
Triptolide Administration Alters Immune Responses to Mitigate Insulin Resistance in Obese States

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
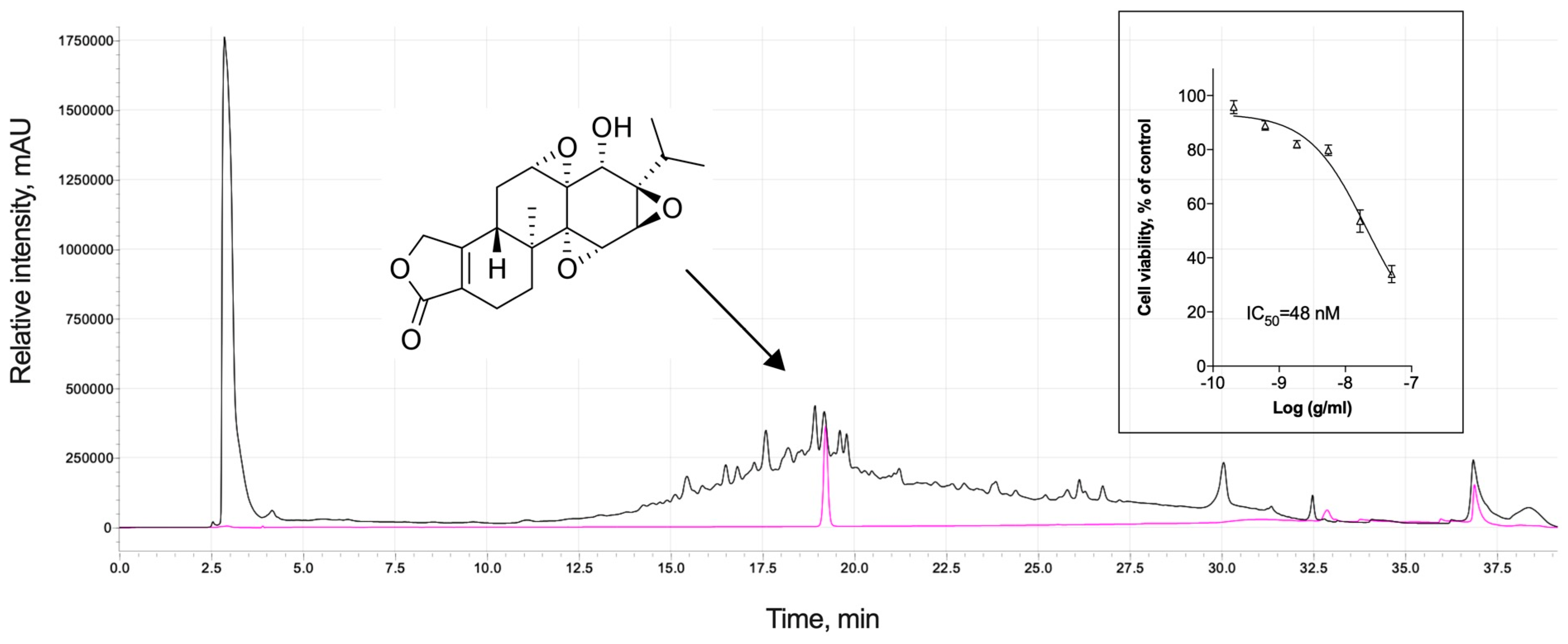
:1. Introduction
2. Materials and Methods
2.1. Reagents and Diets
2.2. Animal Study
2.3. Animal Body Weight, Composition, and Endurance
2.4. Oral Glucose Tolerance and Insulin Tolerance Tests
2.5. Animal Energy Expenditure and Metabolism
2.6. RNA Extraction, Purification, and cDNA Synthesis
2.7. Quantitative PCR Analysis
2.8. Cell Culture
2.9. Analytical Chemistry and HPLC
2.10. Statistical Analysis
3. Results
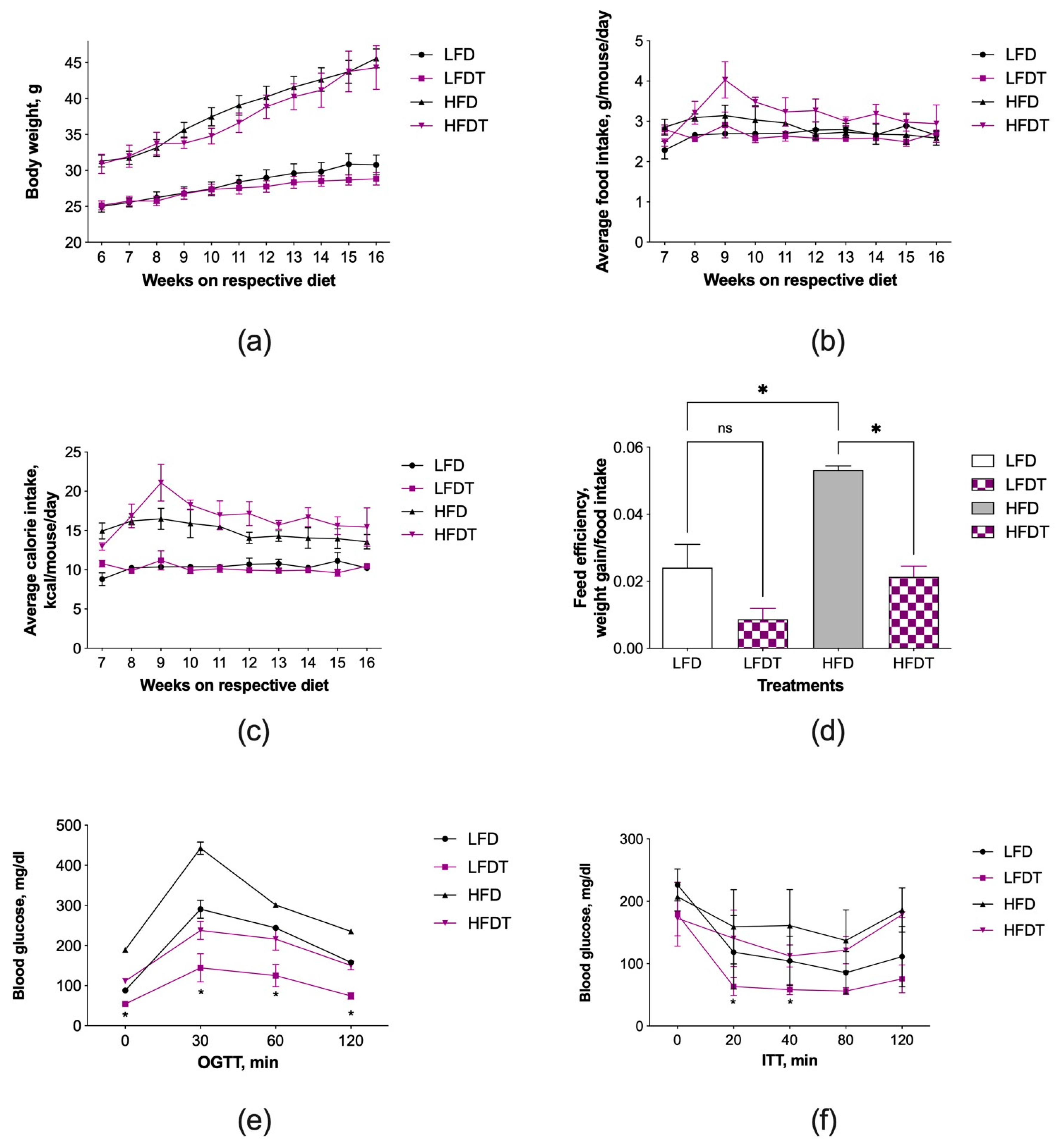
3.1. Triptolide Administration Improves Insulin Resistance without Affecting Obesity
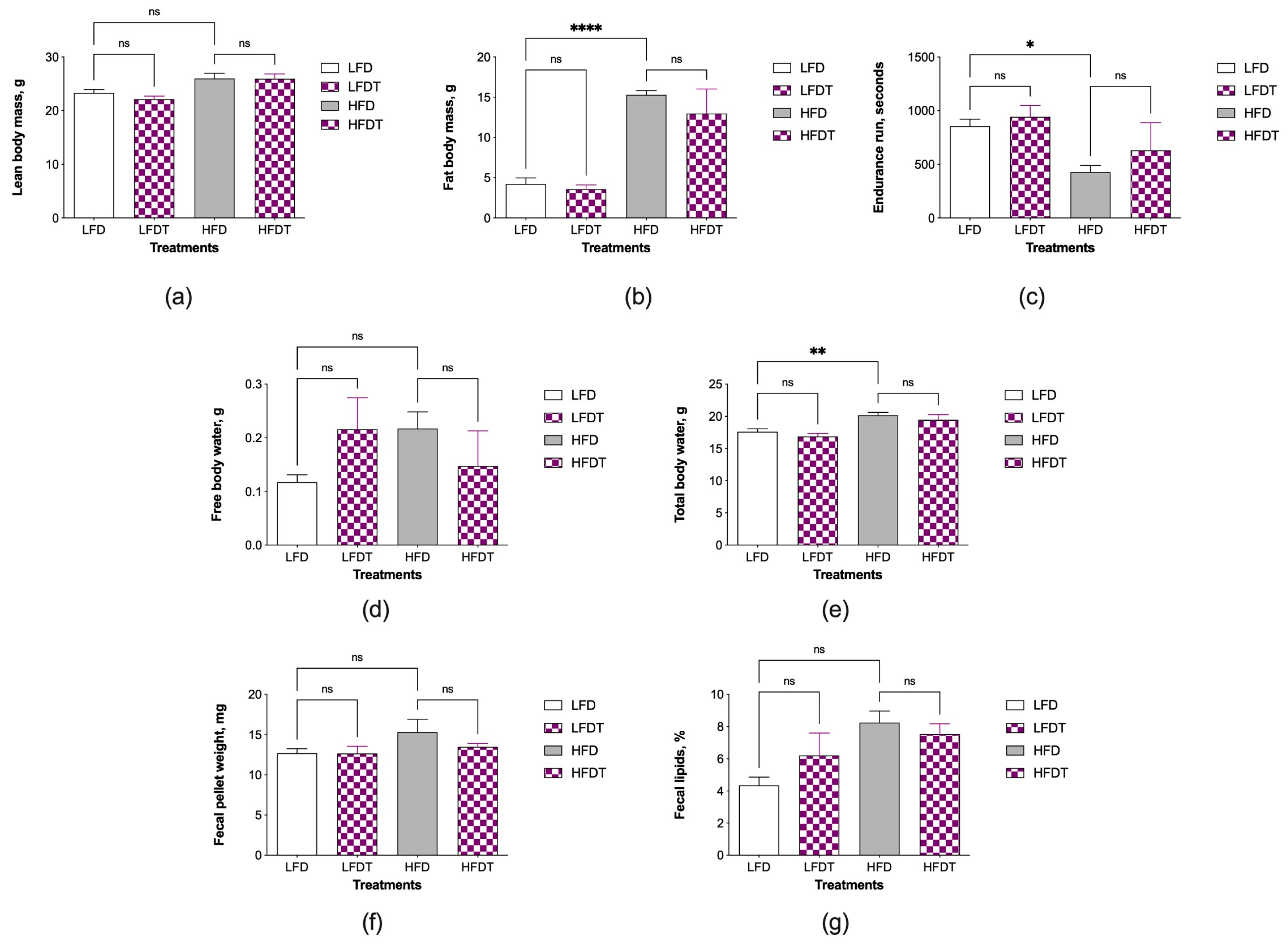
3.2. Changes in Body Composition and Fecal Lipids
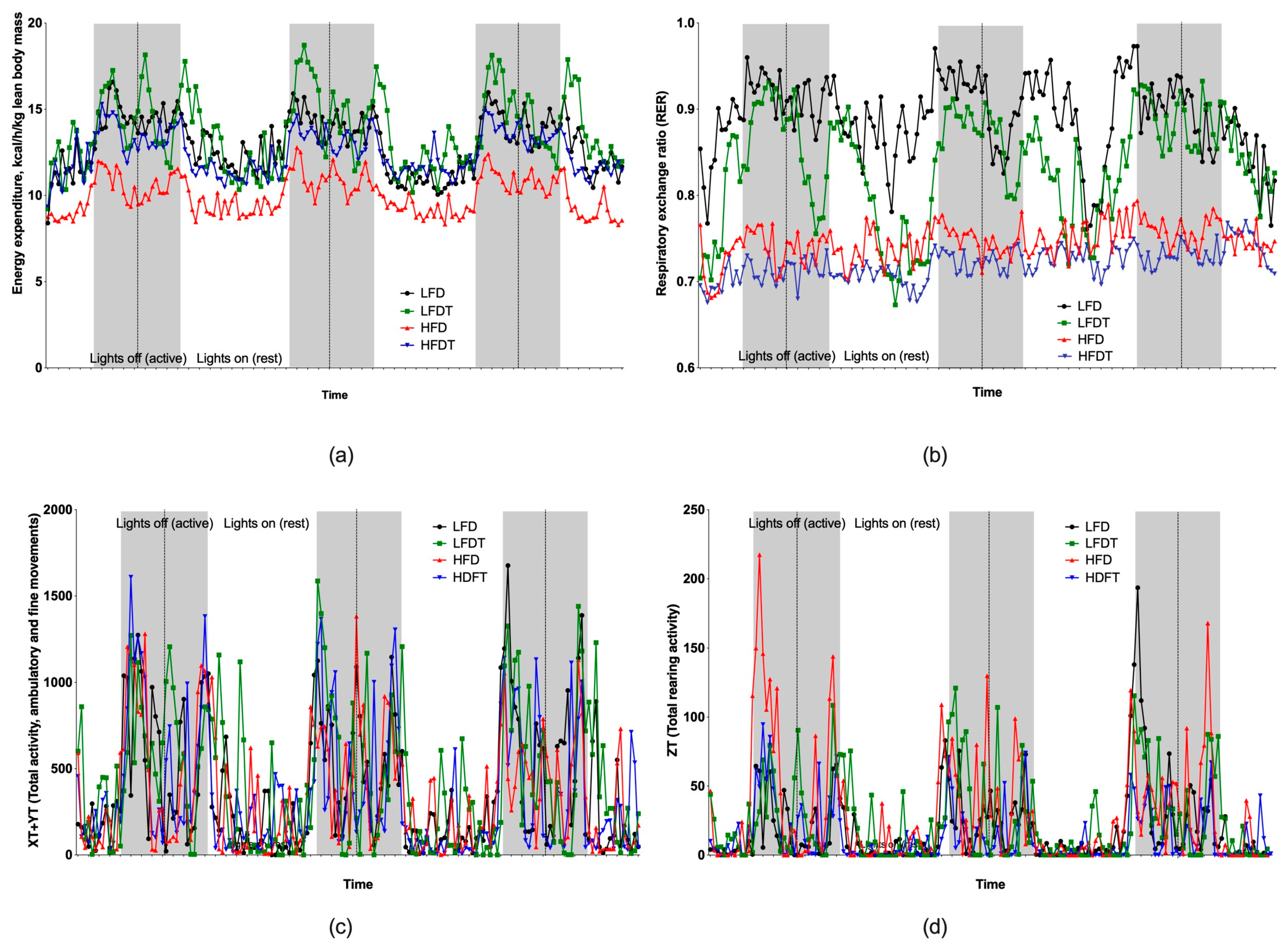
3.3. Changes in Energy Metabolism and Activity
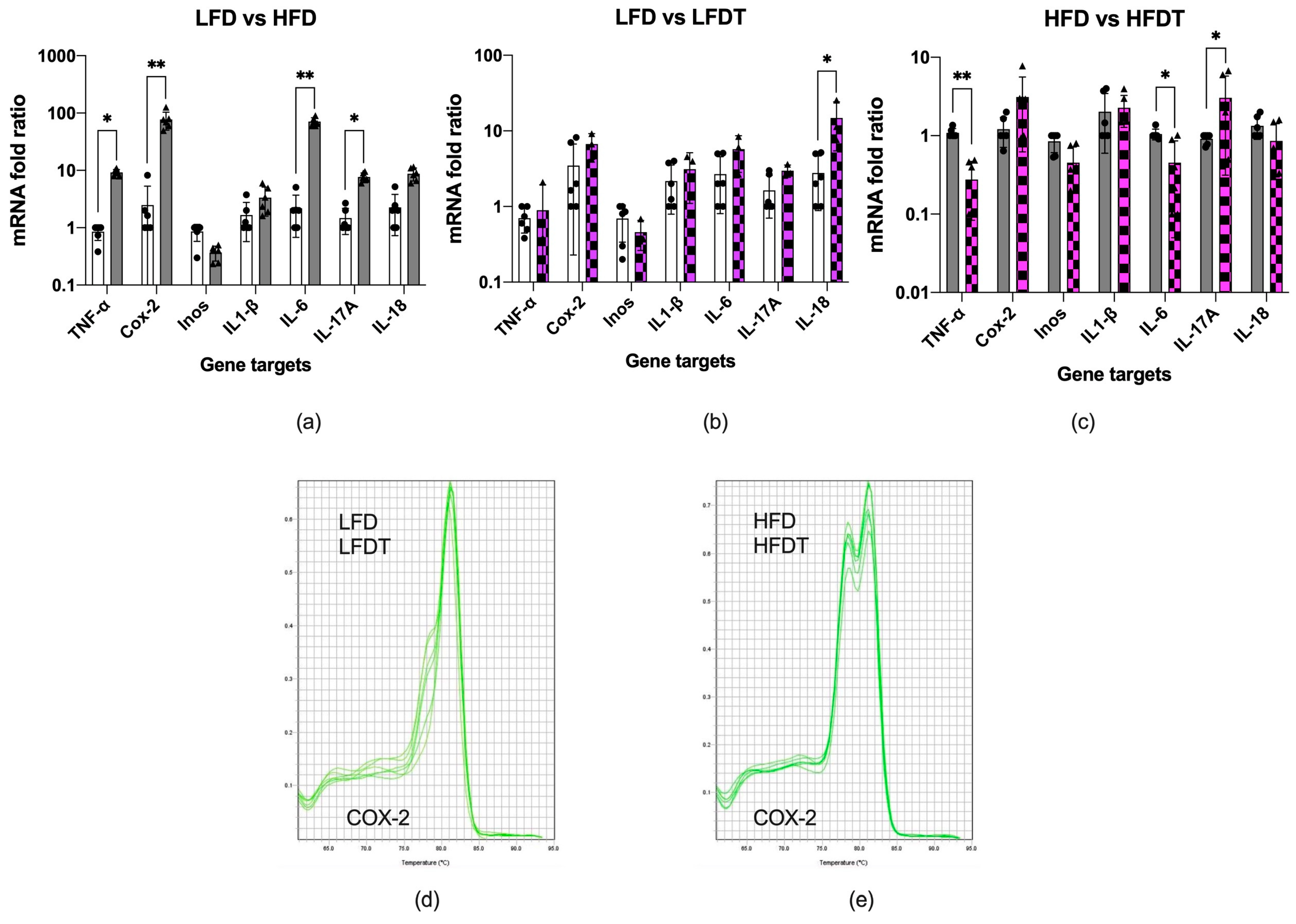
3.4. Inflammation of Adipose Tissue
3.5. Analytical Profile of Triptolide and Cytotoxic Adverse Effects
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Wong, M.C.; McCarthy, C.; Fearnbach, N.; Yang, S.; Shepherd, J.; Heymsfield, S.B. Emergence of the Obesity Epidemic: 6-Decade Visualization with Humanoid Avatars. Am. J. Clin. Nutr. 2022, 115, 1189–1193. [Google Scholar] [CrossRef] [PubMed]
- Hruby, A.; Hu, F.B. The Epidemiology of Obesity: A Big Picture. Pharmacoeconomics 2015, 33, 673–689. [Google Scholar] [CrossRef] [PubMed]
- Cantley, J.; Ashcroft, F.M. Q&A: Insulin Secretion and Type 2 Diabetes: Why Do β-Cells Fail? BMC Biol. 2015, 13, 33. [Google Scholar] [CrossRef]
- Galicia-Garcia, U.; Benito-Vicente, A.; Jebari, S.; Larrea-Sebal, A.; Siddiqi, H.; Uribe, K.B.; Ostolaza, H.; Martín, C. Pathophysiology of Type 2 Diabetes Mellitus. Int. J. Mol. Sci. 2020, 21, 6275. [Google Scholar] [CrossRef] [PubMed]
- Schwartz, S.S.; Epstein, S.; Corkey, B.E.; Grant, S.F.A.; Gavin, J.R.; Aguilar, R.B. The Time Is Right for a New Classification System for Diabetes: Rationale and Implications of the β-Cell–Centric Classification Schema. Diabetes Care 2016, 39, 179–186. [Google Scholar] [CrossRef] [PubMed]
- Choi, J.-Y.; McGregor, R.A.; Kwon, E.-Y.; Kim, Y.J.; Han, Y.; Park, J.H.Y.; Lee, K.W.; Kim, S.-J.; Kim, J.; Yun, J.W.; et al. The Metabolic Response to a High-Fat Diet Reveals Obesity-Prone and -Resistant Phenotypes in Mice with Distinct mRNA-Seq Transcriptome Profiles. Int. J. Obes. 2016, 40, 1452–1460. [Google Scholar] [CrossRef]
- Stefan, N.; Kantartzis, K.; Machann, J.; Schick, F.; Thamer, C.; Rittig, K.; Balletshofer, B.; Machicao, F.; Fritsche, A.; Häring, H.-U. Identification and Characterization of Metabolically Benign Obesity in Humans. Arch. Intern Med. 2008, 168, 1609–1616. [Google Scholar] [CrossRef]
- Muoio, D.M. Metabolic Inflexibility: When Mitochondrial Indecision Leads to Metabolic Gridlock. Cell 2014, 159, 1253–1262. [Google Scholar] [CrossRef]
- Magkos, F.; Fraterrigo, G.; Yoshino, J.; Luecking, C.; Kirbach, K.; Kelly, S.C.; de Las Fuentes, L.; He, S.; Okunade, A.L.; Patterson, B.W.; et al. Effects of Moderate and Subsequent Progressive Weight Loss on Metabolic Function and Adipose Tissue Biology in Humans with Obesity. Cell Metab. 2016, 23, 591–601. [Google Scholar] [CrossRef]
- Rohm, T.V.; Meier, D.T.; Olefsky, J.M.; Donath, M.Y. Inflammation in Obesity, Diabetes, and Related Disorders. Immunity 2022, 55, 31–55. [Google Scholar] [CrossRef]
- Hotamisligil, G.S.; Shargill, N.S.; Spiegelman, B.M. Adipose Expression of Tumor Necrosis Factor-Alpha: Direct Role in Obesity-Linked Insulin Resistance. Science 1993, 259, 87–91. [Google Scholar] [CrossRef] [PubMed]
- Nielsen, S.T.; Janum, S.; Krogh-Madsen, R.; Solomon, T.P.; Møller, K. The Incretin Effect in Critically Ill Patients: A Case–Control Study. Crit. Care 2015, 19, 402. [Google Scholar] [CrossRef] [PubMed]
- Berlanga-Acosta, J.; Mendoza-Marí, Y.; Rodríguez-Rodríguez, N.; García del Barco Herrera, D.; García-Ojalvo, A.; Fernández-Mayola, M.; Guillén-Nieto, G.; Valdés-Sosa, P.A. Burn Injury Insulin Resistance and Central Nervous System Complications: A Review. Burns Open 2020, 4, 41–52. [Google Scholar] [CrossRef]
- Sonagra, A.D.; Biradar, S.M.; Dattatreya, K.; Murthy, D.S.J. Normal Pregnancy- A State of Insulin Resistance. J. Clin. Diagn. Res. 2014, 8, CC01–CC03. [Google Scholar] [CrossRef] [PubMed]
- Williamson, R.T. On the Treatment of Glycosuria and Diabetes Mellitus with Sodium Salicylate. Br. Med. J. 1901, 1, 760–762. [Google Scholar] [CrossRef] [PubMed]
- Yuan, M.; Konstantopoulos, N.; Lee, J.; Hansen, L.; Li, Z.W.; Karin, M.; Shoelson, S.E. Reversal of Obesity- and Diet-Induced Insulin Resistance with Salicylates or Targeted Disruption of Ikkbeta. Science 2001, 293, 1673–1677. [Google Scholar] [CrossRef] [PubMed]
- Hundal, R.S.; Petersen, K.F.; Mayerson, A.B.; Randhawa, P.S.; Inzucchi, S.; Shoelson, S.E.; Shulman, G.I. Mechanism by Which High-Dose Aspirin Improves Glucose Metabolism in Type 2 Diabetes. J. Clin. Investig. 2002, 109, 1321–1326. [Google Scholar] [CrossRef]
- Velikova, T.V.; Kabakchieva, P.P.; Assyov, Y.S.; Georgiev, T.A. Targeting Inflammatory Cytokines to Improve Type 2 Diabetes Control. Biomed. Res. Int. 2021, 2021, 7297419. [Google Scholar] [CrossRef]
- Chu, D.-T.; Malinowska, E.; Jura, M.; Kozak, L.P. C57BL/6J Mice as a Polygenic Developmental Model of Diet-Induced Obesity. Physiol. Rep. 2017, 5, e13093. [Google Scholar] [CrossRef]
- Vispé, S.; DeVries, L.; Créancier, L.; Besse, J.; Bréand, S.; Hobson, D.J.; Svejstrup, J.Q.; Annereau, J.-P.; Cussac, D.; Dumontet, C.; et al. Triptolide Is an Inhibitor of RNA Polymerase I and II-Dependent Transcription Leading Predominantly to down-Regulation of Short-Lived mRNA. Mol. Cancer Ther. 2009, 8, 2780–2790. [Google Scholar] [CrossRef]
- Zeng, R.; Zeng, L.; Chen, Y.; Zhao, F.; Li, R.; Wen, L.; Zhang, C. Triptolide-Induced Apoptosis by Inactivating Nuclear Factor-Kappa B Apoptotic Pathway in Multiple Myeloma in Vitro. J. Huazhong Univ. Sci. Technol. Med. Sci. 2011, 31, 446. [Google Scholar] [CrossRef]
- Huang, J.; Esposito, D.; Kizelsztein, P.; Raskin, I.; Komarnytsky, S. Triptolide Ameliorates Insulin Resistance in Obese Diabetic Mice. Pharm. Biol. 2012, 50, 644. [Google Scholar]
- Skates, E.; Overall, J.; DeZego, K.; Wilson, M.; Esposito, D.; Lila, M.A.; Komarnytsky, S. Berries Containing Anthocyanins with Enhanced Methylation Profiles Are More Effective at Ameliorating High Fat Diet-Induced Metabolic Damage. Food Chem. Toxicol. 2018, 111, 445–453. [Google Scholar] [CrossRef] [PubMed]
- Nair, A.B.; Jacob, S. A Simple Practice Guide for Dose Conversion between Animals and Human. J. Basic Clin. Pharm. 2016, 7, 27–31. [Google Scholar] [CrossRef] [PubMed]
- Milhem, F.; Skates, E.; Wilson, M.; Komarnytsky, S. Obesity-Resistant Mice on a High-Fat Diet Display a Distinct Phenotype Linked to Enhanced Lipid Metabolism. Nutrients 2024, 16, 171. [Google Scholar] [CrossRef] [PubMed]
- Péronnet, F.; Massicotte, D. Table of Nonprotein Respiratory Quotient: An Update. Can. J. Sport Sci. 1991, 16, 23–29. [Google Scholar]
- Lenz, M.; Arts, I.C.W.; Peeters, R.L.M.; de Kok, T.M.; Ertaylan, G. Adipose Tissue in Health and Disease through the Lens of Its Building Blocks. Sci. Rep. 2020, 10, 10433. [Google Scholar] [CrossRef]
- Gao, J.; Zhang, Y.; Liu, X.; Wu, X.; Huang, L.; Gao, W. Triptolide: Pharmacological Spectrum, Biosynthesis, Chemical Synthesis and Derivatives. Theranostics 2021, 11, 7199–7221. [Google Scholar] [CrossRef]
- Yuan, K.; Li, X.; Lu, Q.; Zhu, Q.; Jiang, H.; Wang, T.; Huang, G.; Xu, A. Application and Mechanisms of Triptolide in the Treatment of Inflammatory Diseases—A Review. Front. Pharmacol. 2019, 10, 1469. [Google Scholar] [CrossRef]
- Luo, X.-L.; Shao, Q.; Qu, H.-B.; Cheng, Y.-Y. Simple Method for Determination of Five Terpenoids from Different Parts of Tripterygium Wilfordii and Its Preparations by HPLC Coupled with Evaporative Light Scattering Detection. J. Sep. Sci. 2007, 30, 1284–1291. [Google Scholar] [CrossRef]
- Jiang, M.; Sheng, F.; Zhang, Z.; Ma, X.; Gao, T.; Fu, C.; Li, P. Andrographis Paniculata (Burm.f.) Nees and Its Major Constituent Andrographolide as Potential Antiviral Agents. J. Ethnopharmacol. 2021, 272, 113954. [Google Scholar] [CrossRef]
- Martens, C.R.; Bansal, S.S.; Accornero, F. Cardiovascular Inflammation: RNA Takes the Lead. J. Mol. Cell Cardiol. 2019, 129, 247–256. [Google Scholar] [CrossRef] [PubMed]
- Gao, Q.; Shen, W.; Qin, W.; Zheng, C.; Zhang, M.; Zeng, C.; Wang, S.; Wang, J.; Zhu, X.; Liu, Z. Treatment of Db/Db Diabetic Mice with Triptolide: A Novel Therapy for Diabetic Nephropathy. Nephrol. Dial. Transplant. 2010, 25, 3539–3547. [Google Scholar] [CrossRef] [PubMed]
- Luo, L.; Yang, T. Triptolide Inhibits the Progression of Atherosclerosis in Apolipoprotein E−/− Mice. Exp. Ther. Med. 2016, 12, 2307–2313. [Google Scholar] [CrossRef] [PubMed]
- Huang, R.; Guo, F.; Li, Y.; Liang, Y.; Li, G.; Fu, P.; Ma, L. Activation of AMPK by Triptolide Alleviates Nonalcoholic Fatty Liver Disease by Improving Hepatic Lipid Metabolism, Inflammation and Fibrosis. Phytomedicine 2021, 92, 153739. [Google Scholar] [CrossRef] [PubMed]
- Kizelsztein, P.; Komarnytsky, S.; Raskin, I. Oral Administration of Triptolide Ameliorates the Clinical Signs of Experimental Autoimmune Encephalomyelitis (EAE) by Induction of HSP70 and Stabilization of NF-κB/IκBα Transcriptional Complex. J. Neuroimmunol. 2009, 217, 28–37. [Google Scholar] [CrossRef] [PubMed]
- Fan, D.; Guo, Q.; Shen, J.; Zheng, K.; Lu, C.; Zhang, G.; Lu, A.; He, X. The Effect of Triptolide in Rheumatoid Arthritis: From Basic Research towards Clinical Translation. Int. J. Mol. Sci. 2018, 19, 376. [Google Scholar] [CrossRef]
- Noel, P.; Von Hoff, D.D.; Saluja, A.K.; Velagapudi, M.; Borazanci, E.; Han, H. Triptolide and Its Derivatives as Cancer Therapies. Trends Pharmacol. Sci. 2019, 40, 327–341. [Google Scholar] [CrossRef]
- Durward, C.M.; Hartman, T.J.; Nickols-Richardson, S.M. All-Cause Mortality Risk of Metabolically Healthy Obese Individuals in NHANES III. J. Obes. 2012, 2012, 460321. [Google Scholar] [CrossRef]
- O’Neill, L.A.J.; Kishton, R.J.; Rathmell, J. A Guide to Immunometabolism for Immunologists. Nat. Rev. Immunol. 2016, 16, 553–565. [Google Scholar] [CrossRef]
- Fajgenbaum, D.C.; June, C.H. Cytokine Storm. N. Engl. J. Med. 2020, 383, 2255–2273. [Google Scholar] [CrossRef] [PubMed]
- Chen, G.; Goeddel, D.V. TNF-R1 Signaling: A Beautiful Pathway. Science 2002, 296, 1634–1635. [Google Scholar] [CrossRef]
- Dominguez, H.; Storgaard, H.; Rask-Madsen, C.; Steffen Hermann, T.; Ihlemann, N.; Baunbjerg Nielsen, D.; Spohr, C.; Kober, L.; Vaag, A.; Torp-Pedersen, C. Metabolic and Vascular Effects of Tumor Necrosis Factor-Alpha Blockade with Etanercept in Obese Patients with Type 2 Diabetes. J. Vasc. Res. 2005, 42, 517–525. [Google Scholar] [CrossRef] [PubMed]
- Paquot, N.; Castillo, M.J.; Lefèbvre, P.J.; Scheen, A.J. No Increased Insulin Sensitivity after a Single Intravenous Administration of a Recombinant Human Tumor Necrosis Factor Receptor: Fc Fusion Protein in Obese Insulin-Resistant Patients. J. Clin. Endocrinol. Metab. 2000, 85, 1316–1319. [Google Scholar] [CrossRef] [PubMed]
- Pan, Y.; Cao, S.; Tang, J.; Arroyo, J.P.; Terker, A.S.; Wang, Y.; Niu, A.; Fan, X.; Wang, S.; Zhang, Y.; et al. Cyclooxygenase-2 in Adipose Tissue Macrophages Limits Adipose Tissue Dysfunction in Obese Mice. J. Clin. Investig. 2022, 132, e152391. [Google Scholar] [CrossRef] [PubMed]
- Bechara, R.; McGeachy, M.J.; Gaffen, S.L. The Metabolism-Modulating Activity of IL-17 Signaling in Health and Disease. J. Exp. Med. 2021, 218, e20202191. [Google Scholar] [CrossRef] [PubMed]
- Pietruszewska, W.; Fendler, W.; Podwysocka, M.; Białas, A.J.; Kuna, P.; Kupryś-Lipińska, I.; Borowiec, M. Expression of Transcript Variants of PTGS1 and PTGS2 Genes among Patients with Chronic Rhinosinusitis with Nasal Polyps. Diagnostics 2021, 11, 135. [Google Scholar] [CrossRef]





Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Grodsky, L.; Wilson, M.; Rathinasabapathy, T.; Komarnytsky, S. Triptolide Administration Alters Immune Responses to Mitigate Insulin Resistance in Obese States. Biomolecules 2024, 14, 395. https://doi.org/10.3390/biom14040395
Grodsky L, Wilson M, Rathinasabapathy T, Komarnytsky S. Triptolide Administration Alters Immune Responses to Mitigate Insulin Resistance in Obese States. Biomolecules. 2024; 14(4):395. https://doi.org/10.3390/biom14040395
Chicago/Turabian StyleGrodsky, Lyudmila, Mickey Wilson, Thirumurugan Rathinasabapathy, and Slavko Komarnytsky. 2024. "Triptolide Administration Alters Immune Responses to Mitigate Insulin Resistance in Obese States" Biomolecules 14, no. 4: 395. https://doi.org/10.3390/biom14040395