
Defining Melanoma Immune Biomarkers—Desert, Excluded, and Inflamed Subtypes—Using a Gene Expression Classifier Reflecting Intratumoral Immune Response and Stromal Patterns
 , , , , ,
, , , , ,
Abstract
:1. Introduction
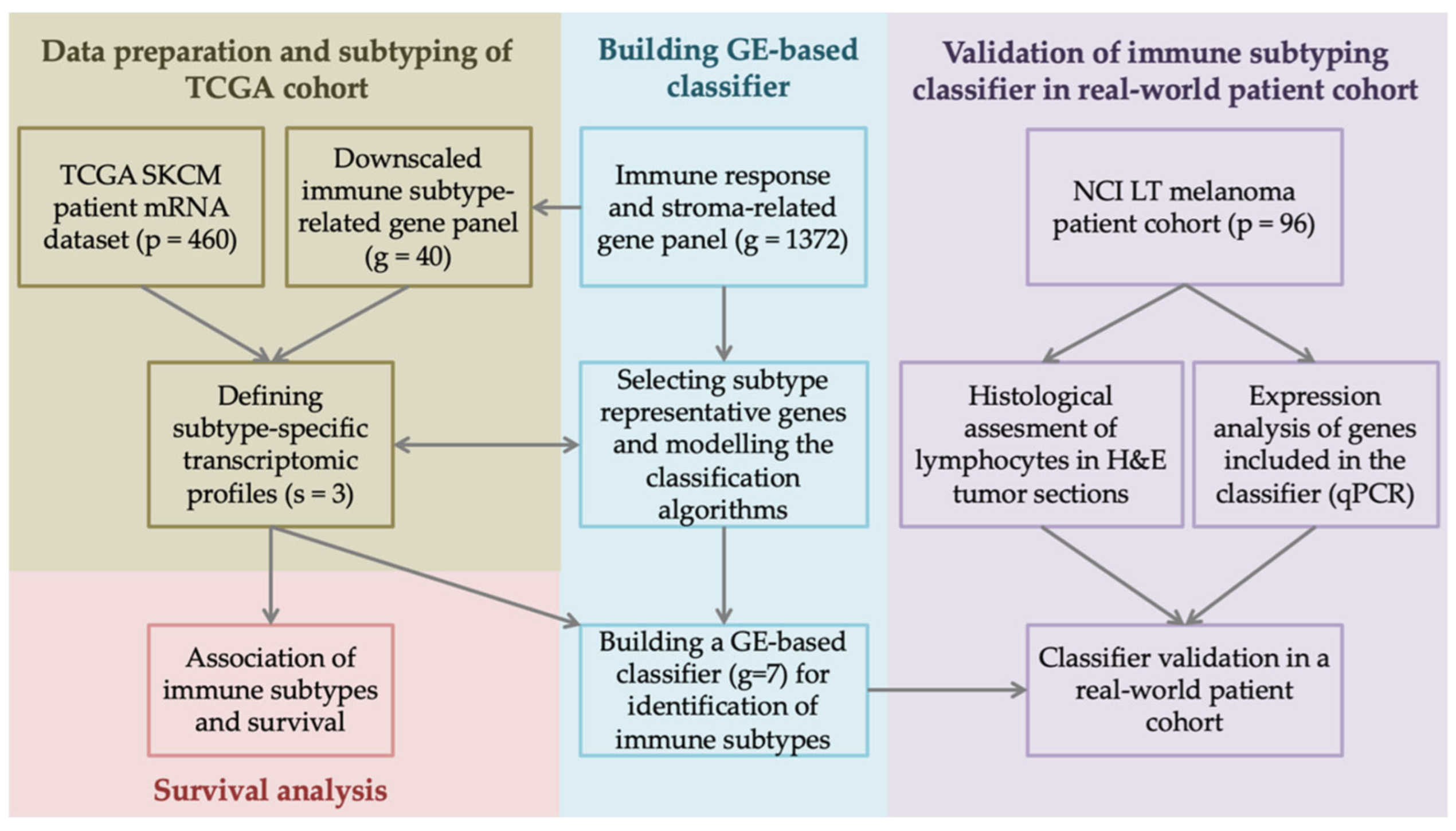
2. Materials and Methods
2.1. Processing of TCGA Dataset and Gene List
2.2. In Silico Immune Subtyping
2.3. Differential Gene Expression Analysis and Building Subtyping Classifier
2.4. Melanoma Patient Cohort
2.5. Histopathological Assessment
2.6. qPCR for Gene Expression Analysis in Real-World Patient Cohort
2.7. Statistical Analysis
3. Results
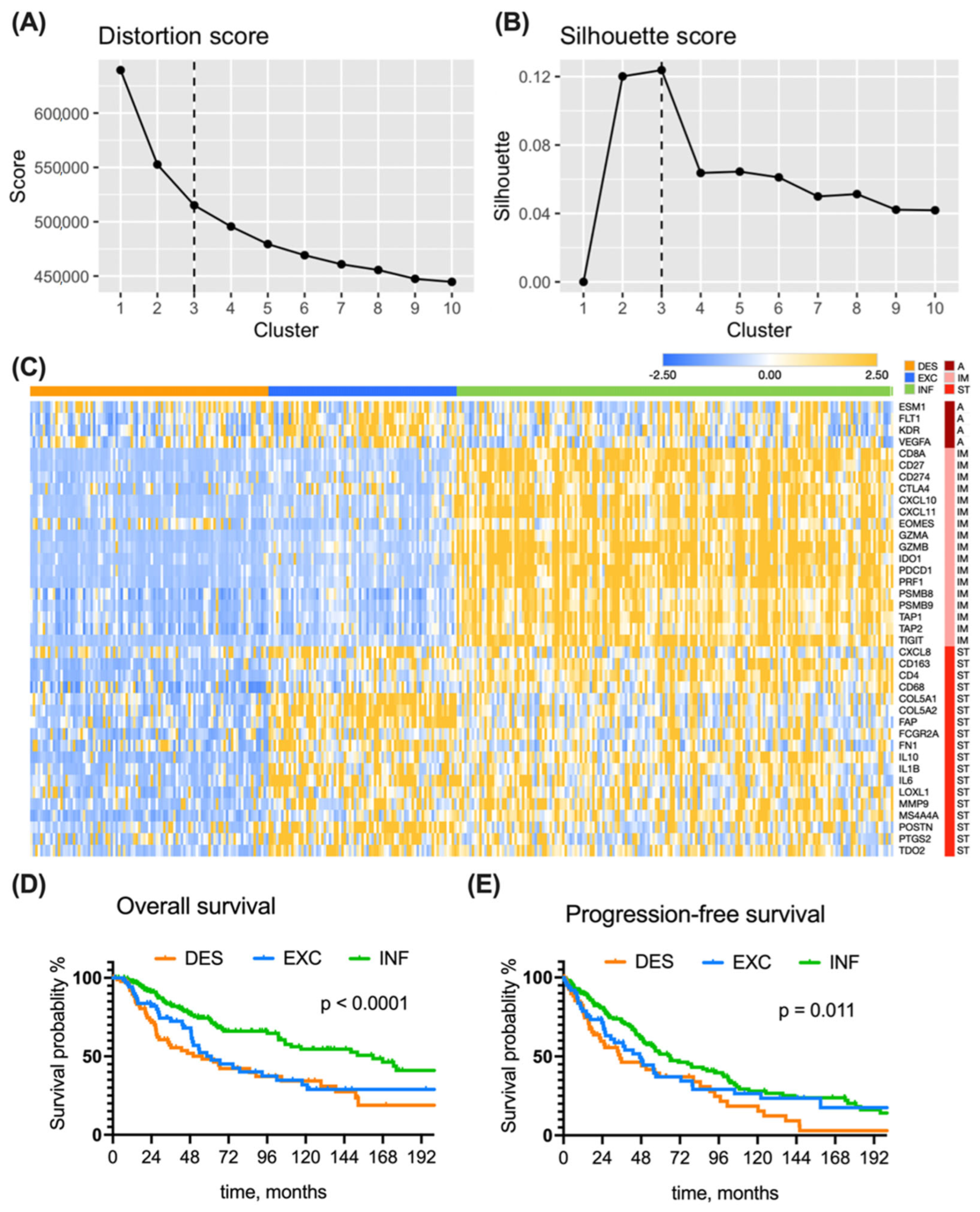
3.1. Immune Subtyping of the TCGA Cohort
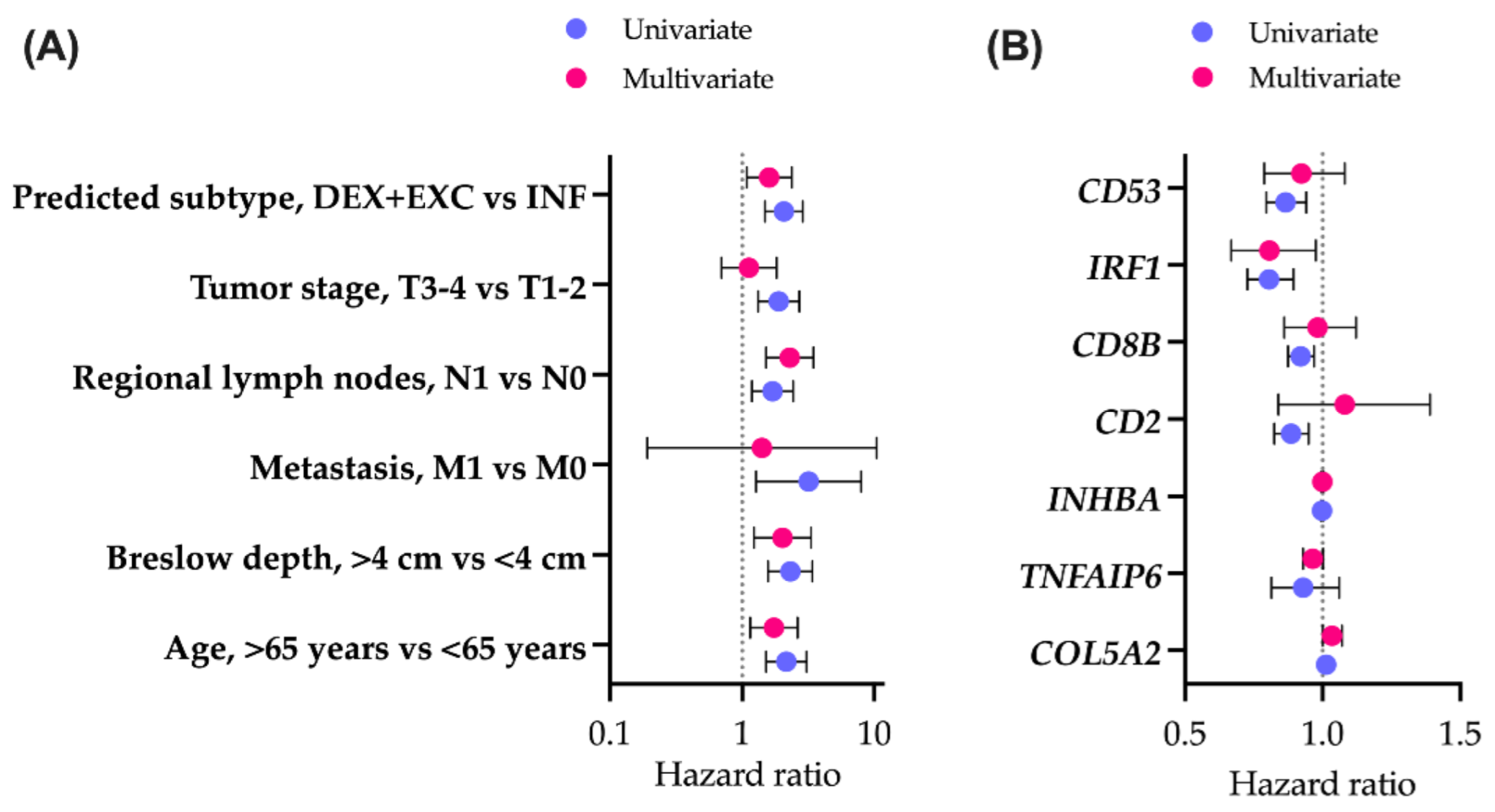
3.2. Clinical Features of the Immune Subtypes in the TCGA Cohort
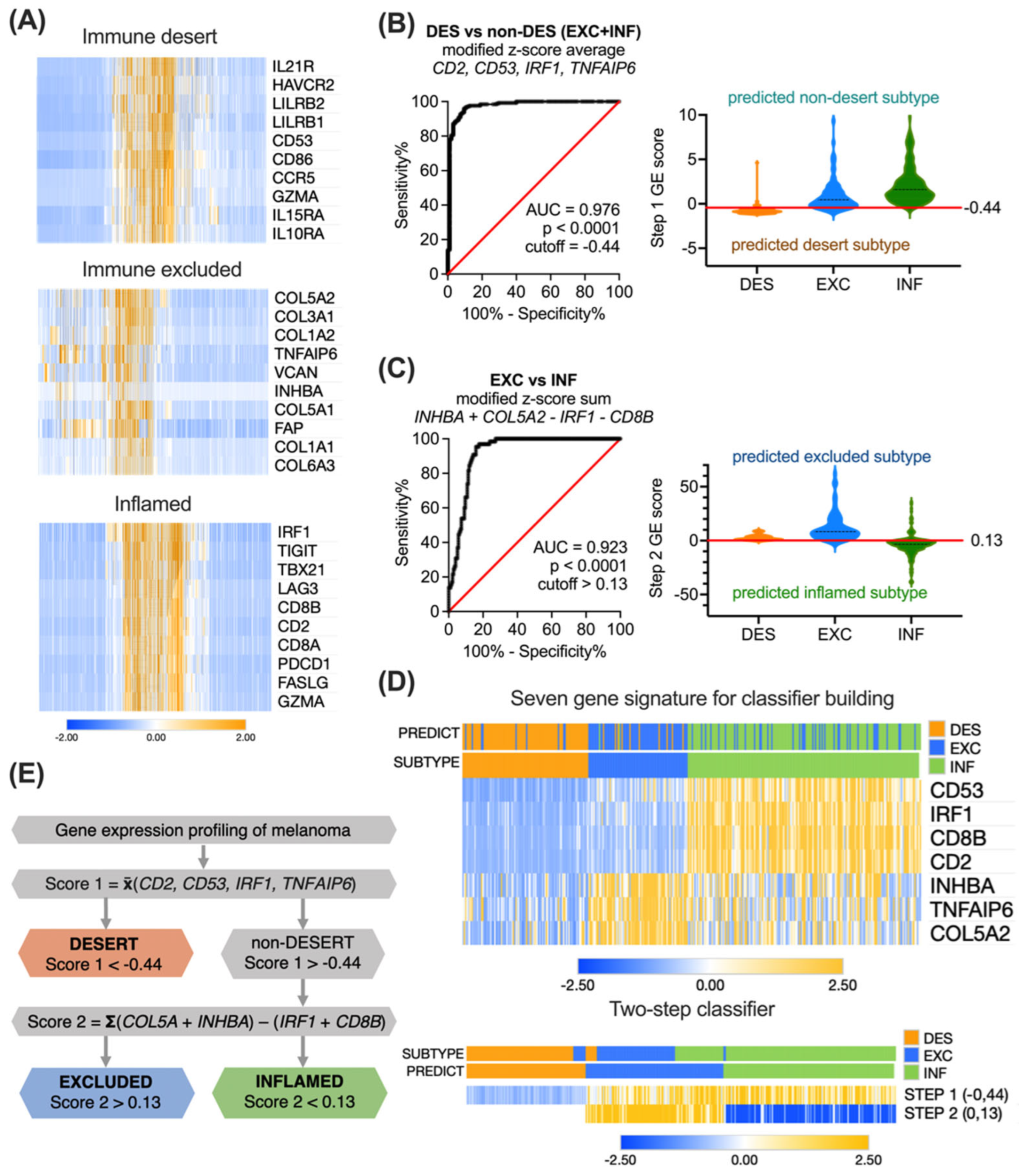
3.3. Building the Immune Subtyping Classifier
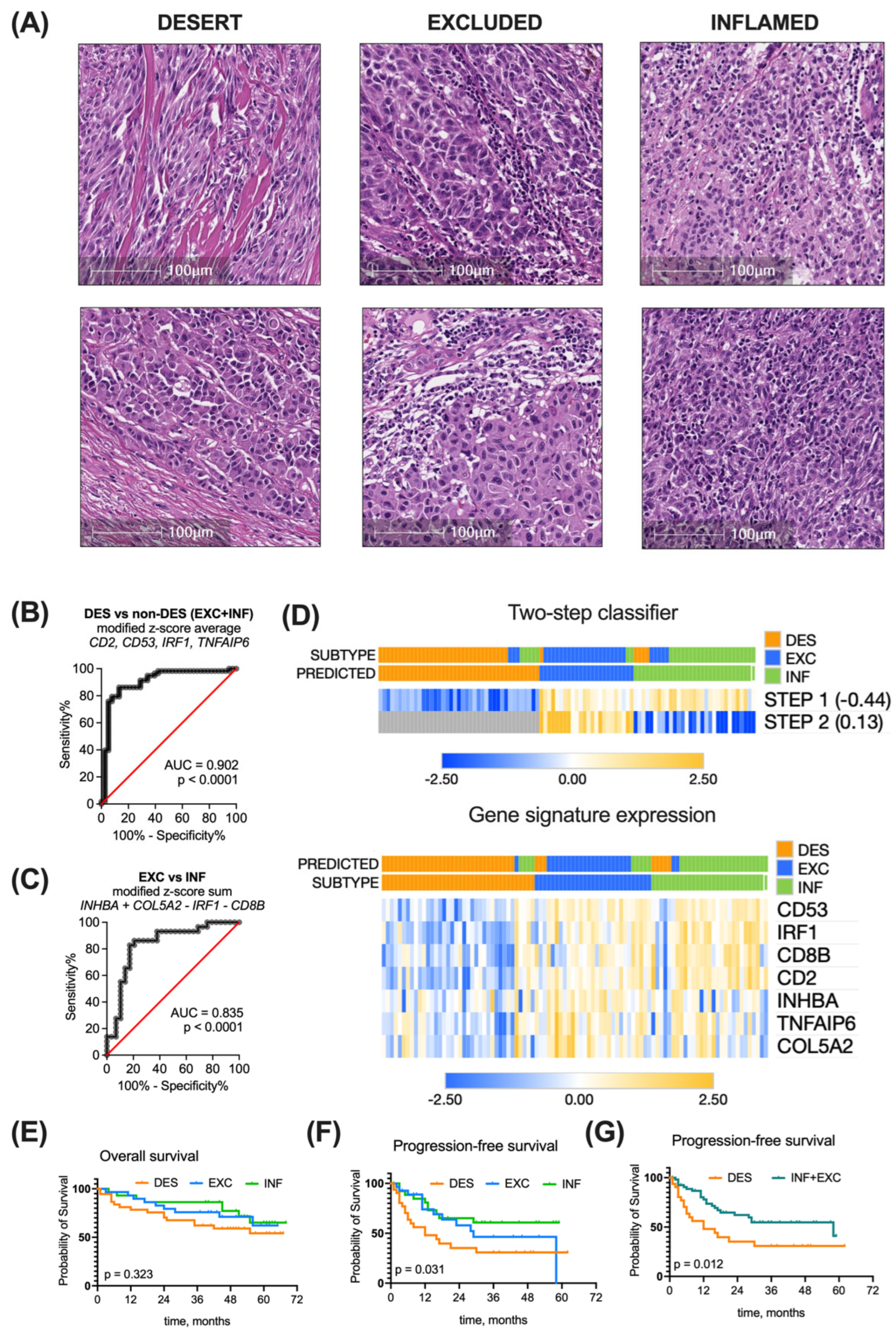
3.4. Validation of the Immune Subtyping Classifier in a Real-World Patient Cohort
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Labani-Motlagh, A.; Ashja-Mahdavi, M.; Loskog, A. The Tumor Microenvironment: A Milieu Hindering and Obstructing Antitumor Immune Responses. Front. Immunol. 2020, 11, 940. [Google Scholar] [CrossRef] [PubMed]
- Chen, D.S.; Mellman, I. Elements of Cancer Immunity and the Cancer-Immune Set Point. Nature 2017, 541, 321–330. [Google Scholar] [CrossRef] [PubMed]
- Anagnostou, V.; Forde, P.M.; White, J.R.; Niknafs, N.; Hruban, C.; Naidoo, J.; Marrone, K.; Ashok Sivakumar, I.K.; Bruhm, D.C.; Rosner, S.; et al. Dynamics of Tumor and Immune Responses during Immune Checkpoint Blockade in Non–Small Cell Lung Cancer. Cancer Res. 2019, 79, 1214–1225. [Google Scholar] [CrossRef] [PubMed]
- Galon, J.; Lanzi, A. Immunoscore and Its Introduction in Clinical Practice. Q. J. Nucl. Med. Mol. Imaging 2020, 64, 152–161. [Google Scholar] [CrossRef] [PubMed]
- Gnjatic, S.; Bronte, V.; Brunet, L.R.; Butler, M.O.; Disis, M.L.; Galon, J.; Hakansson, L.G.; Hanks, B.A.; Karanikas, V.; Khleif, S.N.; et al. Identifying Baseline Immune-Related Biomarkers to Predict Clinical Outcome of Immunotherapy. J. Immunother. Cancer 2017, 5, 44. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Chen, J. Current Status and Future Directions of Cancer Immunotherapy. J. Cancer 2018, 9, 1773–1781. [Google Scholar] [CrossRef] [PubMed]
- Hegde, P.S.; Chen, D.S. Perspective Top 10 Challenges in Cancer Immunotherapy. Immunity 2020, 52, 17–35. [Google Scholar] [CrossRef]
- Krzyszczyk, P.; Acevedo, A.; Davidoff, E.J.; Timmins, L.M.; Marrero-Berrios, I.; Patel, M.; White, C.; Lowe, C.; Sherba, J.J.; Hartmanshenn, C.; et al. The Growing Role of Precision and Personalized Medicine for Cancer Treatment. Technology 2018, 6, 79. [Google Scholar] [CrossRef]
- Liu, Z.; Lin, G.; Yan, Z.; Li, L.; Wu, X.; Shi, J.; He, J.; Zhao, L.; Liang, H.; Wang, W. Predictive Mutation Signature of Immunotherapy Benefits in NSCLC Based on Machine Learning Algorithms. Front. Immunol. 2022, 13, 989275. [Google Scholar] [CrossRef]
- Barker, A.D.; Lee, J.S.H. Translating “Big Data” in Oncology for Clinical Benefit: Progress or Paralysis. Cancer Res 2022, 82, 2072–2075. [Google Scholar] [CrossRef]
- Lanitis, E.; Dangaj, D.; Irving, M.; Coukos, G. Mechanisms Regulating T-Cell Infiltration and Activity in Solid Tumors. Ann. Oncol. 2017, 28, xii18–xii32. [Google Scholar] [CrossRef]
- Kim, J.M.; Chen, D.S. Immune Escape to PD-L1/PD-1 Blockade: Seven Steps to Success (or Failure). Ann. Oncol. 2016, 27, 1492–1504. [Google Scholar] [CrossRef]
- Hegde, P.S.; Karanikas, V.; Evers, S. The Where, the When, and the How of Immune Monitoring for Cancer Immunotherapies in the Era of Checkpoint Inhibition. Clin. Cancer Res. 2016, 22, 1865–1874. [Google Scholar] [CrossRef] [PubMed]
- Mlynska, A.; Vaišnorė, R.; Rafanavičius, V.; Jocys, S.; Janeiko, J.; Petrauskytė, M.; Bijeikis, S.; Cimmperman, P.; Intaitė, B.; Žilionytė, K.; et al. A Gene Signature for Immune Subtyping of Desert, Excluded, and Inflamed Ovarian Tumors. Am. J. Reprod. Immunol. 2020, 84, e13244. [Google Scholar] [CrossRef] [PubMed]
- Skin Cutaneous Melanoma (TCGA, Firehose Legacy). Available online: https://datacatalog.mskcc.org/dataset/10490 (accessed on 28 September 2022).
- Cerami, E.; Gao, J.; Dogrusoz, U.; Gross, B.E.; Sumer, S.O.; Aksoy, B.A.; Jacobsen, A.; Byrne, C.J.; Heuer, M.L.; Larsson, E.; et al. The CBio Cancer Genomics Portal: An Open Platform for Exploring Multidimensional Cancer Genomics Data. Cancer Discov. 2012, 2, 401–404. [Google Scholar] [CrossRef] [PubMed]
- Gao, J.; Aksoy, B.A.; Dogrusoz, U.; Dresdner, G.; Gross, B.; Sumer, S.O.; Sun, Y.; Jacobsen, A.; Sinha, R.; Larsson, E.; et al. Integrative Analysis of Complex Cancer Genomics and Clinical Profiles Using the CBioPortal. Sci. Signal. 2013, 6, pl1. [Google Scholar] [CrossRef] [PubMed]
- Charoentong, P.; Finotello, F.; Angelova, M.; Mayer, C.; Efremova, M.; Rieder, D.; Hackl, H.; Trajanoski, Z. Pan-Cancer Immunogenomic Analyses Reveal Genotype-Immunophenotype Relationships and Predictors of Response to Checkpoint Blockade. Cell Rep. 2017, 18, 248–262. [Google Scholar] [CrossRef] [PubMed]
- Cesano, A. NCounter® PanCancer Immune Profiling Panel (NanoString Technologies, Inc., Seattle, WA). J. Immunother. Cancer 2015, 3, 42. [Google Scholar] [CrossRef] [PubMed]
- Thorndike, R.L. Who Belongs in the Family? Psychometrika 1953, 18, 267–276. [Google Scholar] [CrossRef]
- Rousseeuw, P.J. Silhouettes: A Graphical Aid to the Interpretation and Validation of Cluster Analysis. J. Comput. Appl. Math. 1987, 20, 53–65. [Google Scholar] [CrossRef]
- Bengfort, B.; Gray, L.; Bilbro, R.; Roman, P.; Deziel, P.; McIntyre, K.; Morrison, M.; Ojeda, A.; Schmierer, E.; Morris, A.; et al. Yellowbrick v1.5. Zenodo 2022. [Google Scholar] [CrossRef]
- Mcdermott, D.F.; Huseni, M.A.; Atkins, M.B.; Motzer, R.J.; Rini, B.I.; Escudier, B.; Fong, L.; Joseph, R.W.; Pal, S.K.; Reeves, J.A.; et al. Clinical Activity and Molecular Correlates of Response to Atezolizumab Alone or in Combination with Bevacizumab versus Sunitinib in Renal Cell Carcinoma David. Nat. Med. 2018, 24, 749–757. [Google Scholar] [CrossRef]
- Lloyd, S.P. Least Squares Quantization in PCM. IEEE Trans. Inf. Theory 1982, 28, 129–137. [Google Scholar] [CrossRef]
- Li, B.; Dewey, C.N. RSEM: Accurate Transcript Quantification from RNA-Seq Data with or without a Reference Genome. BMC Bioinform. 2011, 12, 323. [Google Scholar] [CrossRef]
- Foroutan, M.; Bhuva, D.D.; Lyu, R.; Horan, K.; Cursons, J.; Davis, M.J. Single Sample Scoring of Molecular Phenotypes. BMC Bioinform. 2018, 19, 404. [Google Scholar] [CrossRef]
- Obikee, A.C.; Ebuh, G.U.; Obiora-Ilouno, H.O. Comparison of Outlier Techniques Based on Simulated Data. Open J. Stat. 2014, 4, 536–561. [Google Scholar] [CrossRef]
- Youden, W.J. Index for rating diagnostic tests. Cancer 1950, 3, 32–35. [Google Scholar] [CrossRef]
- Salgado, R.; Denkert, C.; Demaria, S.; Sirtaine, N.; Klauschen, F.; Pruneri, G.; Wienert, S.; Van den Eynden, G.; Baehner, F.L.; Penault-Llorca, F.; et al. The Evaluation of Tumor-Infiltrating Lymphocytes (TILs) in Breast Cancer: Recommendations by an International TILs Working Group 2014. Ann. Oncol. 2015, 26, 259–271. [Google Scholar] [CrossRef] [PubMed]
- Hendry, S.; Salgado, R.; Gevaert, T.; Russell, P.A.; John, T.; Thapa, B.; Christie, M.; van de Vijver, K.; Estrada, M.V.; Gonzalez-Ericsson, P.I.; et al. Assessing Tumor-Infiltrating Lymphocytes in Solid Tumors: A Practical Review for Pathologists and Proposal for a Standardized Method from the International Immuno-Oncology Biomarkers Working Group: Part 2: TILs in Melanoma, Gastrointestinal Tract Carcinomas, Non-Small Cell Lung Carcinoma and Mesothelioma, Endometrial and Ovarian Carcinomas, Squamous Cell Carcinoma of the Head and Neck, Genitourinary Carcinomas, and Primary Brain Tumors. Adv. Anat. Pathol. 2017, 24, 311–335. [Google Scholar] [CrossRef] [PubMed]
- Mao, Y.; Qu, Q.; Zhang, Y.; Liu, J.; Chen, X.; Shen, K. The Value of Tumor Infiltrating Lymphocytes (TILs) for Predicting Response to Neoadjuvant Chemotherapy in Breast Cancer: A Systematic Review and Meta-Analysis. PLoS ONE 2014, 9, e115103. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Spandidos, A.; Wang, H.; Seed, B. PrimerBank: A PCR Primer Database for Quantitative Gene Expression Analysis, 2012 Update. Nucleic Acids Res. 2012, 40, D1144–D1149. [Google Scholar] [CrossRef]
- Livak, K.J.; Schmittgen, T.D. Analysis of Relative Gene Expression Data Using Real-Time Quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef]
- Pfaffl, M.W. A New Mathematical Model for Relative Quantification in Real-Time RT-PCR. Nucleic Acids Res. 2001, 29, e45. [Google Scholar] [CrossRef] [PubMed]
- Morpheus. Available online: https://software.broadinstitute.org/morpheus/ (accessed on 28 September 2022).
- Mitchell, A.J. Sensitivity x PPV Is a Recognized Test Called the Clinical Utility Index (CUI+). Eur. J. Epidemiol. 2011, 26, 251–252. [Google Scholar] [CrossRef] [PubMed]
- Benjamini, Y.; Krieger, A.M.; Yekutieli, D. Adaptive Linear Step-up Procedures That Control the False Discovery Rate. Biometrika 2006, 93, 491–507. [Google Scholar] [CrossRef]
- Chen, B.; Khodadoust, M.S.; Liu, C.L.; Newman, A.M.; Alizadeh, A.A. Profiling Tumor Infiltrating Immune Cells with CIBERSORT. Methods Mol. Biol. 2018, 1711, 243. [Google Scholar] [CrossRef]
- Plattner, C.; Finotello, F.; Rieder, D. Deconvoluting Tumor-Infiltrating Immune Cells from RNA-Seq Data Using QuanTIseq. Methods Enzym. 2020, 636, 261–285. [Google Scholar] [CrossRef]
- Chen, D.S.; Mellman, I. Oncology Meets Immunology: The Cancer-Immunity Cycle. Immunity 2013, 39, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Newell, F.; Johansson, P.A.; Wilmott, J.S.; Nones, K.; Lakis, V.; Pritchard, A.L.; Lo, S.N.; Rawson, R.V.; Kazakoff, S.H.; Colebatch, A.J.; et al. Comparative Genomics Provides Etiological and Biological Insights into Melanoma Subtypes. Cancer Discov. 2022, 12, 2856–2879. [Google Scholar] [CrossRef] [PubMed]
- Akbani, R.; Akdemir, K.C.; Aksoy, B.A.; Albert, M.; Ally, A.; Amin, S.B.; Arachchi, H.; Arora, A.; Auman, J.T.; Ayala, B.; et al. Genomic Classification of Cutaneous Melanoma. Cell 2015, 161, 1681. [Google Scholar] [CrossRef]
- Bagaev, A.; Kotlov, N.; Nomie, K.; Svekolkin, V.; Gafurov, A.; Isaeva, O.; Osokin, N.; Kozlov, I.; Frenkel, F.; Gancharova, O.; et al. Conserved Pan-Cancer Microenvironment Subtypes Predict Response to Immunotherapy. Cancer Cell 2021, 39, 845–865.e7. [Google Scholar] [CrossRef]
- Wang, J.; Peng, C.; Dai, W.; Chen, X.; Meng, J.; Jiang, T. Exploring Tumor Immune Microenvironment and Its Associations with Molecular Characteristics in Melanoma. Front. Oncol. 2022, 12, 1443. [Google Scholar] [CrossRef] [PubMed]
- Liu, Q.; Nie, R.; Li, M.; Li, L.; Zhou, H.; Lu, H.; Wang, X. Identification of Subtypes Correlated with Tumor Immunity and Immunotherapy in Cutaneous Melanoma. Comput. Struct. Biotechnol. J. 2021, 19, 4472–4485. [Google Scholar] [CrossRef]
- Liu, L.; Zhu, J.; Jin, T.; Huang, M.; Chen, Y.; Xu, L.; Chen, W.; Jiang, B.; Yan, F. Identification of Immune Function-Related Subtypes in Cutaneous Melanoma. Life 2021, 11, 925. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Hao, S.; Gao, Y.; Sun, W.; Li, Y. Decoding Immune Heterogeneity of Melanoma and Identifying Immune-Prognostic Hub Genes. J. Cancer 2021, 12, 703. [Google Scholar] [CrossRef] [PubMed]
- Sobottka, B.; Nowak, M.; Frei, A.L.; Haberecker, M.; Merki, S.; Aebersold, R.; Ak, M.; Al-Quaddoomi, F.S.; Albinus, J.; Alborelli, I.; et al. Establishing Standardized Immune Phenotyping of Metastatic Melanoma by Digital Pathology. Lab. Investig. 2021, 101, 1561–1570. [Google Scholar] [CrossRef] [PubMed]
- Kather, J.N.; Suarez-Carmona, M.; Charoentong, P.; Weis, C.-A.; Hirsch, D.; Bankhead, P.; Horning, M.; Ferber, D.; Kel, I.; Herpel, E.; et al. Topography of Cancer-Associated Immune Cells in Human Solid Tumors. eLife 2018, 7, e36967. [Google Scholar] [CrossRef]
- Liu, D.; Yang, X.; Wu, X. Tumor Immune Microenvironment Characterization Identifies Prognosis and Immunotherapy-Related Gene Signatures in Melanoma. Front. Immunol. 2021, 12, 1. [Google Scholar] [CrossRef]
- Desbois, M.; Udyavar, A.R.; Ryner, L.; Kozlowski, C.; Guan, Y.; Dürrbaum, M.; Lu, S.; Fortin, J.P.; Koeppen, H.; Ziai, J.; et al. Integrated Digital Pathology and Transcriptome Analysis Identifies Molecular Mediators of T-Cell Exclusion in Ovarian Cancer. Nat. Commun. 2020, 11, 5583. [Google Scholar] [CrossRef]
- Pomponio, R.; Tang, Q.; Mei, A.; Caron, A.; Coulibaly, B.; Theilhaber, J.; Rogers-Grazado, M.; Sanicola-Nadel, M.; Naimi, S.; Olfati-Saber, R.; et al. An Integrative Approach of Digital Image Analysis and Transcriptome Profiling to Explore Potential Predictive Biomarkers for TGFβ Blockade Therapy. Acta Pharm. Sin. B 2022, 12, 3594–3601. [Google Scholar] [CrossRef]
- Banchereau, R.; Chitre, A.S.; Scherl, A.; Wu, T.D.; Patil, N.S.; De Almeida, P.; Kadel, E.E.; Madireddi, S.; Au-Yeung, A.; Takahashi, C.; et al. Intratumoral CD103+ CD8+ T Cells Predict Response to PD-L1 Blockade. J. Immunother. Cancer 2021, 9, 2231. [Google Scholar] [CrossRef] [PubMed]
- Backman, M.; La Fleur, L.; Kurppa, P.; Djureinovic, D.; Elfving, H.; Brunnström, H.; Sofia Margareta Mattsson, J.; Lindberg, A.; Pontén, V.; Eltahir, M.; et al. Infiltration of NK and Plasma Cells Is Associated with a Distinct Immune Subset in Non-Small Cell Lung Cancer. J. Pathol. 2021, 255, 243–256. [Google Scholar] [CrossRef] [PubMed]
- Wu, S.Z.; Roden, D.L.; Wang, C.; Holliday, H.; Harvey, K.; Cazet, A.S.; Murphy, K.J.; Pereira, B.; Al-Eryani, G.; Bartonicek, N.; et al. Stromal Cell Diversity Associated with Immune Evasion in Human Triple-Negative Breast Cancer. EMBO J. 2020, 39, e104063. [Google Scholar] [CrossRef] [PubMed]
- Brooks, J.M.; Menezes, A.N.; Ibrahim, M.; Archer, L.; Lal, N.; Bagnall, C.J.; Von Zeidler, S.V.; Valentine, H.R.; Spruce, R.J.; Batis, N.; et al. Precision Medicine and Imaging Development and Validation of a Combined Hypoxia and Immune Prognostic Classifier for Head and Neck Cancer. Clin. Cancer Res. 2019, 25, 5315–5328. [Google Scholar] [CrossRef] [PubMed]
- Echarti, A.; Hecht, M.; Büttner-Herold, M.; Haderlein, M.; Hartmann, A.; Fietkau, R.; Distel, L. CD8+ and Regulatory T Cells Differentiate Tumor Immune Phenotypes and Predict Survival in Locally Advanced Head and Neck Cancer. Cancers 2019, 11, 1398. [Google Scholar] [CrossRef] [PubMed]
- Hammerl, D.; Martens, J.W.M.; Timmermans, M.; Smid, M.; Trapman-Jansen, A.M.; Foekens, R.; Isaeva, O.I.; Voorwerk, L.; Balcioglu, H.E.; Wijers, R.; et al. Spatial Immunophenotypes Predict Response to Anti-PD1 Treatment and Capture Distinct Paths of T Cell Evasion in Triple Negative Breast Cancer. Nat. Commun. 2021, 12, 5668. [Google Scholar] [CrossRef]
- Di Blasi, D.; Boldanova, T.; Mori, L.; Terracciano, L.; Heim, M.H.; De Libero, G. Unique T-Cell Populations Define Immune-Inflamed Hepatocellular Carcinoma. Cell. Mol. Gastroenterol. Hepatol. 2020, 9, 195–218. [Google Scholar] [CrossRef]
- Lenzo, F.L.; Kato, S.; Pabla, S.; DePietro, P.; Nesline, M.K.; Conroy, J.M.; Burgher, B.; Glenn, S.T.; Kuvshinoff, B.; Kurzrock, R.; et al. Immune Profiling and Immunotherapeutic Targets in Pancreatic Cancer. Ann. Transl. Med. 2021, 9, 119. [Google Scholar] [CrossRef]
- Ohe, C.; Yoshida, T.; Ikeda, J.; Tsuzuki, T.; Ohashi, R.; Ohsugi, H.; Atsumi, N.; Yamaka, R.; Saito, R.; Yasukochi, Y.; et al. Histologic-Based Tumor-Associated Immune Cells Status in Clear Cell Renal Cell Carcinoma Correlates with Gene Signatures Related to Cancer Immunity and Clinical Outcomes. Biomedicines 2022, 10, 323. [Google Scholar] [CrossRef] [PubMed]
- Trujillo, J.A.; Sweis, R.F.; Bao, R.; Luke, J.J. T Cell–Inflamed versus Non-T Cell–Inflamed Tumors: A Conceptual Framework for Cancer Immunotherapy Drug Development and Combination Therapy Selection. Cancer Immunol. Res. 2018, 6, 990. [Google Scholar] [CrossRef] [PubMed]
- Gooden, M.J.M.; De Bock, G.H.; Leffers, N.; Daemen, T.; Nijman, H.W. The Prognostic Influence of Tumour-Infiltrating Lymphocytes in Cancer: A Systematic Review with Meta-Analysis. Br. J. Cancer 2011, 105, 93. [Google Scholar] [CrossRef]
- Raskov, H.; Orhan, A.; Christensen, J.P.; Gögenur, I. Cytotoxic CD8+ T Cells in Cancer and Cancer Immunotherapy. Br. J. Cancer 2020, 124, 359–367. [Google Scholar] [CrossRef] [PubMed]
- Binder, C.; Cvetkovski, F.; Sellberg, F.; Berg, S.; Paternina Visbal, H.; Sachs, D.H.; Berglund, E.; Berglund, D. CD2 Immunobiology. Front. Immunol. 2020, 11, 1090. [Google Scholar] [CrossRef]
- Dunlock, V.M.E.; Arp, A.B.; Singh, S.P.; Charrin, S.; Nguyen, V.; Jansen, E.; Schaper, F.; Ter Beest, M.; Zuidscherwoude, M.; van Deventer, S.J.; et al. Tetraspanin CD53 Controls T Cell Immunity through Regulation of CD45RO Stability, Mobility, and Function. Cell Rep. 2022, 39, 111006. [Google Scholar] [CrossRef]
- Kröger, A. IRFs as Competing Pioneers in T-Cell Differentiation. Cell. Mol. Immunol. 2017, 14, 649–651. [Google Scholar] [CrossRef]
- Ghislat, G.; Cheema, A.S.; Baudoin, E.; Verthuy, C.; Ballester, P.J.; Crozat, K.; Attaf, N.; Dong, C.; Milpied, P.; Malissen, B.; et al. NF-KB–Dependent IRF1 Activation Programs CDC1 Dendritic Cells to Drive Antitumor Immunity. Sci. Immunol. 2021, 6, eabg3570. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Schaafsma, E.; Gorlov, I.P.; Hernando, E.; Thomas, N.E.; Shen, R.; Turk, M.J.; Berwick, M.; Amos, C.I.; Cheng, C. A Leukocyte Infiltration Score Defined by a Gene Signature Predicts Melanoma Patient Prognosis. Mol. Cancer Res. 2019, 17, 109–119. [Google Scholar] [CrossRef] [PubMed]
- Yokoyama, S.; Takahashi, A.; Kikuchi, R.; Nishibu, S.; Lo, J.A.; Hejna, M.; Moon, W.M.; Kato, S.; Zhou, Y.; Hodi, F.S.; et al. SOX10 Regulates Melanoma Immunogenicity through an IRF4-IRF1 Axis. Cancer Res. 2021, 81, 6131–6141. [Google Scholar] [CrossRef]
- Zhao, K.; Yi, Y.; Ma, Z.; Zhang, W. INHBA Is a Prognostic Biomarker and Correlated with Immune Cell Infiltration in Cervical Cancer. Front. Genet. 2022, 12, 2675. [Google Scholar] [CrossRef]
- Wang, Y.; Ryner, L.; Udyavar, A.R.; Desbois, M.; Kozlowski, C.; Chang, C.-W.; Guan, Y.; Lu, S.; Koeppen, H.; Ziai, J.; et al. Reactive Stroma Mediates CD8+ T Cell Spatial Distribution and Function in Ovarian Cancer. Ann. Oncol. 2017, 28, v349. [Google Scholar] [CrossRef]
- Ilieva, K.M.; Correa, I.; Josephs, D.H.; Karagiannis, P.; Egbuniwe, I.U.; Cafferkey, M.J.; Spicer, J.F.; Harries, M.; Nestle, F.O.; Lacy, K.E.; et al. Effects of BRAF Mutations and BRAF Inhibition on Immune Responses to Melanoma. Mol. Cancer Ther. 2014, 13, 2769. [Google Scholar] [CrossRef] [PubMed]
- Reddy, S.M.; Reuben, A.; Wargo, J.A. Influences of BRAF Inhibitors on the Immune Microenvironment and the Rationale for Combined Molecular and Immune Targeted Therapy. Curr. Oncol. Rep. 2016, 18, 42. [Google Scholar] [CrossRef] [PubMed]
- Cen, S.; Liu, K.; Zheng, Y.; Shan, J.; Jing, C.; Gao, J.; Pan, H.; Bai, Z.; Liu, Z. BRAF Mutation as a Potential Therapeutic Target for Checkpoint Inhibitors: A Comprehensive Analysis of Immune Microenvironment in BRAF Mutated Colon Cancer. Front. Cell Dev. Biol. 2021, 9, 1988. [Google Scholar] [CrossRef] [PubMed]
- Lee, E.; Chuang, H.Y.; Kim, J.W.; Ideker, T.; Lee, D. Inferring Pathway Activity toward Precise Disease Classification. PLoS Comput. Biol. 2008, 4, e1000217. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Feature | Desert | Excluded | Inflamed | p-Value |
|---|---|---|---|---|
| N | 95 | 75 | 176 | |
| Age, median (range) | 58 (15–86) | 62 (20–90) | 56 (18–90) | 0.031 |
| BMI, median (range) | 26.1 (17.8–42.5) | 27.3 (17.6–55.5) | 28.7 (18.3–49.1) | 0.045 |
| TMB mut/Mb, median (range) | 12.3 (0.6–129.9) | 16.4 (0.3–102.2) | 25.2 (0.2–1060.3) | 0.403 |
| Stage | 0.003 | |||
| I | 8 (9%) | 10 (14%) | 40 (25%) | |
| II | 41 (47%) | 34 (48%) | 46 (29%) | |
| III | 33 (38%) | 23 (32%) | 68 (42%) | |
| IV | 5 (6%) | 4 (6%) | 6 (4%) | |
| 28 undocumented | ||||
| Clark level | 0.402 | |||
| 1 | 2 (3%) | 1 (2%) | 2 (2%) | |
| 2 | 2 (3%) | 1 (2%) | 11 (10%) | |
| 3 | 9 (13%) | 13 (22%) | 36 (31%) | |
| 4 | 44 (62%) | 35 (60%) | 52 (45%) | |
| 5 | 14 (20%) | 8 (14%) | 14 (12%) | |
| 102 undocumented | ||||
| Breslow in mm, median (range) | 5.8 (0.0–75.0) | 7.3 (0.5–29.0) | 4.2 (0.0–74.0) | 0.032 |
| BRAF status | <0.001 | |||
| wild type | 34 (72%) | 14 (30%) | 56 (47%) | |
| mutated | 13 (28%) | 32 (70%) | 63 (53%) | |
| 134 undocumented | ||||
| NRAS status | 0.049 | |||
| wild type | 25 (53%) | 33 (72%) | 87 (72%) | |
| mutated | 22 (47%) | 13 (28%) | 22 (28%) | |
| 134 undocumented | ||||
| KIT status | 0.010 | |||
| wild type | 42 (89%) | 39 (85%) | 116 (98%) | |
| mutated | 5 (11%) | 7 (15%) | 3 (2%) | |
| 134 undocumented | ||||
| Group | mOS, Months (95% CI) | Estimated Survival, % (95% CI) | Univariate Analysis | Multivariate Analysis * | |||
|---|---|---|---|---|---|---|---|
| HR (95% CI) | padj | HR (95% CI) | padj | ||||
| OVERALL SURVIVAL | |||||||
| 3 years | 5 years | ||||||
| INF | 162.0 (72.8–133.5) | 82.7 (77.4–90.0) | 73.4 (66.7–81.9) | 1.00 (ref.) | 1.00 (ref.) | ||
| EXC | 61.2 (37.1–85.4) | 72.4 (62.5–85.7) | 50.1 (37.2–64.7) | 1.81 (1.20–2.72) | 0.004 | 1.62 (1.03–2.57) | 0.036 |
| DES | 55.6 (23.1–87.9) | 57.2 (46.5–69.7) | 48.2 (36.7–60.8) | 2.38 (1.60–3.41) | 0.002 | 1.57 (0.97–2.55) | 0.056 |
| PROGRESSION-FREE SURVIVAL | |||||||
| 3 years | 5 years | ||||||
| INF | 65.9 (47.6–84.3) | 72.2 (64.0–78.8) | 51.5 (42.2–60.0) | 1.00 (ref.) | 1.00 (ref.) | ||
| EXC | 47.1 (32.8–61.5) | 58.8 (44.4–60.6) | 37.2 (23.5–50.9) | 1.19 (0.81–1.72) | 0.012 | 1.56 (1.02–2.38) | 0.041 |
| DES | 35.0 (8.8–61.2) | 46.4 (32.5–59.2) | 37.2 (23.9–50.5) | 1.68 (1.18–2.39) | 0.377 | 1.13 (0.72–1.78) | 0.586 |
| Feature | Desert | Excluded | Inflamed | p-Value |
|---|---|---|---|---|
| N | 38 | 29 | 29 | |
| Age, median (range) | 72 (36–88) | 65 (32–92) | 69 (30–88) | 0.456 |
| Stage | 0.985 | |||
| I | 2 (5%) | 1 (3%) | 2 (7%) | |
| II | 19 (50%) | 17 (59%) | 14 (48%) | |
| III | 16 (42%) | 10 (35%) | 12 (41%) | |
| IV | 1 (3%) | 1 (3%) | 1 (3%) | |
| 28 undocumented | ||||
| Clark level | 0.378 | |||
| 3 | 9 (24%) | 13 (46%) | 11 (41%) | |
| 4 | 22 (60%) | 14 (50%) | 14 (52%) | |
| 5 | 6 (16%) | 1 (4%) | 2 (7%) | |
| 4 undocumented | ||||
| Breslow depth | 0.300 | |||
| 1–4 mm | 15 (41%) | 13 (50%) | 17 (63%) | |
| >4 mm | 22 (59%) | 13 (50%) | 10 (37%) | |
| 6 undocumented | ||||
| BRAF status | 0.067 | |||
| wild-type | 28 (74%) | 14 (48%) | 15 (52%) | |
| mutated | 10 (26%) | 15 (52%) | 14 (48%) | |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mlynska, A.; Gibavičienė, J.; Kutanovaitė, O.; Senkus, L.; Mažeikaitė, J.; Kerševičiūtė, I.; Maskoliūnaitė, V.; Rupeikaitė, N.; Sabaliauskaitė, R.; Gaiževska, J.; et al. Defining Melanoma Immune Biomarkers—Desert, Excluded, and Inflamed Subtypes—Using a Gene Expression Classifier Reflecting Intratumoral Immune Response and Stromal Patterns. Biomolecules 2024, 14, 171. https://doi.org/10.3390/biom14020171
Mlynska A, Gibavičienė J, Kutanovaitė O, Senkus L, Mažeikaitė J, Kerševičiūtė I, Maskoliūnaitė V, Rupeikaitė N, Sabaliauskaitė R, Gaiževska J, et al. Defining Melanoma Immune Biomarkers—Desert, Excluded, and Inflamed Subtypes—Using a Gene Expression Classifier Reflecting Intratumoral Immune Response and Stromal Patterns. Biomolecules. 2024; 14(2):171. https://doi.org/10.3390/biom14020171
Chicago/Turabian StyleMlynska, Agata, Jolita Gibavičienė, Otilija Kutanovaitė, Linas Senkus, Julija Mažeikaitė, Ieva Kerševičiūtė, Vygantė Maskoliūnaitė, Neda Rupeikaitė, Rasa Sabaliauskaitė, Justina Gaiževska, and et al. 2024. "Defining Melanoma Immune Biomarkers—Desert, Excluded, and Inflamed Subtypes—Using a Gene Expression Classifier Reflecting Intratumoral Immune Response and Stromal Patterns" Biomolecules 14, no. 2: 171. https://doi.org/10.3390/biom14020171