
Identifying OGN as a Biomarker Covering Multiple Pathogenic Pathways for Diagnosing Heart Failure: From Machine Learning to Mechanism Interpretation


Abstract
:1. Introduction
2. Materials and Methods
2.1. Gathering and Processing of Gene Expression Profiling Datasets
2.2. Partial Least Squares Discriminant Analysis, DEGs Identification, and KEGG Pathway Enrichment Analysis
2.3. Weighted Gene Co-Expression Network Analysis on DEGs
2.4. Multiscale Embedded Gene Co-Expression Network Analysis on DEGs
2.5. DO, GO, and KEGG Enrichment Analyses on Crucial Gene Modules
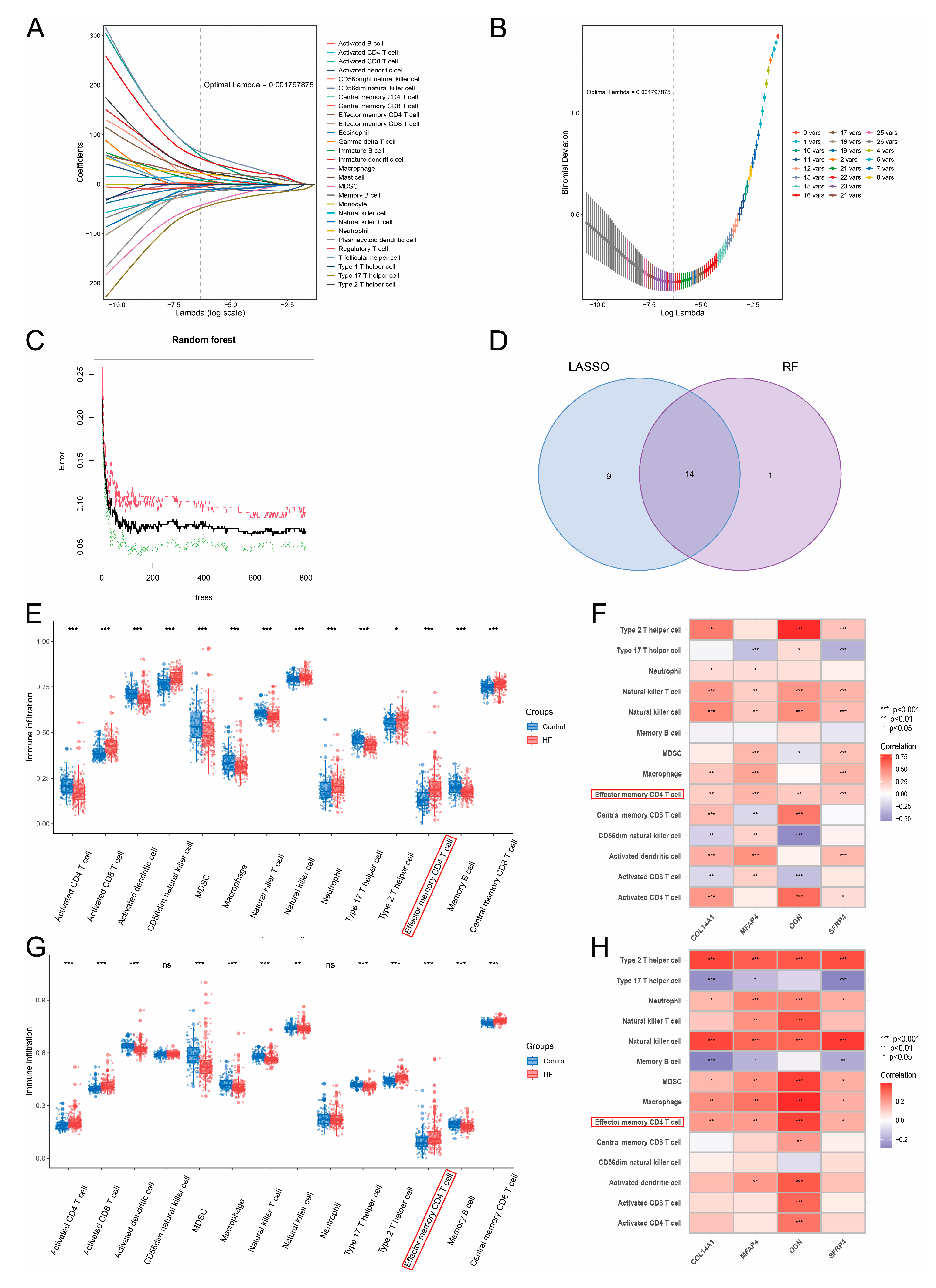
2.6. Feature Selection via Machine Learning-Based Integration
2.7. Construction of Protein-Protein Interaction Network
2.8. Diagnostic Abilities, Expression Levels, and Correlation Pattern of Hub Genes
2.9. Single-Cell RNA-seq Data Processing
2.10. GSVA and GSEA of Hub Genes
2.11. Expression Levels of Predicted TFs and Their Interaction with Hub Gene-Related Pathways
2.12. Evaluation of Immune Cell Characteristics in HF via ssGSEA and Identifying Immune Cell Types Highly Correlated with Hub Genes
2.13. Molecular Docking of Predictive Drugs Targeting Hub Genes and TFs
2.14. Quantitative Reverse Transcription PCR
2.15. Two-Sample Mendel Randomization Analysis
2.16. Statistical Analysis
3. Results
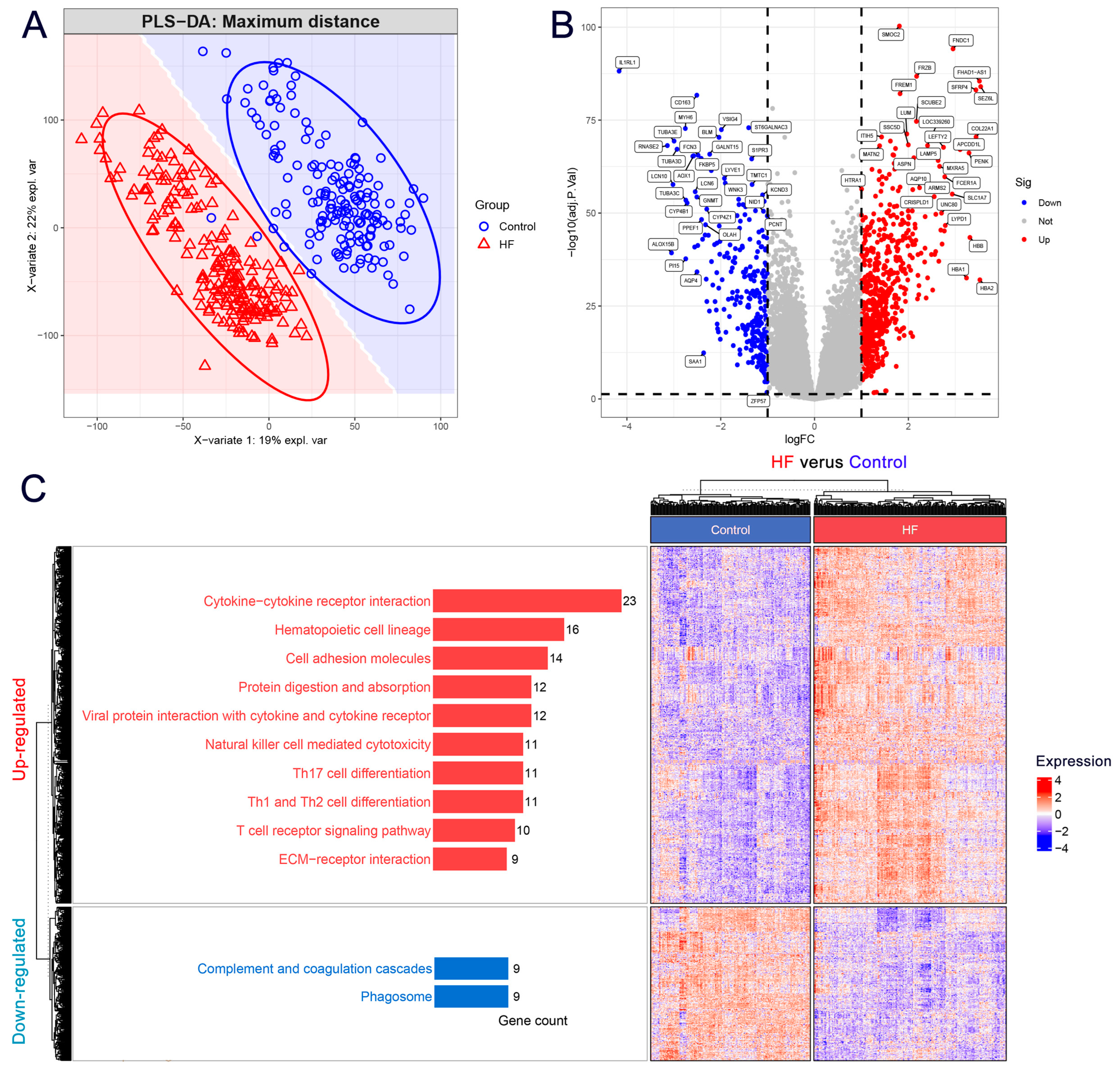
3.1. DEGs Mainly Participated in the Extracellular Matrix and Immune-Related Pathways in HF
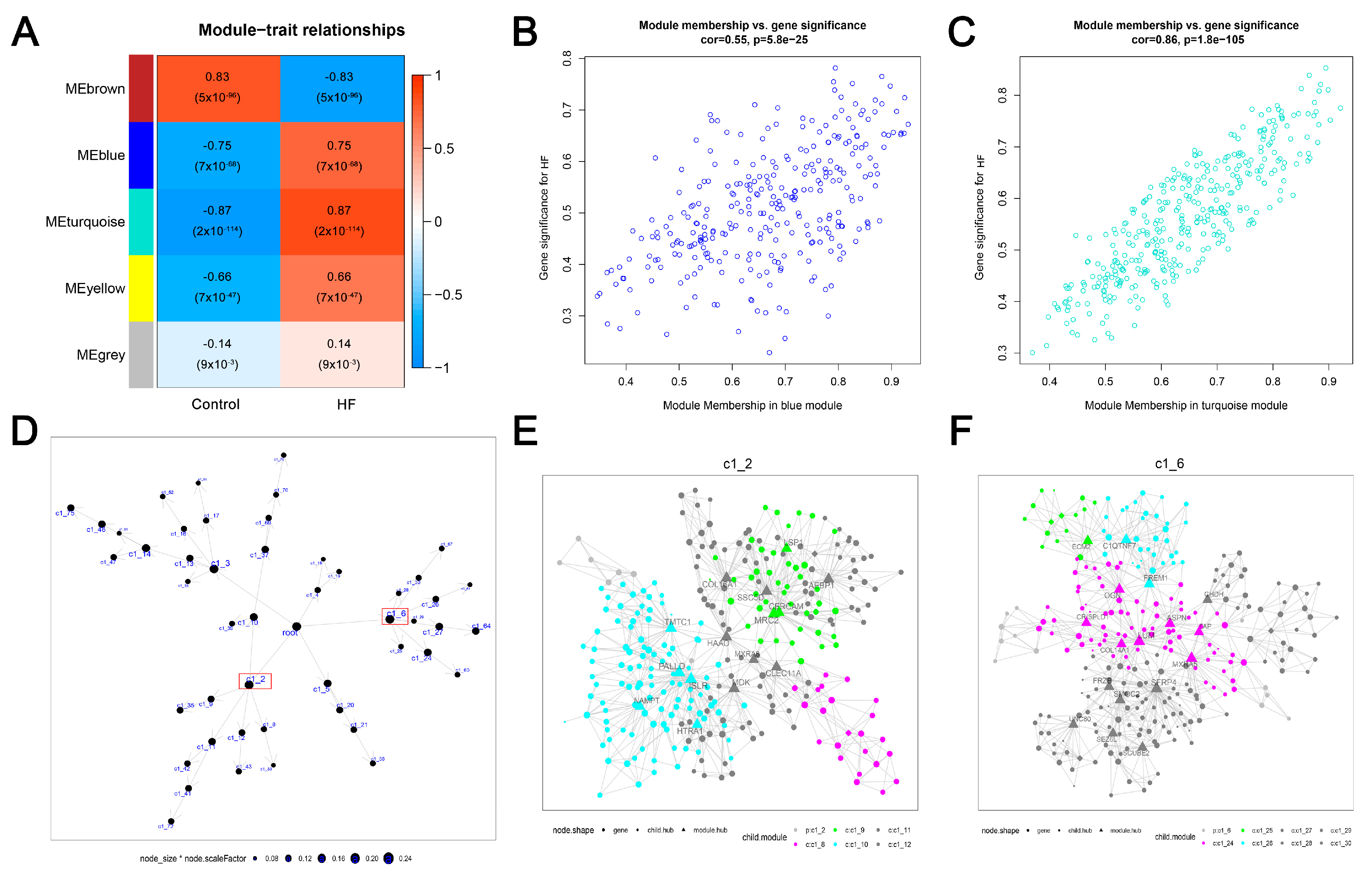
3.2. Two Crucial Modules Strongly Related to HF Were Identified by WGCNA on DEGs
3.3. Two Largest Crucial Modules of HF Were Identified by MEGENA on DEGs
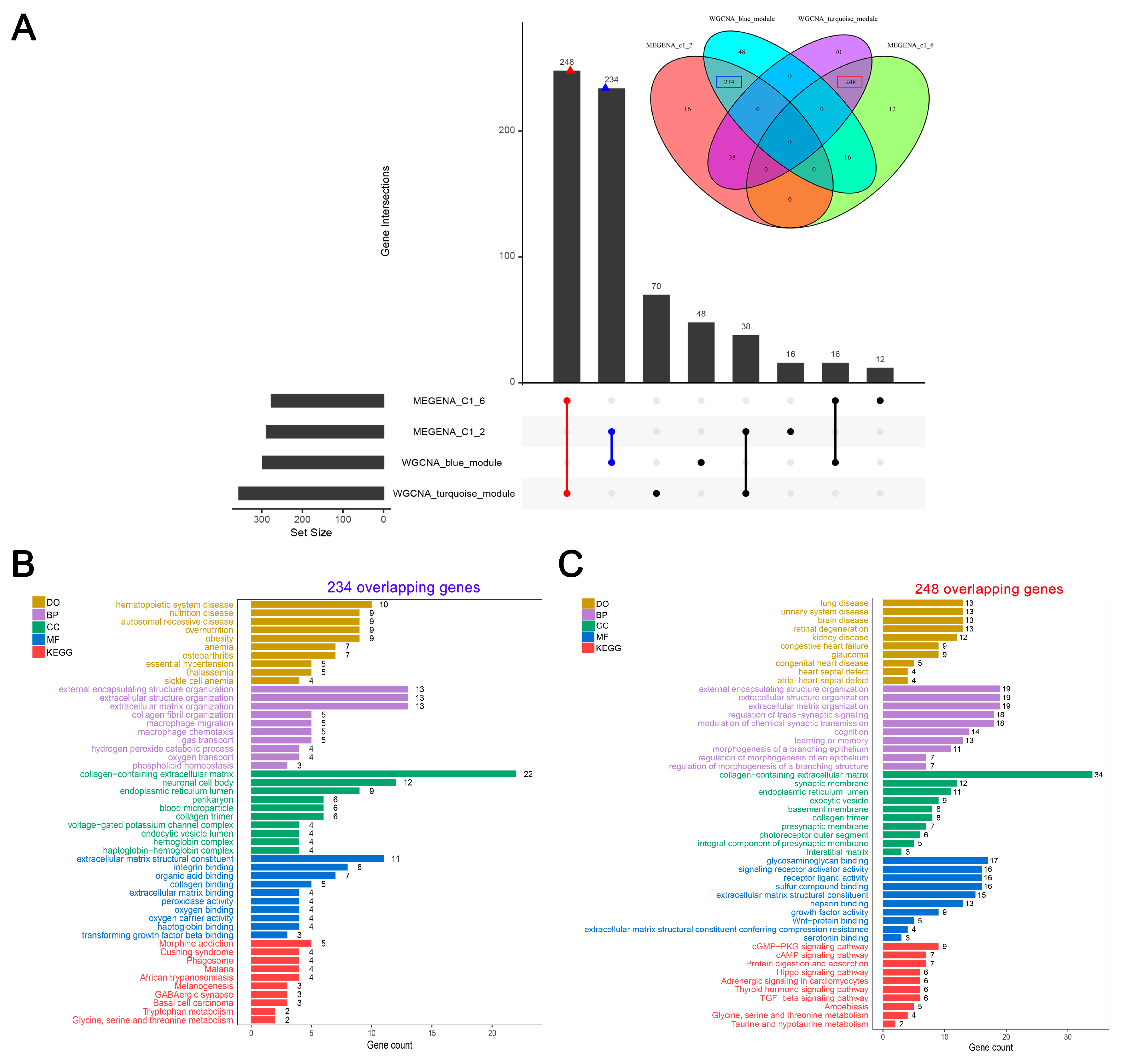
3.4. A Large Set of Overlaps between Crucial Modules Showed Close Associations with HF-Related Biological Functions and Pathways
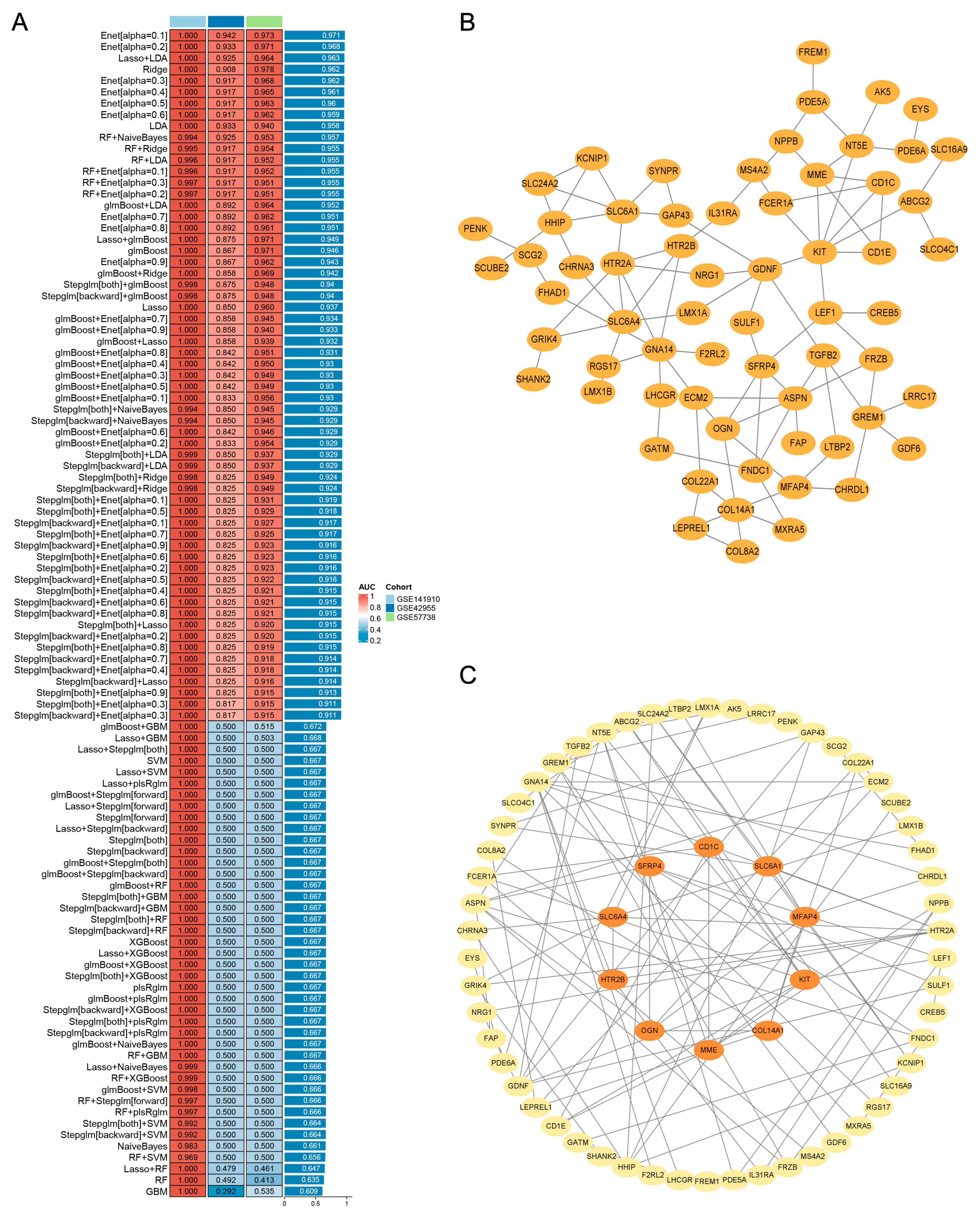
3.5. Machine Learning-Based Integration and PPI Network Analysis Screened 10 Pivotal Genes of HF
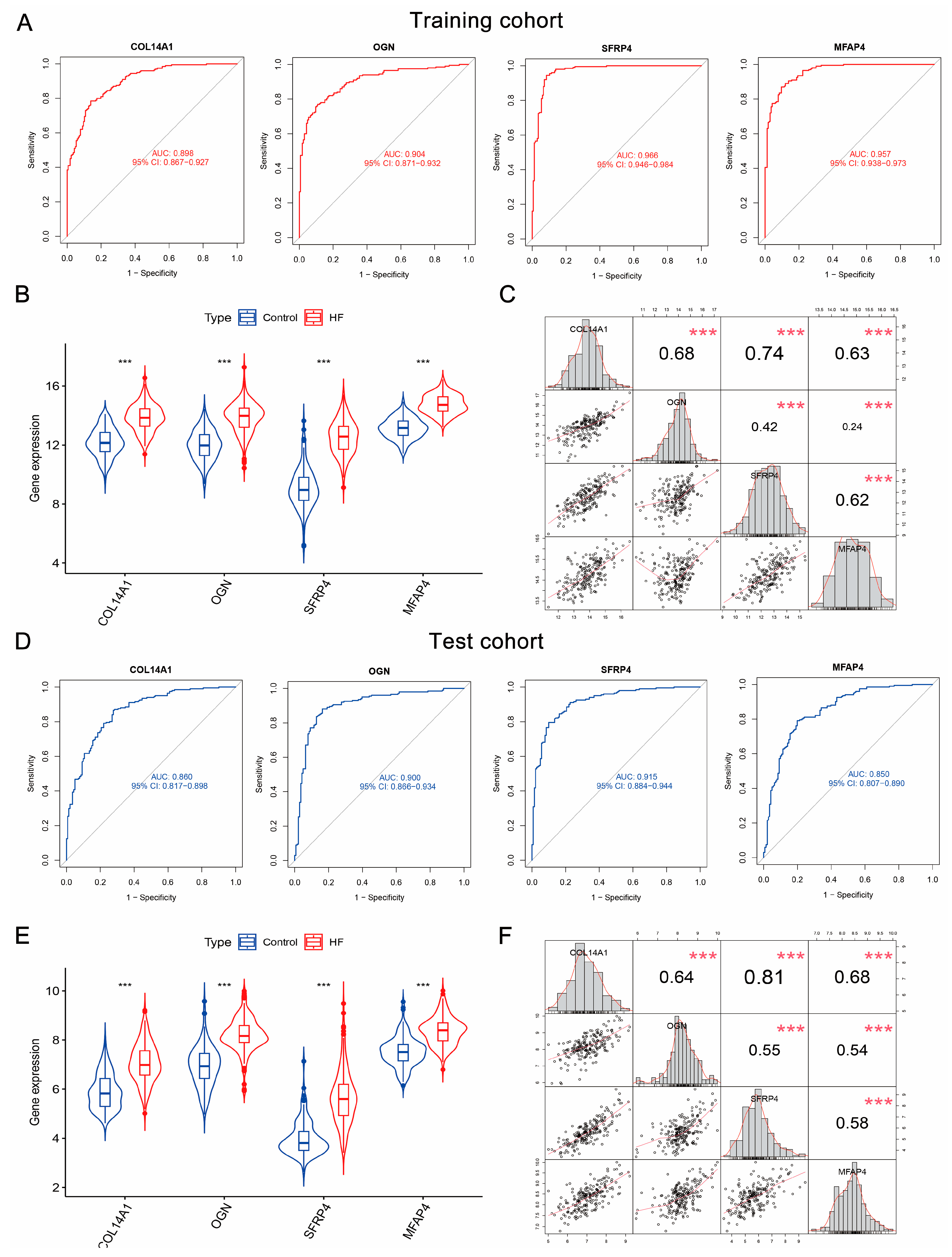
3.6. COL14A1, OGN, MFAP4, and SFRP4 Were Hub Genes as Candidate Diagnostic Biomarkers of HF
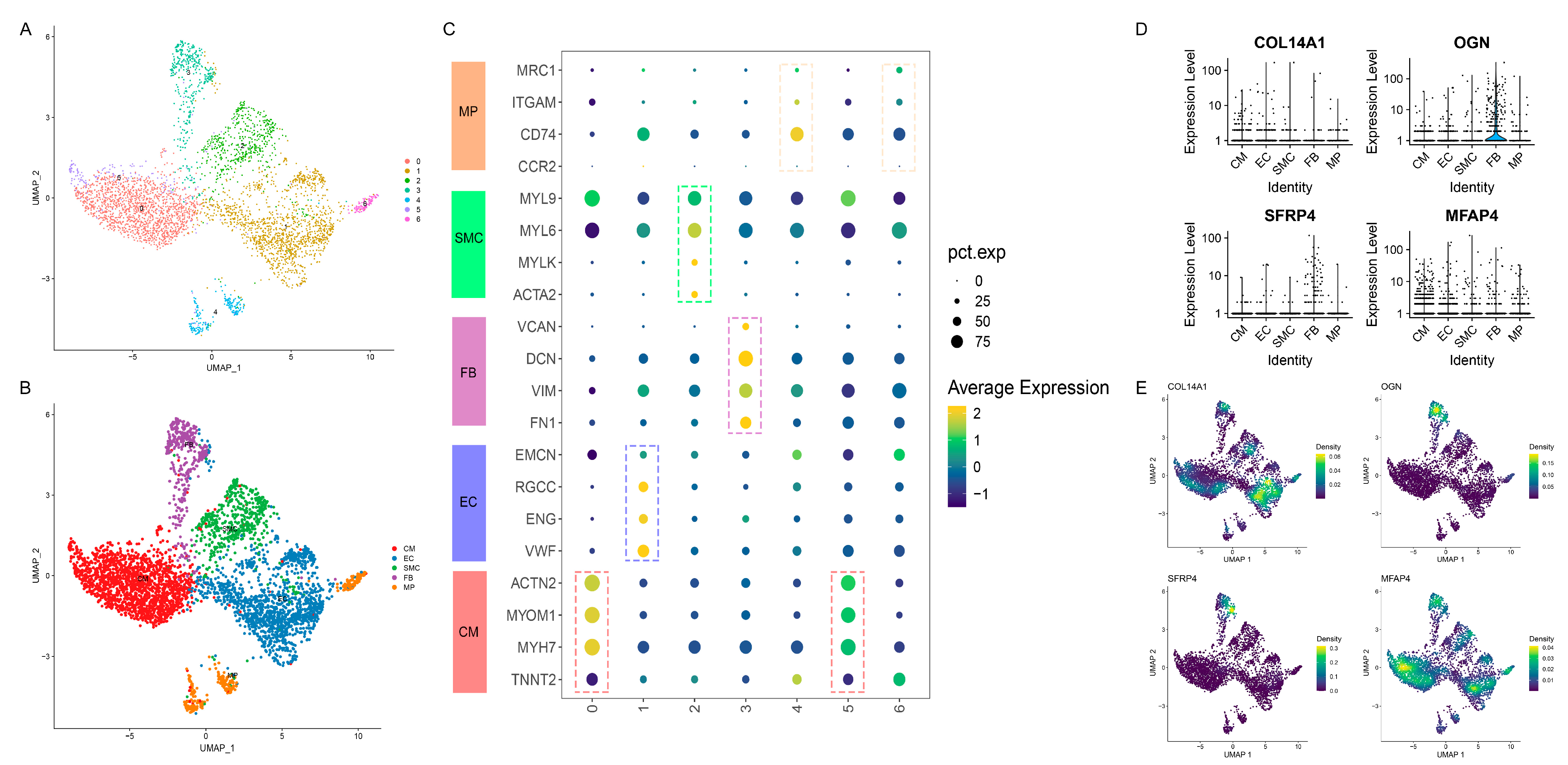
3.7. Distinct Cell-Specific Expression Patterns of Hub Genes in HF via scRNA-seq Analysis
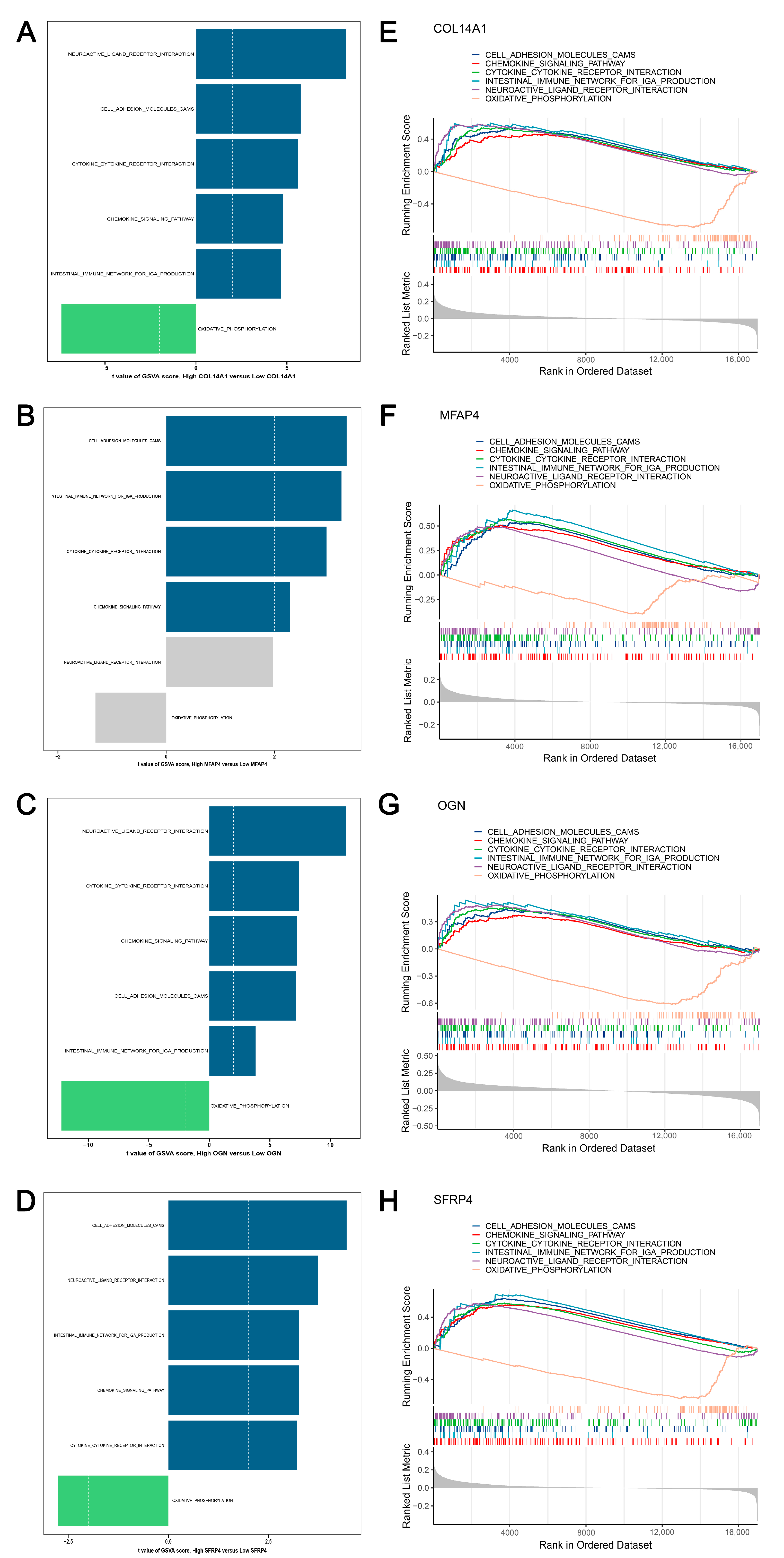
3.8. GSVA and GSEA Revealed the Activated and Suppressed Pathways Co-Regulated by Hub Genes in HF
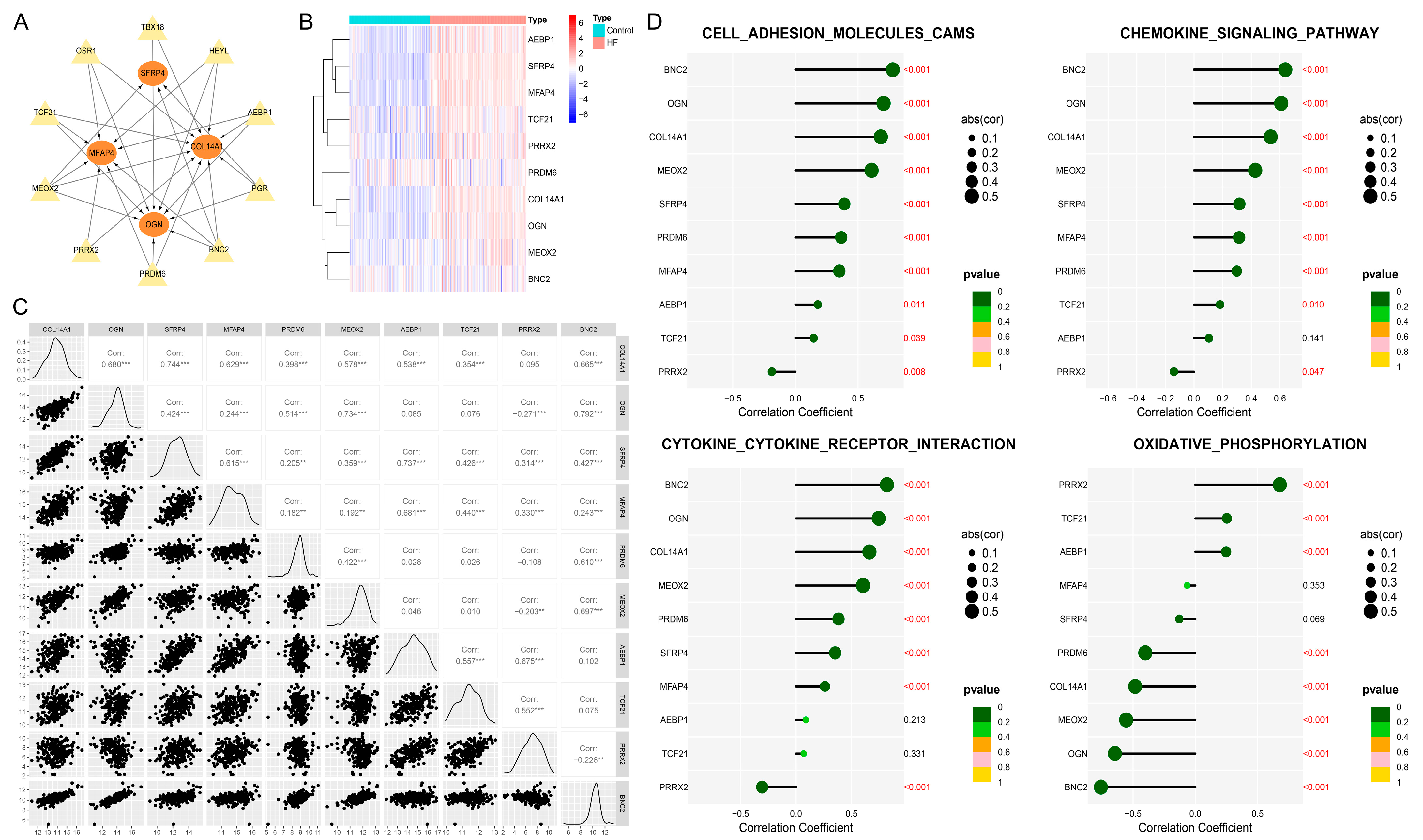
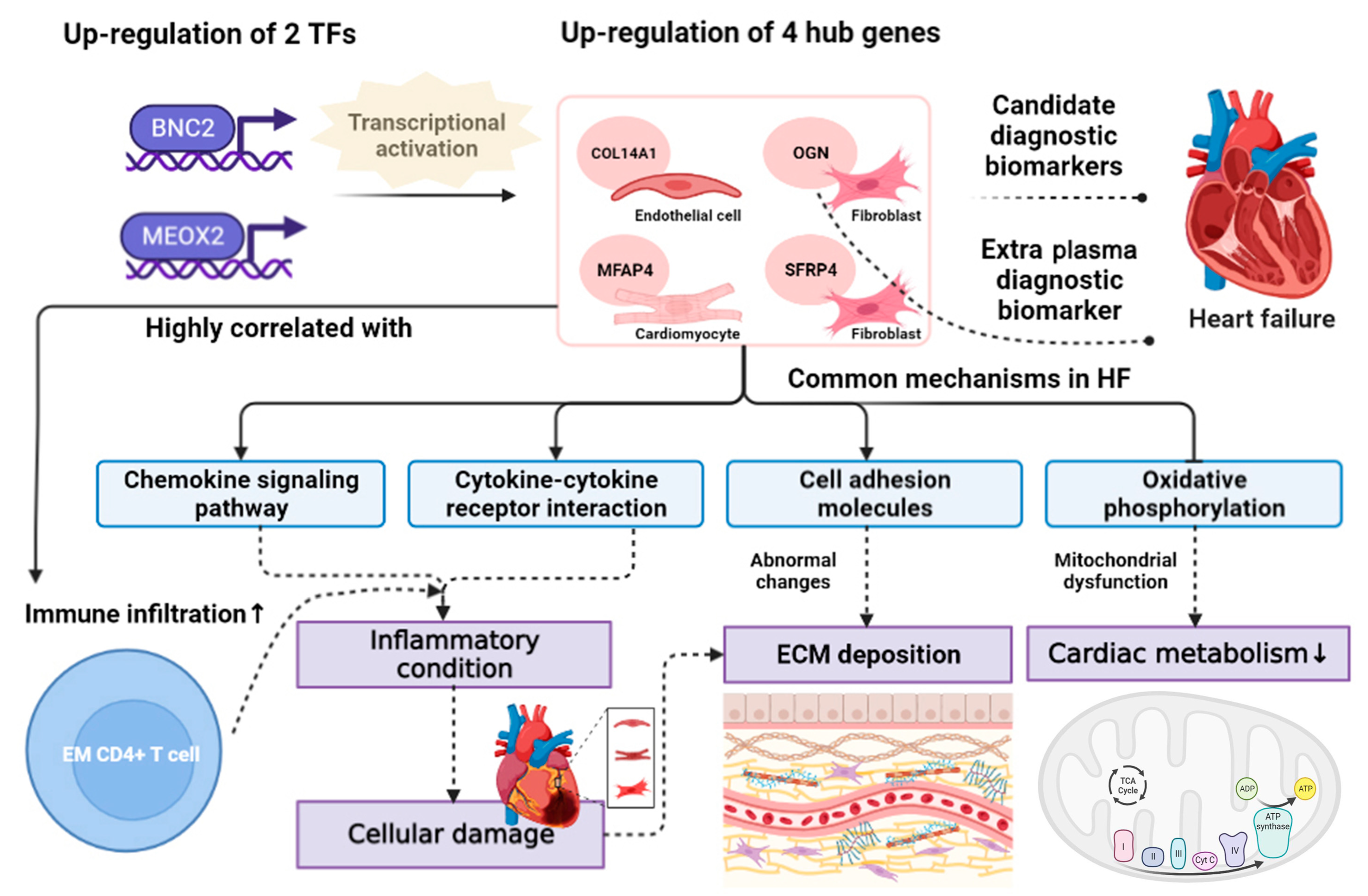
3.9. BNC2 and MEOX2 Were TFs Actively Targeting Hub Genes
3.10. Elevated Infiltration Levels of Effector Memory CD4+ T Cells Were Highly Related to Hub Genes
3.11. Small-Molecule Agents Targeting Active TFs and Hub Genes Could Serve as Candidate Drugs for Alleviating HF
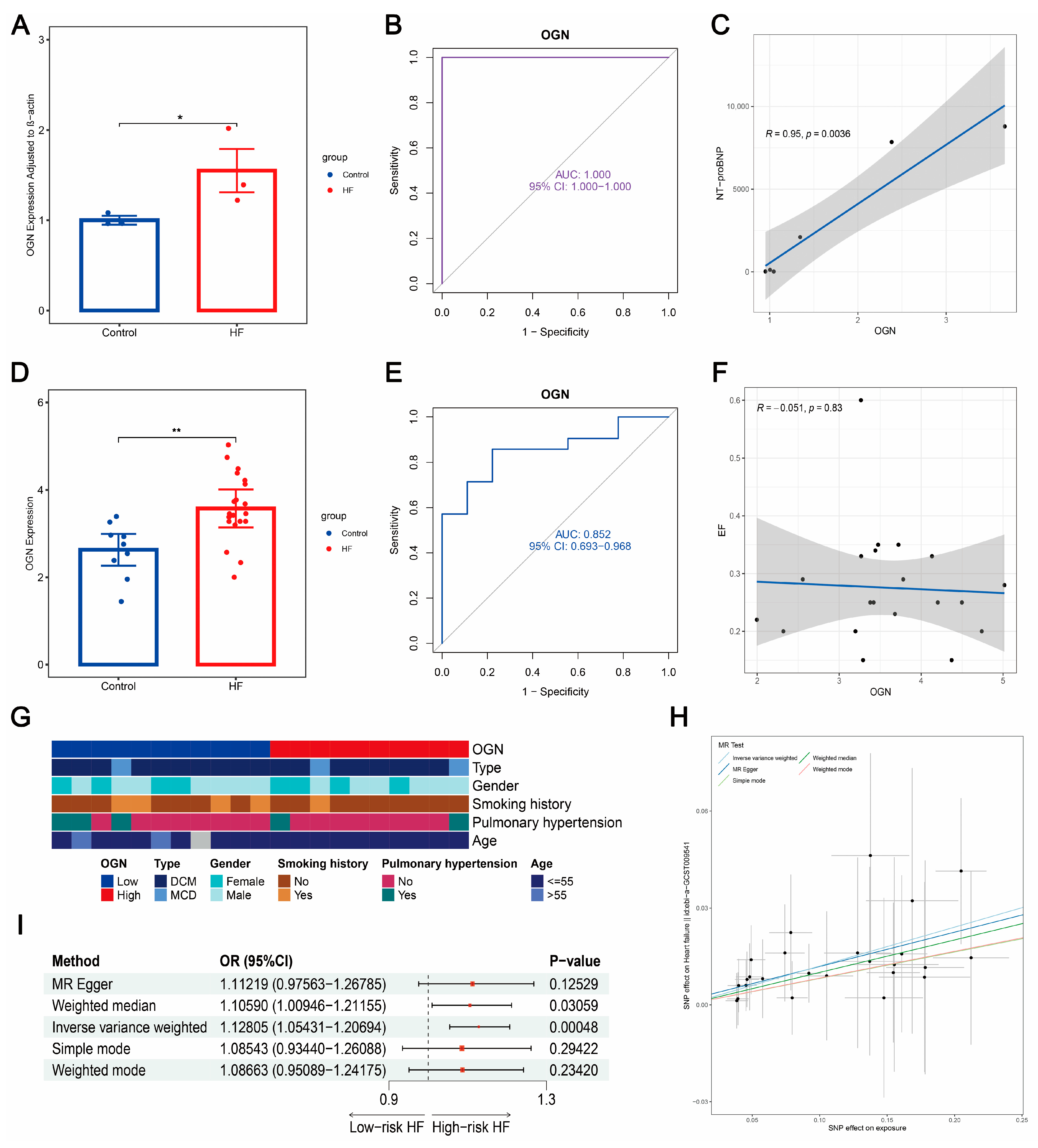
3.12. Plasma OGN Elevated in HF with Robust Diagnostic Value and Positive Causal Correlation to HF Risk
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- McDonagh, T.A.; Metra, M.; Adamo, M.; Gardner, R.S.; Baumbach, A.; Böhm, M.; Burri, H.; Butler, J.; Čelutkienė, J.; Chioncel, O.; et al. 2021 ESC Guidelines for the diagnosis and treatment of acute and chronic heart failure: Developed by the Task Force for the diagnosis and treatment of acute and chronic heart failure of the European Society of Cardiology (ESC). With the special contribution of the Heart Failure Association (HFA) of the ESC. Eur. J. Heart Fail. 2022, 24, 4–131. [Google Scholar] [PubMed]
- Castiglione, V.; Aimo, A.; Vergaro, G.; Saccaro, L.; Passino, C.; Emdin, M. Biomarkers for the diagnosis and management of heart failure. Heart Fail. Rev. 2022, 27, 625–643. [Google Scholar] [CrossRef] [PubMed]
- Savarese, G.; Lund, L.H. Global Public Health Burden of Heart Failure. Card. Fail. Rev. 2017, 3, 7–11. [Google Scholar] [CrossRef] [PubMed]
- Bozkurt, B.; Coats, A.J.; Tsutsui, H.; Abdelhamid, M.; Adamopoulos, S.; Albert, N.; Anker, S.D.; Atherton, J.; Böhm, M.; Butler, J.; et al. Universal Definition and Classification of Heart Failure: A Report of the Heart Failure Society of America, Heart Failure Association of the European Society of Cardiology, Japanese Heart Failure Society and Writing Committee of the Universal Definition of Heart Failure. J. Card. Fail. 2021, 27, 387–413. [Google Scholar]
- Sarhene, M.; Wang, Y.; Wei, J.; Huang, Y.; Li, M.; Li, L.; Acheampong, E.; Zhengcan, Z.; Xiaoyan, Q.; Yunsheng, X.; et al. Biomarkers in heart failure: The past, current and future. Heart Fail. Rev. 2019, 24, 867–903. [Google Scholar] [CrossRef]
- Chow, S.L.; Maisel, A.S.; Anand, I.; Bozkurt, B.; de Boer, R.A.; Felker, G.M.; Fonarow, G.C.; Greenberg, B.; Januzzi, J.J.; Kiernan, M.S.; et al. Role of Biomarkers for the Prevention, Assessment, and Management of Heart Failure: A Scientific Statement From the American Heart Association. Circulation 2017, 135, e1054–e1091. [Google Scholar] [CrossRef]
- Sun, C.; Ni, M.; Song, B.; Cao, L. Circulating Circular RNAs: Novel Biomarkers for Heart Failure. Front. Pharmacol. 2020, 11, 560537. [Google Scholar] [CrossRef]
- Takase, H.; Dohi, Y. Kidney function crucially affects B-type natriuretic peptide (BNP), N-terminal proBNP and their relationship. Eur. J. Clin. Investig. 2014, 44, 303–308. [Google Scholar] [CrossRef]
- Kucher, N.; Printzen, G.; Goldhaber, S.Z. Prognostic role of brain natriuretic peptide in acute pulmonary embolism. Circulation 2003, 107, 2545–2547. [Google Scholar] [CrossRef]
- Madamanchi, C.; Alhosaini, H.; Sumida, A.; Runge, M.S. Obesity and natriuretic peptides, BNP and NT-proBNP: Mechanisms and diagnostic implications for heart failure. Int. J. Cardiol. 2014, 176, 611–617. [Google Scholar] [CrossRef]
- Braunwald, E. Biomarkers in heart failure. N. Engl. J. Med. 2008, 358, 2148–2159. [Google Scholar] [CrossRef]
- Ahmad, T.; Fiuzat, M.; Pencina, M.J.; Geller, N.L.; Zannad, F.; Cleland, J.G.; Snider, J.V.; Blankenberg, S.; Adams, K.F.; Redberg, R.F.; et al. Charting a roadmap for heart failure biomarker studies. JACC Heart Fail. 2014, 2, 477–488. [Google Scholar] [CrossRef]
- Piek, A.; Du, W.; de Boer, R.A.; Silljé, H. Novel heart failure biomarkers: Why do we fail to exploit their potential? Crit. Rev. Clin. Lab. Sci. 2018, 55, 246–263. [Google Scholar] [CrossRef]
- Savic-Radojevic, A.; Pljesa-Ercegovac, M.; Matic, M.; Simic, D.; Radovanovic, S.; Simic, T. Novel Biomarkers of Heart Failure. Adv. Clin. Chem. 2017, 79, 93–152. [Google Scholar]
- Weinberg, E.O.; Shimpo, M.; Hurwitz, S.; Tominaga, S.; Rouleau, J.L.; Lee, R.T. Identification of serum soluble ST2 receptor as a novel heart failure biomarker. Circulation 2003, 107, 721–726. [Google Scholar] [CrossRef] [PubMed]
- Yu, Y.D.; Xue, Y.T.; Li, Y. Identification and verification of feature biomarkers associated in heart failure by bioinformatics analysis. Sci. Rep. 2023, 13, 3488. [Google Scholar] [CrossRef]
- Kolur, V.; Vastrad, B.; Vastrad, C.; Kotturshetti, S.; Tengli, A. Identification of candidate biomarkers and therapeutic agents for heart failure by bioinformatics analysis. BMC Cardiovasc. Disord. 2021, 21, 329. [Google Scholar] [CrossRef] [PubMed]
- Fan, S.; Hu, Y. Integrative analyses of biomarkers and pathways for heart failure. BMC Med. Genom. 2022, 15, 72. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.; Zhang, W.; Wei, C.; Zhang, Z.; Yi, D.; Peng, X.; Peng, J.; Yin, R.; Zheng, Z.; Qi, H.; et al. Weighted correlation network bioinformatics uncovers a key molecular biosignature driving the left-sided heart failure. BMC Med. Genom. 2020, 13, 93. [Google Scholar] [CrossRef] [PubMed]
- Flam, E.; Jang, C.; Murashige, D.; Yang, Y.; Morley, M.P.; Jung, S.; Kantner, D.S.; Pepper, H.; Bedi, K.J.; Brandimarto, J.; et al. Integrated landscape of cardiac metabolism in end-stage human nonischemic dilated cardiomyopathy. Nat. Cardiovasc. Res. 2022, 1, 817–829. [Google Scholar] [CrossRef] [PubMed]
- Molina-Navarro, M.M.; Roselló-Lletí, E.; Ortega, A.; Tarazón, E.; Otero, M.; Martínez-Dolz, L.; Lago, F.; González-Juanatey, J.R.; España, F.; García-Pavía, P.; et al. Differential gene expression of cardiac ion channels in human dilated cardiomyopathy. PLoS ONE 2013, 8, e79792. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Morley, M.; Brandimarto, J.; Hannenhalli, S.; Hu, Y.; Ashley, E.A.; Tang, W.H.; Moravec, C.S.; Margulies, K.B.; Cappola, T.P.; et al. RNA-Seq identifies novel myocardial gene expression signatures of heart failure. Genomics 2015, 105, 83–89. [Google Scholar] [CrossRef] [PubMed]
- Hua, X.; Wang, Y.Y.; Jia, P.; Xiong, Q.; Hu, Y.; Chang, Y.; Lai, S.; Xu, Y.; Zhao, Z.; Song, J. Multi-level transcriptome sequencing identifies COL1A1 as a candidate marker in human heart failure progression. BMC Med. 2020, 18, 2. [Google Scholar] [CrossRef] [PubMed]
- Kerr, M.K. Linear models for microarray data analysis: Hidden similarities and differences. J. Comput. Biol. 2003, 10, 891–901. [Google Scholar] [CrossRef] [PubMed]
- Rohart, F.; Gautier, B.; Singh, A.; Lê, C.K. mixOmics: An R package for ‘omics feature selection and multiple data integration. PLoS Comput. Biol. 2017, 13, e1005752. [Google Scholar] [CrossRef]
- Iqbal, N.; Kumar, P. Integrated COVID-19 Predictor: Differential expression analysis to reveal potential biomarkers and prediction of coronavirus using RNA-Seq profile data. Comput. Biol. Med. 2022, 147, 105684. [Google Scholar] [CrossRef]
- Yu, G.; Wang, L.G.; Han, Y.; He, Q.Y. clusterProfiler: An R package for comparing biological themes among gene clusters. OMICS 2012, 16, 284–287. [Google Scholar] [CrossRef]
- Langfelder, P.; Horvath, S. WGCNA: An R package for weighted correlation network analysis. BMC Bioinform. 2008, 9, 559. [Google Scholar] [CrossRef] [PubMed]
- Song, W.M.; Zhang, B. Multiscale Embedded Gene Co-expression Network Analysis. PLoS Comput. Biol. 2015, 11, e1004574. [Google Scholar] [CrossRef] [PubMed]
- Cao, J.; Liu, Z.; Liu, J.; Li, C.; Zhang, G.; Shi, R. Bioinformatics Analysis and Identification of Genes and Pathways in Ischemic Cardiomyopathy. Int. J. Gen. Med. 2021, 14, 5927–5937. [Google Scholar] [CrossRef]
- Yu, G.; Wang, L.G.; Yan, G.R.; He, Q.Y. DOSE: An R/Bioconductor package for disease ontology semantic and enrichment analysis. Bioinformatics 2015, 31, 608–609. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Liu, L.; Weng, S.; Guo, C.; Dang, Q.; Xu, H.; Wang, L.; Lu, T.; Zhang, Y.; Sun, Z.; et al. Machine learning-based integration develops an immune-derived lncRNA signature for improving outcomes in colorectal cancer. Nat. Commun. 2022, 13, 816. [Google Scholar] [CrossRef] [PubMed]
- Shannon, P.; Markiel, A.; Ozier, O.; Baliga, N.S.; Wang, J.T.; Ramage, D.; Amin, N.; Schwikowski, B.; Ideker, T. Cytoscape: A software environment for integrated models of biomolecular interaction networks. Genome Res. 2003, 13, 2498–2504. [Google Scholar] [CrossRef]
- Szklarczyk, D.; Franceschini, A.; Wyder, S.; Forslund, K.; Heller, D.; Huerta-Cepas, J.; Simonovic, M.; Roth, A.; Santos, A.; Tsafou, K.P.; et al. STRING v10: Protein-protein interaction networks, integrated over the tree of life. Nucleic Acids Res. 2015, 43, D447–D452. [Google Scholar] [CrossRef]
- Chin, C.H.; Chen, S.H.; Wu, H.H.; Ho, C.W.; Ko, M.T.; Lin, C.Y. cytoHubba: Identifying hub objects and sub-networks from complex interactome. BMC Syst. Biol. 2014, 8 (Suppl. S4), S11. [Google Scholar] [CrossRef]
- Robin, X.; Turck, N.; Hainard, A.; Tiberti, N.; Lisacek, F.; Sanchez, J.C.; Müller, M. pROC: An open-source package for R and S+ to analyze and compare ROC curves. BMC Bioinform. 2011, 12, 77. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Yu, P.; Zhou, B.; Song, J.; Li, Z.; Zhang, M.; Guo, G.; Wang, Y.; Chen, X.; Han, L.; et al. Single-cell reconstruction of the adult human heart during heart failure and recovery reveals the cellular landscape underlying cardiac function. Nat. Cell Biol. 2020, 22, 108–119. [Google Scholar] [CrossRef]
- Hao, Y.; Hao, S.; Andersen-Nissen, E.; Mauck, W.R.; Zheng, S.; Butler, A.; Lee, M.J.; Wilk, A.J.; Darby, C.; Zager, M.; et al. Integrated analysis of multimodal single-cell data. Cell 2021, 184, 3573–3587. [Google Scholar] [CrossRef]
- Aibar, S.; González-Blas, C.B.; Moerman, T.; Huynh-Thu, V.A.; Imrichova, H.; Hulselmans, G.; Rambow, F.; Marine, J.C.; Geurts, P.; Aerts, J.; et al. SCENIC: Single-cell regulatory network inference and clustering. Nat. Methods 2017, 14, 1083–1086. [Google Scholar] [CrossRef]
- Alquicira-Hernandez, J.; Powell, J.E. Nebulosa recovers single-cell gene expression signals by kernel density estimation. Bioinformatics 2021, 37, 2485–2487. [Google Scholar] [CrossRef]
- Liberzon, A.; Birger, C.; Thorvaldsdóttir, H.; Ghandi, M.; Mesirov, J.P.; Tamayo, P. The Molecular Signatures Database (MSigDB) hallmark gene set collection. Cell Syst. 2015, 1, 417–425. [Google Scholar] [CrossRef]
- Hänzelmann, S.; Castelo, R.; Guinney, J. GSVA: Gene set variation analysis for microarray and RNA-seq data. BMC Bioinform. 2013, 14, 7. [Google Scholar] [CrossRef]
- Cao, R.; Yuan, L.; Ma, B.; Wang, G.; Qiu, W.; Tian, Y. An EMT-related gene signature for the prognosis of human bladder cancer. J. Cell. Mol. Med. 2020, 24, 605–617. [Google Scholar] [CrossRef]
- Subramanian, A.; Tamayo, P.; Mootha, V.K.; Mukherjee, S.; Ebert, B.L.; Gillette, M.A.; Paulovich, A.; Pomeroy, S.L.; Golub, T.R.; Lander, E.S.; et al. Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. USA 2005, 102, 15545–15550. [Google Scholar] [CrossRef]
- Sato, N.; Tamada, Y.; Yu, G.; Okuno, Y. CBNplot: Bayesian network plots for enrichment analysis. Bioinformatics 2022, 38, 2959–2960. [Google Scholar] [CrossRef]
- Han, H.; Cho, J.W.; Lee, S.; Yun, A.; Kim, H.; Bae, D.; Yang, S.; Kim, C.Y.; Lee, M.; Kim, E.; et al. TRRUST v2: An expanded reference database of human and mouse transcriptional regulatory interactions. Nucleic Acids Res. 2018, 46, D380–D386. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Wang, X. Identification of gastric cancer subtypes based on pathway clustering. NPJ Precis. Oncol. 2021, 5, 46. [Google Scholar] [CrossRef]
- Yu, H.; Yu, M.; Li, Z.; Zhang, E.; Ma, H. Identification and analysis of mitochondria-related key genes of heart failure. J. Transl. Med. 2022, 20, 410. [Google Scholar] [CrossRef] [PubMed]
- Wang, T.; Tian, J.; Jin, Y. VCAM1 expression in the myocardium is associated with the risk of heart failure and immune cell infiltration in myocardium. Sci. Rep. 2021, 11, 19488. [Google Scholar] [CrossRef] [PubMed]
- Tu, P.; Li, X.; Cao, L.; Zhong, M.; Xie, Z.; Wu, Z. Machine learning and BP neural network revealed abnormal B cell infiltration predicts the survival of lung cancer patients. Front. Oncol. 2022, 12, 882018. [Google Scholar] [CrossRef]
- Engebretsen, S.; Bohlin, J. Statistical predictions with glmnet. Clin. Epigenet. 2019, 11, 123. [Google Scholar] [CrossRef]
- Statnikov, A.; Wang, L.; Aliferis, C.F. A comprehensive comparison of random forests and support vector machines for microarray-based cancer classification. BMC Bioinform. 2008, 9, 319. [Google Scholar] [CrossRef]
- Yoo, M.; Shin, J.; Kim, J.; Ryall, K.A.; Lee, K.; Lee, S.; Jeon, M.; Kang, J.; Tan, A.C. DSigDB: Drug signatures database for gene set analysis. Bioinformatics 2015, 31, 3069–3071. [Google Scholar] [CrossRef]
- Pagadala, N.S.; Syed, K.; Tuszynski, J. Software for molecular docking: A review. Biophys. Rev. 2017, 9, 91–102. [Google Scholar] [CrossRef]
- Trott, O.; Olson, A.J. AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem. 2010, 31, 455–461. [Google Scholar] [CrossRef]
- Emdin, C.A.; Khera, A.V.; Kathiresan, S. Mendelian Randomization. JAMA 2017, 318, 1925–1926. [Google Scholar] [CrossRef] [PubMed]
- Xie, J.; Huang, H.; Liu, Z.; Li, Y.; Yu, C.; Xu, L.; Xu, C. The associations between modifiable risk factors and nonalcoholic fatty liver disease: A comprehensive Mendelian randomization study. Hepatology 2023, 77, 949–964. [Google Scholar] [CrossRef] [PubMed]
- Fang, Y.; Huang, S.; Han, L.; Wang, S.; Xiong, B. Comprehensive Analysis of Peritoneal Metastasis Sequencing Data to Identify LINC00924 as a Prognostic Biomarker in Gastric Cancer. Cancer Manag. Res. 2021, 13, 5599–5611. [Google Scholar] [CrossRef]
- Francis, S.H.; Busch, J.L.; Corbin, J.D.; Sibley, D. cGMP-dependent protein kinases and cGMP phosphodiesterases in nitric oxide and cGMP action. Pharmacol. Rev. 2010, 62, 525–563. [Google Scholar] [CrossRef] [PubMed]
- Müller, F.U.; Neumann, J.; Schmitz, W. Transcriptional regulation by cAMP in the heart. Mol. Cell. Biochem. 2000, 212, 11–17. [Google Scholar] [CrossRef] [PubMed]
- Leach, J.P.; Heallen, T.; Zhang, M.; Rahmani, M.; Morikawa, Y.; Hill, M.C.; Segura, A.; Willerson, J.T.; Martin, J.F. Hippo pathway deficiency reverses systolic heart failure after infarction. Nature 2017, 550, 260–264. [Google Scholar] [CrossRef]
- Lohse, M.J.; Engelhardt, S.; Eschenhagen, T. What is the role of beta-adrenergic signaling in heart failure? Circ. Res. 2003, 93, 896–906. [Google Scholar] [CrossRef]
- Dobaczewski, M.; Chen, W.; Frangogiannis, N.G. Transforming growth factor (TGF)-β signaling in cardiac remodeling. J. Mol. Cell. Cardiol. 2011, 51, 600–606. [Google Scholar] [CrossRef] [PubMed]
- Rajagopalan, S.; Pitt, B. Aldosterone as a target in congestive heart failure. Med. Clin. N. Am. 2003, 87, 441–457. [Google Scholar] [CrossRef]
- Jankowska, E.A.; Rozentryt, P.; Ponikowska, B.; Hartmann, O.; Kustrzycka-Kratochwil, D.; Reczuch, K.; Nowak, J.; Borodulin-Nadzieja, L.; Polonski, L.; Banasiak, W.; et al. Circulating estradiol and mortality in men with systolic chronic heart failure. JAMA 2009, 301, 1892–1901. [Google Scholar] [CrossRef] [PubMed]
- Crawford, R.J.; Allman, S.; Gibson, W.; Kitchen, S.; Richards, H.H. A comparative study of frusemide-amiloride and cyclopenthiazide-potassium chloride in the treatment of congestive cardiac failure in general practice. J. Int. Med. Res. 1988, 16, 143–149. [Google Scholar] [CrossRef]
- Matori, H.; Umar, S.; Nadadur, R.D.; Sharma, S.; Partow-Navid, R.; Afkhami, M.; Amjedi, M.; Eghbali, M. Genistein, a soy phytoestrogen, reverses severe pulmonary hypertension and prevents right heart failure in rats. Hypertension 2012, 60, 425–430. [Google Scholar] [CrossRef]
- Sommer, C.M.; Radeleff, B.A. A novel approach for percutaneous treatment of massive nonocclusive mesenteric ischemia: Tolazoline and glycerol trinitrate as effective local vasodilators. Catheter. Cardiovasc. Interv. 2009, 73, 152–155. [Google Scholar] [CrossRef]
- Pfeffer, M.A.; McMurray, J.J.; Velazquez, E.J.; Rouleau, J.L.; Køber, L.; Maggioni, A.P.; Solomon, S.D.; Swedberg, K.; Van de Werf, F.; White, H.; et al. Valsartan, captopril, or both in myocardial infarction complicated by heart failure, left ventricular dysfunction, or both. N. Engl. J. Med. 2003, 349, 1893–1906. [Google Scholar] [CrossRef] [PubMed]
- Baumgarten, G.; Knuefermann, P.; Kalra, D.; Gao, F.; Taffet, G.E.; Michael, L.; Blackshear, P.J.; Carballo, E.; Sivasubramanian, N.; Mann, D.L. Load-dependent and -independent regulation of proinflammatory cytokine and cytokine receptor gene expression in the adult mammalian heart. Circulation 2002, 105, 2192–2197. [Google Scholar] [CrossRef]
- Salvador, A.M.; Nevers, T.; Velázquez, F.; Aronovitz, M.; Wang, B.; Abadía, M.A.; Jaffe, I.Z.; Karas, R.H.; Blanton, R.M.; Alcaide, P. Intercellular Adhesion Molecule 1 Regulates Left Ventricular Leukocyte Infiltration, Cardiac Remodeling, and Function in Pressure Overload-Induced Heart Failure. J. Am. Heart Assoc. 2016, 5, e3126. [Google Scholar] [CrossRef]
- Garlapati, V.; Molitor, M.; Michna, T.; Harms, G.S.; Finger, S.; Jung, R.; Lagrange, J.; Efentakis, P.; Wild, J.; Knorr, M.; et al. Targeting myeloid cell coagulation signaling blocks MAP kinase/TGF-β1-driven fibrotic remodeling in ischemic heart failure. J. Clin. Investig. 2023, 133, e156436. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Zhang, Y.; Wang, M.; Song, W.M.; Shen, Q.; McKenzie, A.; Choi, I.; Zhou, X.; Pan, P.Y.; Yue, Z.; et al. The landscape of multiscale transcriptomic networks and key regulators in Parkinson’s disease. Nat. Commun. 2019, 10, 5234. [Google Scholar] [CrossRef] [PubMed]
- Chella, K.K.; Kurt, Z.; Barrere-Cain, R.; Sabir, S.; Das, A.; Floyd, R.; Vergnes, L.; Zhao, Y.; Che, N.; Charugundla, S.; et al. Integration of Multi-omics Data from Mouse Diversity Panel Highlights Mitochondrial Dysfunction in Non-alcoholic Fatty Liver Disease. Cell Syst. 2018, 6, 103–115. [Google Scholar] [CrossRef] [PubMed]
- Ma, Z.; Wang, X.; Lv, Q.; Gong, Y.; Xia, M.; Zhuang, L.; Lu, X.; Yang, Y.; Zhang, W.; Fu, G.; et al. Identification of Underlying Hub Genes Associated with Hypertrophic Cardiomyopathy by Integrated Bioinformatics Analysis. Pharmgenomics Pers. Med. 2021, 14, 823–837. [Google Scholar] [CrossRef] [PubMed]
- Gil-Cayuela, C.; Rivera, M.; Ortega, A.; Tarazón, E.; Triviño, J.C.; Lago, F.; González-Juanatey, J.R.; Almenar, L.; Martínez-Dolz, L.; Portolés, M. RNA sequencing analysis identifies new human collagen genes involved in cardiac remodeling. J. Am. Coll. Cardiol. 2015, 65, 1265–1267. [Google Scholar] [CrossRef] [PubMed]
- Tao, G.; Levay, A.K.; Peacock, J.D.; Huk, D.J.; Both, S.N.; Purcell, N.H.; Pinto, J.R.; Galantowicz, M.L.; Koch, M.; Lucchesi, P.A.; et al. Collagen XIV is important for growth and structural integrity of the myocardium. J. Mol. Cell Cardiol. 2012, 53, 626–638. [Google Scholar] [CrossRef] [PubMed]
- Petretto, E.; Sarwar, R.; Grieve, I.; Lu, H.; Kumaran, M.K.; Muckett, P.J.; Mangion, J.; Schroen, B.; Benson, M.; Punjabi, P.P.; et al. Integrated genomic approaches implicate osteoglycin (Ogn) in the regulation of left ventricular mass. Nat. Genet. 2008, 40, 546–552. [Google Scholar] [CrossRef] [PubMed]
- Van Aelst, L.N.; Voss, S.; Carai, P.; Van Leeuwen, R.; Vanhoutte, D.; Sanders-van, W.S.; Eurlings, L.; Swinnen, M.; Verheyen, F.K.; Verbeken, E.; et al. Osteoglycin prevents cardiac dilatation and dysfunction after myocardial infarction through infarct collagen strengthening. Circ. Res. 2015, 116, 425–436. [Google Scholar] [CrossRef]
- Deckx, S.; Heggermont, W.; Carai, P.; Rienks, M.; Dresselaers, T.; Himmelreich, U.; van Leeuwen, R.; Lommen, W.; van der Velden, J.; Gonzalez, A.; et al. Osteoglycin prevents the development of age-related diastolic dysfunction during pressure overload by reducing cardiac fibrosis and inflammation. Matrix Biol. 2018, 66, 110–124. [Google Scholar] [CrossRef]
- Fang, Y.; Chang, Z.; Xu, Z.; Hu, J.; Zhou, H.; Yu, S.; Wan, X. Osteoglycin silencing exerts inhibitory effects on myocardial fibrosis and epithelial/endothelial-mesenchymal transformation in a mouse model of myocarditis. Biofactors 2020, 46, 1018–1030. [Google Scholar] [CrossRef]
- Humeres, C.; Frangogiannis, N.G. Fibroblasts in the Infarcted, Remodeling, and Failing Heart. JACC Basic Transl. Sci. 2019, 4, 449–467. [Google Scholar] [CrossRef]
- Lindholt, J.S.; Madsen, M.; Kirketerp-Møller, K.L.; Schlosser, A.; Kristensen, K.L.; Andersen, C.B.; Sorensen, G.L. High plasma microfibrillar-associated protein 4 is associated with reduced surgical repair in abdominal aortic aneurysms. J. Vasc. Surg. 2020, 71, 1921–1929. [Google Scholar] [CrossRef]
- Kanaan, R.; Medlej-Hashim, M.; Jounblat, R.; Pilecki, B.; Sorensen, G.L. Microfibrillar-associated protein 4 in health and disease. Matrix Biol. 2022, 111, 1–25. [Google Scholar] [CrossRef] [PubMed]
- Sækmose, S.G.; Mössner, B.; Christensen, P.B.; Lindvig, K.; Schlosser, A.; Holst, R.; Barington, T.; Holmskov, U.; Sorensen, G.L. Microfibrillar-Associated Protein 4: A Potential Biomarker for Screening for Liver Fibrosis in a Mixed Patient Cohort. PLoS ONE 2015, 10, e140418. [Google Scholar] [CrossRef]
- Wang, H.B.; Yang, J.; Shuai, W.; Yang, J.; Liu, L.B.; Xu, M.; Tang, Q.Z. Deletion of Microfibrillar-Associated Protein 4 Attenuates Left Ventricular Remodeling and Dysfunction in Heart Failure. J. Am. Heart Assoc. 2020, 9, e15307. [Google Scholar] [CrossRef]
- Dorn, L.E.; Lawrence, W.; Petrosino, J.M.; Xu, X.; Hund, T.J.; Whitson, B.A.; Stratton, M.S.; Janssen, P.; Mohler, P.J.; Schlosser, A.; et al. Microfibrillar-Associated Protein 4 Regulates Stress-Induced Cardiac Remodeling. Circ. Res. 2021, 128, 723–737. [Google Scholar] [CrossRef]
- Pawar, N.M.; Rao, P. Secreted frizzled related protein 4 (sFRP4) update: A brief review. Cell. Signal. 2018, 45, 63–70. [Google Scholar] [CrossRef] [PubMed]
- Zeng, W.; Cao, Y.; Jiang, W.; Kang, G.; Huang, J.; Xie, S. Knockdown of Sfrp4 attenuates apoptosis to protect against myocardial ischemia/reperfusion injury. J. Pharmacol. Sci. 2019, 140, 14–19. [Google Scholar] [CrossRef] [PubMed]
- Matsushima, K.; Suyama, T.; Takenaka, C.; Nishishita, N.; Ikeda, K.; Ikada, Y.; Sawa, Y.; Jakt, L.M.; Mori, H.; Kawamata, S. Secreted frizzled related protein 4 reduces fibrosis scar size and ameliorates cardiac function after ischemic injury. Tissue Eng. Part. A 2010, 16, 3329–3341. [Google Scholar] [CrossRef] [PubMed]
- Ji, Q.; Zhang, J.; Du, Y.; Zhu, E.; Wang, Z.; Que, B.; Miao, H.; Shi, S.; Qin, X.; Zhao, Y.; et al. Human epicardial adipose tissue-derived and circulating secreted frizzled-related protein 4 (SFRP4) levels are increased in patients with coronary artery disease. Cardiovasc. Diabetol. 2017, 16, 133. [Google Scholar] [CrossRef] [PubMed]
- Zannad, F.; Rossignol, P.; Iraqi, W. Extracellular matrix fibrotic markers in heart failure. Heart Fail. Rev. 2010, 15, 319–329. [Google Scholar] [CrossRef] [PubMed]
- Hanna, A.; Frangogiannis, N.G. Inflammatory Cytokines and Chemokines as Therapeutic Targets in Heart Failure. Cardiovasc. Drugs Ther. 2020, 34, 849–863. [Google Scholar] [CrossRef] [PubMed]
- Lemieux, H.; Semsroth, S.; Antretter, H.; Höfer, D.; Gnaiger, E. Mitochondrial respiratory control and early defects of oxidative phosphorylation in the failing human heart. Int. J. Biochem. Cell Biol. 2011, 43, 1729–1738. [Google Scholar] [CrossRef]
- Bobowski-Gerard, M.; Boulet, C.; Zummo, F.P.; Dubois-Chevalier, J.; Gheeraert, C.; Bou, S.M.; Strub, J.M.; Farce, A.; Ploton, M.; Guille, L.; et al. Functional genomics uncovers the transcription factor BNC2 as required for myofibroblastic activation in fibrosis. Nat. Commun. 2022, 13, 5324. [Google Scholar] [CrossRef] [PubMed]
- Lin, J.; Friesen, M.T.; Bocangel, P.; Cheung, D.; Rawszer, K.; Wigle, J.T. Characterization of Mesenchyme Homeobox 2 (MEOX2) transcription factor binding to RING finger protein 10. Mol. Cell. Biochem. 2005, 275, 75–84. [Google Scholar] [CrossRef]
- Douville, J.M.; Cheung, D.Y.; Herbert, K.L.; Moffatt, T.; Wigle, J.T. Mechanisms of MEOX1 and MEOX2 regulation of the cyclin dependent kinase inhibitors p21 and p16 in vascular endothelial cells. PLoS ONE 2011, 6, e29099. [Google Scholar] [CrossRef]
- Adamo, L.; Rocha-Resende, C.; Prabhu, S.D.; Mann, D.L. Reappraising the role of inflammation in heart failure. Nat. Rev. Cardiol. 2020, 17, 269–285. [Google Scholar] [CrossRef]
- Sciomer, S.; Moscucci, F.; Salvioni, E.; Marchese, G.; Bussotti, M.; Corrà, U.; Piepoli, M.F. Role of gender, age and BMI in prognosis of heart failure. Eur. J. Prev. Cardiol. 2020, 27 (Suppl. S2), 46–51. [Google Scholar] [CrossRef]
- Cediel, G.; Codina, P.; Spitaleri, G.; Domingo, M.; Santiago-Vacas, E.; Lupón, J.; Bayes-Genis, A. Gender-Related Differences in Heart Failure Biomarkers. Front. Cardiovasc. Med. 2020, 7, 617705. [Google Scholar] [CrossRef]












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Accession | Sample Source | Sequencing Type | Control Samples | HF Samples | Dataset Usage |
|---|---|---|---|---|---|
| GSE141910 | left ventricle | RNA-seq | 166 | 200 | Training dataset |
| GSE57338 | left ventricle | Array | 136 | 177 | Testing dataset |
| GSE42955 | left ventricle near the apex | Array | 5 | 24 | Testing dataset |
| GSE135055 | left ventricle | RNA-seq | 9 | 21 | External validation dataset |
| Drugs | Targets | Affinity (kcal/mol) | Bond | Protein’s Residues |
|---|---|---|---|---|
| Captopril | BNC2 | −3.9 | H-bond | GLN-199 |
| Aldosterone | MEOX2 | −7.1 | H-bond | GLU-203 LYS-241 |
| Cyclopenthiazide | MEOX2 | −5.7 | H-bond | THR-192 ASN-237 |
| Estradiol | COL14A1 | −8.2 | H-bond | ARG-171 VAL-930 |
| Tolazoline | COL14A1 | −5.9 | H-bond | ASP-1148 |
| Genistein | SFRP4 | −3.8 | H-bond | GLU-127 ARG-262 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhu, Y.; Chen, B.; Zu, Y. Identifying OGN as a Biomarker Covering Multiple Pathogenic Pathways for Diagnosing Heart Failure: From Machine Learning to Mechanism Interpretation. Biomolecules 2024, 14, 179. https://doi.org/10.3390/biom14020179
Zhu Y, Chen B, Zu Y. Identifying OGN as a Biomarker Covering Multiple Pathogenic Pathways for Diagnosing Heart Failure: From Machine Learning to Mechanism Interpretation. Biomolecules. 2024; 14(2):179. https://doi.org/10.3390/biom14020179
Chicago/Turabian StyleZhu, Yihao, Bin Chen, and Yao Zu. 2024. "Identifying OGN as a Biomarker Covering Multiple Pathogenic Pathways for Diagnosing Heart Failure: From Machine Learning to Mechanism Interpretation" Biomolecules 14, no. 2: 179. https://doi.org/10.3390/biom14020179