] [PubMed]
- Feinberg, M.B.; Silvestri, G. T(S) cells and immune tolerance induction: A regulatory renaissance? Nat. Immunol. 2002, 3, 215–217. [Google Scholar] [CrossRef]
- Marin-Acevedo, J.A.; Kimbrough, E.O.; Lou, Y. Next generation of immune checkpoint inhibitors and beyond. J. Hematol. Oncol. 2021, 14, 45. [Google Scholar] [CrossRef]
- Chang, C.C.; Ciubotariu, R.; Manavalan, J.S.; Yuan, J.; Colovai, A.I.; Piazza, F.; Lederman, S.; Colonna, M.; Cortesini, R.; Dalla-Favera, R.; et al. Tolerization of dendritic cells by T(S) cells: The crucial role of inhibitory receptors ILT3 and ILT4. Nat. Immunol. 2002, 3, 237–243. [Google Scholar] [CrossRef]
- Banchereau, J.; Zurawski, S.; Thompson-Snipes, L.; Blanck, J.P.; Clayton, S.; Munk, A.; Cao, Y.; Wang, Z.; Khandelwal, S.; Hu, J.; et al. Immunoglobulin-like transcript receptors on human dermal CD14+ dendritic cells act as a CD8-antagonist to control cytotoxic T cell priming. Proc. Natl. Acad. Sci. USA 2012, 109, 18885–18890. [Google Scholar] [CrossRef]
- Young, N.T.; Waller, E.C.; Patel, R.; Roghanian, A.; Austyn, J.M.; Trowsdale, J. The inhibitory receptor LILRB1 modulates the differentiation and regulatory potential of human dendritic cells. Blood 2008, 111, 3090–3096. [Google Scholar] [CrossRef]
- Brenk, M.; Scheler, M.; Koch, S.; Neumann, J.; Takikawa, O.; Häcker, G.; Bieber, T.; von Bubnoff, D. Tryptophan deprivation induces inhibitory receptors ILT3 and ILT4 on dendritic cells favoring the induction of human CD4+CD25+ Foxp3+ T regulatory cells. J. Immunol. 2009, 183, 145–154. [Google Scholar] [CrossRef]
- Sheu, J.; Shih Ie, M. HLA-G and immune evasion in cancer cells. J. Formos. Med. Assoc. 2010, 109, 248–257. [Google Scholar] [CrossRef]
- Kuroki, K.; Maenaka, K. Immune modulation of HLA-G dimer in maternal-fetal interface. Eur. J. Immunol. 2007, 37, 1727–1729. [Google Scholar] [CrossRef] [PubMed]
- Yeboah, M.; Papagregoriou, C.; Jones, D.C.; Chan, H.T.C.; Hu, G.; McPartlan, J.S.; Schiött, T.; Mattson, U.; Mockridge, C.I.; Tornberg, U.C.; et al. LILRB3 (ILT5) is a myeloid cell checkpoint that elicits profound immunomodulation. JCI Insight 2020, 5, e141593. [Google Scholar] [CrossRef]
- Liu, J.; Wu, Q.; Shi, J.; Guo, W.; Jiang, X.; Zhou, B.; Ren, C. LILRB4, from the immune system to the disease target. Am. J. Transl. Res. 2020, 12, 3149–3166. [Google Scholar]
- Itagaki, F.; Nakatsuka, K.; Sakai, H.; Endo, S.; Su, M.T.; Takai, T. Fibronectin on target cells attenuates natural cytotoxicity of NK cells via myeloid immune checkpoint ILT3/LILRB4/gp49B. Int. Immunol. 2023, 35, 339–348. [Google Scholar] [CrossRef] [PubMed]
- Vlad, G.; Chang, C.C.; Colovai, A.I.; Vasilescu, E.R.; Cortesini, R.; Suciu-Foca, N. Membrane and soluble ILT3 are critical to the generation of T suppressor cells and induction of immunological tolerance. Int. Rev. Immunol. 2010, 29, 119–132. [Google Scholar] [CrossRef] [PubMed]
- Xu, Z.; Ho, S.; Chang, C.C.; Liu, Z.; Li, M.; Vasilescu, E.R.; Clynes, R.A.; Vlad, G.; Suciu-Foca, N. ILT3.Fc inhibits the production of exosomes containing inflammatory microRNA in supernatants of alloactivated T cells. Hum. Immunol. 2014, 75, 756–759. [Google Scholar] [CrossRef]
- Vlad, G.; Chang, C.C.; Colovai, A.I.; Berloco, P.; Cortesini, R.; Suciu-Foca, N. Immunoglobulin-like transcript 3: A crucial regulator of dendritic cell function. Hum. Immunol. 2009, 70, 340–344. [Google Scholar] [CrossRef]
- Chang, C.C.; Vlad, G.; D’Agati, V.D.; Liu, Z.; Zhang, Q.Y.; Witkowski, P.; Torkamani, A.A.; Stokes, M.B.; Ho, E.K.; Cortesini, R.; et al. BCL6 is required for differentiation of Ig-like transcript 3-Fc-induced CD8+ T suppressor cells. J. Immunol. 2010, 185, 5714–5722. [Google Scholar] [CrossRef]
- Wang, L.L.; Blasioli, J.; Plas, D.R.; Thomas, M.L.; Yokoyama, W.M. Specificity of the SH2 domains of SHP-1 in the interaction with the immunoreceptor tyrosine-based inhibitory motif-bearing receptor gp49B. J. Immunol. 1999, 162, 1318–1323. [Google Scholar] [CrossRef]
- Su, M.T.; Ono, K.; Kezuka, D.; Miyamoto, S.; Mori, Y.; Takai, T. Fibronectin-LILRB4/gp49B interaction negatively regulates osteoclastogenesis through inhibition of RANKL-induced TRAF6/TAK1/NF-kB/MAPK signaling. Int. Immunol. 2023, 35, 135–145. [Google Scholar] [CrossRef] [PubMed]
- Truong, A.D.; Hong, Y.; Tran, H.T.T.; Dang, H.V.; Nguyen, V.K.; Pham, T.T.; Lillehoj, H.S.; Hong, Y.H. Characterization and functional analyses of novel chicken leukocyte immunoglobulin-like receptor subfamily B members 4 and 5. Poult. Sci. 2019, 98, 6989–7002. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Su, R.; Deng, X.; Chen, Y.; Chen, J. FTO in cancer: Functions, molecular mechanisms, and therapeutic implications. Trends Cancer 2022, 8, 598–614. [Google Scholar] [CrossRef] [PubMed]
- Su, R.; Dong, L.; Li, Y.; Gao, M.; Han, L.; Wunderlich, M.; Deng, X.; Li, H.; Huang, Y.; Gao, L.; et al. Targeting FTO Suppresses Cancer Stem Cell Maintenance and Immune Evasion. Cancer Cell 2020, 38, 79–96.e11. [Google Scholar] [CrossRef] [PubMed]
- Owen, K.A.; Grammer, A.C.; Lipsky, P.E. Deconvoluting the heterogeneity of SLE: The contribution of ancestry. J. Allergy Clin. Immunol. 2022, 149, 12–23. [Google Scholar] [CrossRef]
- Wong, Y.L.; Su, M.T.; Sugahara-Tobinai, A.; Itoi, S.; Kezuka, D.; Endo, S.; Inui, M.; Takai, T. Gp49B is a pathogenic marker for auto-antibody-producing plasma cells in lupus-prone BXSB/Yaa mice. Int. Immunol. 2019, 31, 397–406. [Google Scholar] [CrossRef]
- Thompson, A.J.; Baranzini, S.E.; Geurts, J.; Hemmer, B.; Ciccarelli, O. Multiple sclerosis. Lancet 2018, 391, 1622–1636. [Google Scholar] [CrossRef] [PubMed]
- Rodríguez Murúa, S.; Farez, M.F.; Quintana, F.J. The Immune Response in Multiple Sclerosis. Annu. Rev. Pathol. 2022, 17, 121–139. [Google Scholar] [CrossRef]
- Jensen, M.A.; Yanowitch, R.N.; Reder, A.T.; White, D.M.; Arnason, B.G. Immunoglobulin-like transcript 3, an inhibitor of T cell activation, is reduced on blood monocytes during multiple sclerosis relapses and is induced by interferon beta-1b. Mult. Scler. 2010, 16, 30–38. [Google Scholar] [CrossRef] [PubMed]
- Waschbisch, A.; Sanderson, N.; Krumbholz, M.; Vlad, G.; Theil, D.; Schwab, S.; Mäurer, M.; Derfuss, T. Interferon beta and vitamin D synergize to induce immunoregulatory receptors on peripheral blood monocytes of multiple sclerosis patients. PLoS ONE 2014, 9, e115488. [Google Scholar] [CrossRef] [PubMed]
- Xu, Z.; Lin, C.C.; Ho, S.; Vlad, G.; Suciu-Foca, N. Suppression of Experimental Autoimmune Encephalomyelitis by ILT3.Fc. J. Immunol. 2021, 206, 554–565. [Google Scholar] [CrossRef] [PubMed]
- Bisgaard, T.H.; Allin, K.H.; Keefer, L.; Ananthakrishnan, A.N.; Jess, T. Depression and anxiety in inflammatory bowel disease: Epidemiology, mechanisms and treatment. Nat. Rev. Gastroenterol. Hepatol. 2022, 19, 717–726. [Google Scholar] [CrossRef] [PubMed]
- Hodson, R. Inflammatory bowel disease. Nature 2016, 540, S97. [Google Scholar] [CrossRef] [PubMed]
- Munitz, A.; Cole, E.T.; Beichler, A.; Groschwitz, K.; Ahrens, R.; Steinbrecher, K.; Willson, T.; Han, X.; Denson, L.; Rothenberg, M.E.; et al. Paired immunoglobulin-like receptor B (PIR-B) negatively regulates macrophage activation in experimental colitis. Gastroenterology 2010, 139, 530–541. [Google Scholar] [CrossRef] [PubMed]
- Cortesini, R.; Suciu-Foca, N. ILT3+ ILT4+ tolerogenic endothelial cells in transplantation. Transplantation 2006, 82, S30–S32. [Google Scholar] [CrossRef] [PubMed]
- Kim-Schulze, S.; Seki, T.; Vlad, G.; Scotto, L.; Fan, J.; Colombo, P.C.; Liu, J.; Cortesini, R.; Suciu-Foca, N. Regulation of ILT3 gene expression by processing of precursor transcripts in human endothelial cells. Am. J. Transplant. 2006, 6, 76–82. [Google Scholar] [CrossRef]
- Manavalan, J.S.; Kim-Schulze, S.; Scotto, L.; Naiyer, A.J.; Vlad, G.; Colombo, P.C.; Marboe, C.; Mancini, D.; Cortesini, R.; Suciu-Foca, N. Alloantigen specific CD8+CD28- FOXP3+ T suppressor cells induce ILT3+ ILT4+ tolerogenic endothelial cells, inhibiting alloreactivity. Int. Immunol. 2004, 16, 1055–1068. [Google Scholar] [CrossRef]
- Karimi, M.; Ahmadpoor, P.; Nafar, M.; Pourrezagholi, F.; Jamali, S.; Eteghadi, A.; Yekaninejad, M.S.; Amirzargar, A.A. Frequency of dendritic cell subsets and ILT3, ILT4 gene expression in two different immunosuppressive protocols in kidney transplant recipients. A cohort report. Mol. Biol. Rep. 2020, 47, 123–128. [Google Scholar] [CrossRef]
- Rosborough, B.R.; Hackstein, H.; Turnquist, H.R. A window into immunosuppressant immunoregulation: Recipient conversion to rapamycin increases potentially tolerogenic immune cells. Kidney Int. 2014, 85, 743–745. [Google Scholar] [CrossRef]
- Stallone, G.; Pontrelli, P.; Infante, B.; Gigante, M.; Netti, G.S.; Ranieri, E.; Grandaliano, G.; Gesualdo, L. Rapamycin induces ILT3(high)ILT4(high) dendritic cells promoting a new immunoregulatory pathway. Kidney Int. 2014, 85, 888–897. [Google Scholar] [CrossRef]
- Suciu-Foca, N.; Manavalan, J.S.; Cortesini, R. Generation and function of antigen-specific suppressor and regulatory T cells. Transpl. Immunol. 2003, 11, 235–244. [Google Scholar] [CrossRef]
- Penna, G.; Roncari, A.; Amuchastegui, S.; Daniel, K.C.; Berti, E.; Colonna, M.; Adorini, L. Expression of the inhibitory receptor ILT3 on dendritic cells is dispensable for induction of CD4+Foxp3+ regulatory T cells by 1,25-dihydroxyvitamin D3. Blood 2005, 106, 3490–3497. [Google Scholar] [CrossRef]
- Kasai, S.; Inui, M.; Nakamura, K.; Kakizaki, Y.; Endo, S.; Nakamura, A.; Ito, S.; Takai, T. A novel regulatory role of gp49B on dendritic cells in T-cell priming. Eur. J. Immunol. 2008, 38, 2426–2437. [Google Scholar] [CrossRef]
- Vlad, G.; Liu, Z.; Zhang, Q.Y.; Cortesini, R.; Suciu-Foca, N. Immunosuppressive activity of recombinant ILT3. Int. Immunopharmacol. 2006, 6, 1889–1894. [Google Scholar] [CrossRef]
- Kim-Schulze, S.; Scotto, L.; Vlad, G.; Piazza, F.; Lin, H.; Liu, Z.; Cortesini, R.; Suciu-Foca, N. Recombinant Ig-like transcript 3-Fc modulates T cell responses via induction of Th anergy and differentiation of CD8+ T suppressor cells. J. Immunol. 2006, 176, 2790–2798. [Google Scholar] [CrossRef] [PubMed]
- Vlad, G.; D’Agati, V.D.; Zhang, Q.Y.; Liu, Z.; Ho, E.K.; Mohanakumar, T.; Hardy, M.A.; Cortesini, R.; Suciu-Foca, N. Immunoglobulin-like transcript 3-Fc suppresses T-cell responses to allogeneic human islet transplants in hu-NOD/SCID mice. Diabetes 2008, 57, 1878–1886. [Google Scholar] [CrossRef] [PubMed]
- Vlad, G.; Suciu-Foca, N. Induction of antigen-specific human T suppressor cells by membrane and soluble ILT3. Exp. Mol. Pathol. 2012, 93, 294–301. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Guo, J.; Zhang, H.; Li, Z.; Ren, Y.; Jiang, Y.; Liu, X.; Hu, X. LILRB4 regulates the function of decidual MDSCs via the SHP-2/STAT6 pathway during Toxoplasma gondii infection. Parasit. Vectors 2023, 16, 237. [Google Scholar] [CrossRef]
- Ferreira, L.M.R.; Meissner, T.B.; Tilburgs, T.; Strominger, J.L. HLA-G: At the Interface of Maternal-Fetal Tolerance. Trends Immunol. 2017, 38, 272–286. [Google Scholar] [CrossRef] [PubMed]
- LeMaoult, J.; Zafaranloo, K.; Le Danff, C.; Carosella, E.D. HLA-G up-regulates ILT2, ILT3, ILT4, and KIR2DL4 in antigen presenting cells, NK cells, and T cells. FASEB J 2005, 19, 662–664. [Google Scholar] [CrossRef]
- Zhou, H.; Li, W.M.; Zhang, M.; Liu, Z.R.; Zou, P. Induction of tolerogenic dendritic cells by membrane-bound HLA-G in vitro. Zhongguo Shi Yan Xue Ye Xue Za Zhi 2007, 15, 369–372. [Google Scholar]
- Rochat, M.K.; Ege, M.J.; Plabst, D.; Steinle, J.; Bitter, S.; Braun-Fahrländer, C.; Dalphin, J.C.; Riedler, J.; Roponen, M.; Hirvonen, M.R.; et al. Maternal vitamin D intake during pregnancy increases gene expression of ILT3 and ILT4 in cord blood. Clin. Exp. Allergy 2010, 40, 786–794. [Google Scholar] [CrossRef]
- Lourido, S. Toxoplasma gondii. Trends Parasitol. 2019, 35, 944–945. [Google Scholar] [CrossRef] [PubMed]
- Zhao, X.Y.; Ewald, S.E. The molecular biology and immune control of chronic Toxoplasma gondii infection. J. Clin. Investig. 2020, 130, 3370–3380. [Google Scholar] [CrossRef]
- Li, Z.; Zhao, M.; Li, T.; Zheng, J.; Liu, X.; Jiang, Y.; Zhang, H.; Hu, X. Decidual Macrophage Functional Polarization during Abnormal Pregnancy due to Toxoplasma gondii: Role for LILRB4. Front. Immunol. 2017, 8, 1013. [Google Scholar] [CrossRef] [PubMed]
- Faral-Tello, P.; Pagotto, R.; Bollati-Fogolín, M.; Francia, M.E. Modeling the human placental barrier to understand Toxoplasma gondii’s vertical transmission. Front. Cell Infect. Microbiol. 2023, 13, 1130901. [Google Scholar] [CrossRef]
- Sharma, N.; Atolagbe, O.T.; Ge, Z.; Allison, J.P. LILRB4 suppresses immunity in solid tumors and is a potential target for immunotherapy. J. Exp. Med. 2021, 218, e20201811. [Google Scholar] [CrossRef]
- de Goeje, P.L.; Bezemer, K.; Heuvers, M.E.; Dingemans, A.C.; Groen, H.J.; Smit, E.F.; Hoogsteden, H.C.; Hendriks, R.W.; Aerts, J.G.; Hegmans, J.P. Immunoglobulin-like transcript 3 is expressed by myeloid-derived suppressor cells and correlates with survival in patients with non-small cell lung cancer. Oncoimmunology 2015, 4, e1014242. [Google Scholar] [CrossRef]
- Li, J.; Gao, A.; Zhang, F.; Wang, S.; Wang, J.; Wang, J.; Han, S.; Yang, Z.; Chen, X.; Fang, Y.; et al. ILT3 promotes tumor cell motility and angiogenesis in non-small cell lung cancer. Cancer Lett. 2021, 501, 263–276. [Google Scholar] [CrossRef]
- Kumata, S.; Notsuda, H.; Su, M.T.; Saito-Koyama, R.; Tanaka, R.; Suzuki, Y.; Funahashi, J.; Endo, S.; Yokota, I.; Takai, T.; et al. Prognostic impact of LILRB4 expression on tumor-infiltrating cells in resected non-small cell lung cancer. Thorac. Cancer 2023, 14, 2057–2068. [Google Scholar] [CrossRef]
- Zurli, V.; Wimmer, G.; Cattaneo, F.; Candi, V.; Cencini, E.; Gozzetti, A.; Raspadori, D.; Campoccia, G.; Sanseviero, F.; Bocchia, M.; et al. Ectopic ILT3 controls BCR-dependent activation of Akt in B-cell chronic lymphocytic leukemia. Blood 2017, 130, 2006–2017. [Google Scholar] [CrossRef] [PubMed]
- Paavola, K.J.; Roda, J.M.; Lin, V.Y.; Chen, P.; O’Hollaren, K.P.; Ventura, R.; Crawley, S.C.; Li, B.; Chen, H.H.; Malmersjö, S.; et al. The Fibronectin-ILT3 Interaction Functions as a Stromal Checkpoint that Suppresses Myeloid Cells. Cancer Immunol. Res. 2021, 9, 1283–1297. [Google Scholar] [CrossRef]
- Gui, X.; Deng, M.; Song, H.; Chen, Y.; Xie, J.; Li, Z.; He, L.; Huang, F.; Xu, Y.; Anami, Y.; et al. Disrupting LILRB4/APOE Interaction by an Efficacious Humanized Antibody Reverses T-cell Suppression and Blocks AML Development. Cancer Immunol. Res. 2019, 7, 1244–1257. [Google Scholar] [CrossRef]
- Blackburn, S.D.; Shin, H.; Haining, W.N.; Zou, T.; Workman, C.J.; Polley, A.; Betts, M.R.; Freeman, G.J.; Vignali, D.A.; Wherry, E.J. Coregulation of CD8+ T cell exhaustion by multiple inhibitory receptors during chronic viral infection. Nat. Immunol. 2009, 10, 29–37. [Google Scholar] [CrossRef]
- Zhai, Y.; Franco, L.M.; Atmar, R.L.; Quarles, J.M.; Arden, N.; Bucasas, K.L.; Wells, J.M.; Niño, D.; Wang, X.; Zapata, G.E.; et al. Host Transcriptional Response to Influenza and Other Acute Respiratory Viral Infections--A Prospective Cohort Study. PLoS Pathog. 2015, 11, e1004869. [Google Scholar] [CrossRef]
- Bost, P.; Giladi, A.; Liu, Y.; Bendjelal, Y.; Xu, G.; David, E.; Blecher-Gonen, R.; Cohen, M.; Medaglia, C.; Li, H.; et al. Host-Viral Infection Maps Reveal Signatures of Severe COVID-19 Patients. Cell 2020, 181, 1475–1488.e1412. [Google Scholar] [CrossRef] [PubMed]
- Gu, X.; Laouar, A.; Wan, J.; Daheshia, M.; Lieberman, J.; Yokoyama, W.M.; Katz, H.R.; Manjunath, N. The gp49B1 inhibitory receptor regulates the IFN-gamma responses of T cells and NK cells. J. Immunol. 2003, 170, 4095–4101. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.L.; Chu, D.T.; Dokun, A.O.; Yokoyama, W.M. Inducible expression of the gp49B inhibitory receptor on NK cells. J. Immunol. 2000, 164, 5215–5220. [Google Scholar] [CrossRef]
- Lu, H.K.; Rentero, C.; Raftery, M.J.; Borges, L.; Bryant, K.; Tedla, N. Leukocyte Ig-like receptor B4 (LILRB4) is a potent inhibitor of FcgammaRI-mediated monocyte activation via dephosphorylation of multiple kinases. J. Biol. Chem. 2009, 284, 34839–34848. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Z.; Qin, J.J.; Zhang, Y.; Cheng, W.L.; Ji, Y.X.; Gong, F.H.; Zhu, X.Y.; Zhang, Y.; She, Z.G.; Huang, Z.; et al. LILRB4 deficiency aggravates the development of atherosclerosis and plaque instability by increasing the macrophage inflammatory response via NF-κB signaling. Clin. Sci. 2017, 131, 2275–2288. [Google Scholar] [CrossRef] [PubMed]
- Lu, Y.; Jiang, Z.; Dai, H.; Miao, R.; Shu, J.; Gu, H.; Liu, X.; Huang, Z.; Yang, G.; Chen, A.F.; et al. Hepatic leukocyte immunoglobulin-like receptor B4 (LILRB4) attenuates nonalcoholic fatty liver disease via SHP1-TRAF6 pathway. Hepatology 2018, 67, 1303–1319. [Google Scholar] [CrossRef] [PubMed]
- Mitsune, A.; Yamada, M.; Fujino, N.; Numakura, T.; Ichikawa, T.; Suzuki, A.; Matsumoto, S.; Mitsuhashi, Y.; Itakura, K.; Makiguchi, T.; et al. Upregulation of leukocyte immunoglobulin-like receptor B4 on interstitial macrophages in COPD; their possible protective role against emphysema formation. Respir. Res. 2021, 22, 232. [Google Scholar] [CrossRef] [PubMed]
- Qiu, T.; Zhou, J.; Wang, T.; Chen, Z.; Ma, X.; Zhang, L.; Zou, J. Leukocyte immunoglobulin-like receptor B4 deficiency exacerbates acute lung injury via NF-κB signaling in bone marrow-derived macrophages. Biosci. Rep. 2019, 39, BSR20181888. [Google Scholar] [CrossRef] [PubMed]
- Zöller, T.; Attaai, A.; Potru, P.S.; Ruß, T.; Spittau, B. Aged Mouse Cortical Microglia Display an Activation Profile Suggesting Immunotolerogenic Functions. Int. J. Mol. Sci. 2018, 19, 706. [Google Scholar] [CrossRef] [PubMed]
- Baik, S.H.; Kang, S.; Lee, W.; Choi, H.; Chung, S.; Kim, J.I.; Mook-Jung, I. A Breakdown in Metabolic Reprogramming Causes Microglia Dysfunction in Alzheimer’s Disease. Cell Metab. 2019, 30, 493–507.e496. [Google Scholar] [CrossRef] [PubMed]
- Sarlus, H.; Heneka, M.T. Microglia in Alzheimer’s disease. J. Clin. Investig. 2017, 127, 3240–3249. [Google Scholar] [CrossRef]
- Katz, H.R. Inhibition of anaphylactic inflammation by the gp49B1 receptor on mast cells. Mol. Immunol. 2002, 38, 1301–1305. [Google Scholar] [CrossRef]
- Katz, H.R. Inhibition of pathologic inflammation by leukocyte Ig-like receptor B4 and related inhibitory receptors. Immunol. Rev. 2007, 217, 222–230. [Google Scholar] [CrossRef] [PubMed]
- Malbec, O.; Daëron, M. The mast cell IgG receptors and their roles in tissue inflammation. Immunol. Rev. 2007, 217, 206–221. [Google Scholar] [CrossRef] [PubMed]
- Daheshia, M.; Friend, D.S.; Grusby, M.J.; Austen, K.F.; Katz, H.R. Increased severity of local and systemic anaphylactic reactions in gp49B1-deficient mice. J. Exp. Med. 2001, 194, 227–234. [Google Scholar] [CrossRef] [PubMed]
- Hitomi, K.; Tahara-Hanaoka, S.; Someya, S.; Fujiki, A.; Tada, H.; Sugiyama, T.; Shibayama, S.; Shibuya, K.; Shibuya, A. An immunoglobulin-like receptor, Allergin-1, inhibits immunoglobulin E-mediated immediate hypersensitivity reactions. Nat. Immunol. 2010, 11, 601–607. [Google Scholar] [CrossRef] [PubMed]
- Norris, H.H.; Peterson, M.E.; Stebbins, C.C.; McConchie, B.W.; Bundoc, V.G.; Trivedi, S.; Hodges, M.G.; Anthony, R.M.; Urban, J.F., Jr.; Long, E.O.; et al. Inhibitory receptor gp49B regulates eosinophil infiltration during allergic inflammation. J. Leukoc. Biol. 2007, 82, 1531–1541. [Google Scholar] [CrossRef] [PubMed]
- Kolkhir, P.; Elieh-Ali-Komi, D.; Metz, M.; Siebenhaar, F.; Maurer, M. Understanding human mast cells: Lesson from therapies for allergic and non-allergic diseases. Nat. Rev. Immunol. 2022, 22, 294–308. [Google Scholar] [CrossRef]
- Kanagaratham, C.; El Ansari, Y.S.; Lewis, O.L.; Oettgen, H.C. IgE and IgG Antibodies as Regulators of Mast Cell and Basophil Functions in Food Allergy. Front. Immunol. 2020, 11, 603050. [Google Scholar] [CrossRef]
- Katz, H.R. Inhibitory receptors and allergy. Curr. Opin. Immunol. 2002, 14, 698–704. [Google Scholar] [CrossRef]
- Oka, T.; Akazawa, H.; Naito, A.T.; Komuro, I. Angiogenesis and cardiac hypertrophy: Maintenance of cardiac function and causative roles in heart failure. Circ. Res. 2014, 114, 565–571. [Google Scholar] [CrossRef]
- Rafiq, K.; Kolpakov, M.A.; Seqqat, R.; Guo, J.; Guo, X.; Qi, Z.; Yu, D.; Mohapatra, B.; Zutshi, N.; An, W.; et al. c-Cbl inhibition improves cardiac function and survival in response to myocardial ischemia. Circulation 2014, 129, 2031–2043. [Google Scholar] [CrossRef]
- Zhou, H.; Li, N.; Yuan, Y.; Jin, Y.G.; Wu, Q.; Yan, L.; Bian, Z.Y.; Deng, W.; Shen, D.F.; Li, H.; et al. Leukocyte immunoglobulin-like receptor B4 protects against cardiac hypertrophy via SHP-2-dependent inhibition of the NF-κB pathway. J. Mol. Med. 2020, 98, 691–705. [Google Scholar] [CrossRef] [PubMed]





Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Xiang, Z.; Yin, X.; Wei, L.; Peng, M.; Zhu, Q.; Lu, X.; Guo, J.; Zhang, J.; Li, X.; Zou, Y. LILRB4 Checkpoint for Immunotherapy: Structure, Mechanism and Disease Targets. Biomolecules 2024, 14, 187. https://doi.org/10.3390/biom14020187
Xiang Z, Yin X, Wei L, Peng M, Zhu Q, Lu X, Guo J, Zhang J, Li X, Zou Y. LILRB4 Checkpoint for Immunotherapy: Structure, Mechanism and Disease Targets. Biomolecules. 2024; 14(2):187. https://doi.org/10.3390/biom14020187
Chicago/Turabian StyleXiang, Zhiqing, Xiangli Yin, Leiyan Wei, Manqing Peng, Quan Zhu, Xiaofang Lu, Junshuang Guo, Jing Zhang, Xin Li, and Yizhou Zou. 2024. "LILRB4 Checkpoint for Immunotherapy: Structure, Mechanism and Disease Targets" Biomolecules 14, no. 2: 187. https://doi.org/10.3390/biom14020187