
Ca2+ Influx through TRPC Channels Is Regulated by Homocysteine–Copper Complexes
 , ,
, , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cell Culture and Transfection
2.2. Electrophysiological Recordings and Ca2+ Measurements
2.3. RT-PCR
2.4. Cell Proliferation, Migration, and Angiogenesis Assays
2.5. Reagents and Drugs
2.6. Statistics
3. Results
3.1. Ca2+ Influx Induced by Hcy in HAECs
3.2. Hcy-Induced Ca2+ Influx through TRPC4 and TRPC5 Channels
3.3. Activation of TRPC4 and TRPC5 by Divalent Cu2+ and the Interference by Hcy
3.4. No Effect of Monovalent Cu+ on TRPC Channel
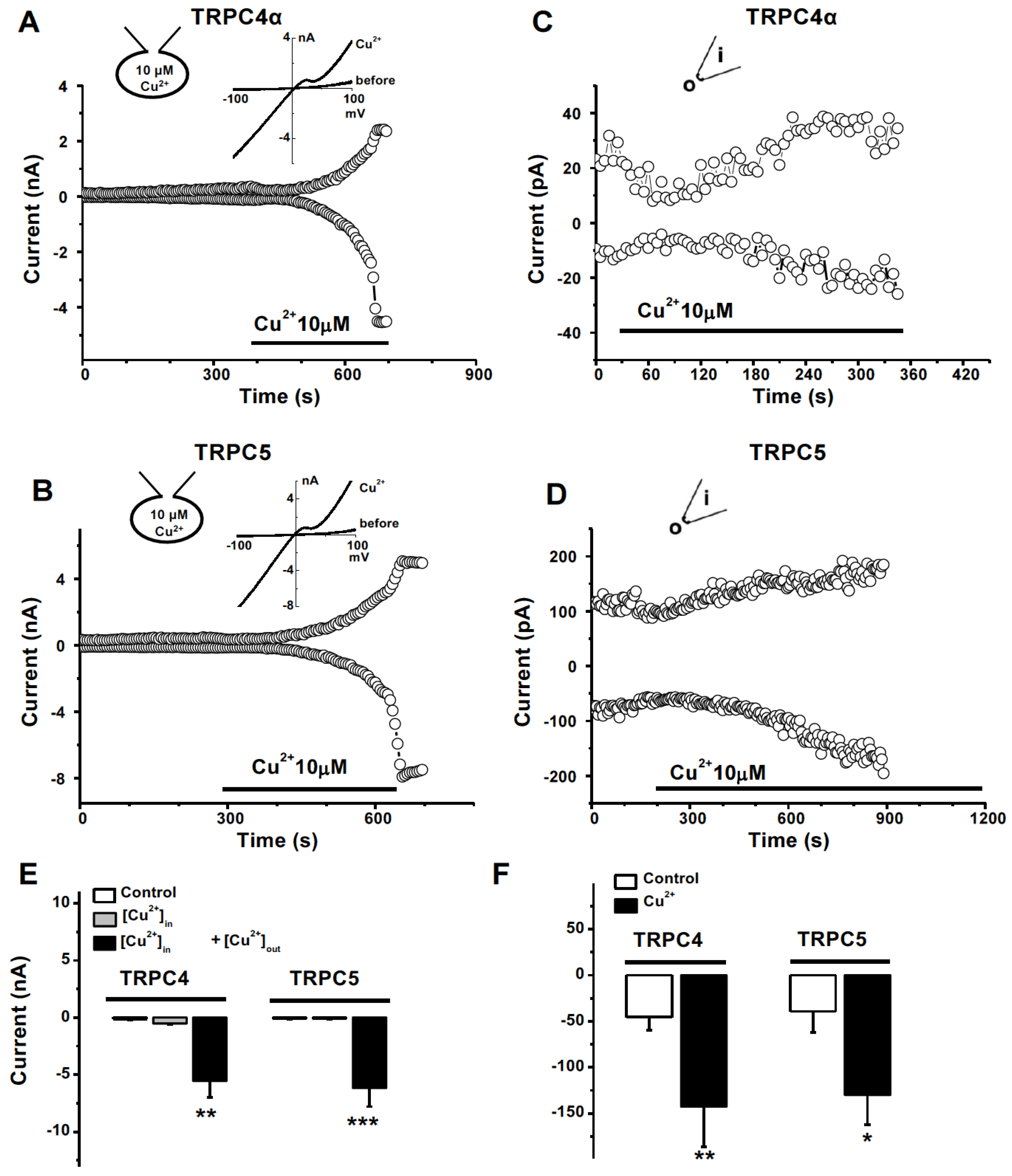
3.5. Extracellular Activation of Cu2+ on TRPC4 and 5 Channels
3.6. Amino acid Residues of TRPC4 Involved in Copper Activation
3.7. TRPC and Homocysteine-Copper Complexes in the Regulation of Endothelial Cell Proliferation
3.8. Hcy–Copper Complexes in the Regulation of Cell Migration and Angiogenesis
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- McCully, K.S. Vascular pathology of homocysteinemia: Implications for the pathogenesis of arteriosclerosis. Am. J. Pathol. 1969, 56, 111–128. [Google Scholar] [PubMed]
- Jensen, M.K.; Bertoia, M.L.; Cahill, L.E.; Agarwal, I.; Rimm, E.B.; Mukamal, K.J. Novel metabolic biomarkers of cardiovascular disease. Nat. Rev. Endocrinol. 2014, 10, 659–672. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.-C.; Su, H.-M.; Chang, J.-M.; Liu, W.-C.; Tsai, J.-C.; Tsai, Y.-C.; Lin, M.-Y.; Hwang, S.-J.; Chen, H.-C. Increasing prevalence of peripheral artery occlusive disease in hemodialysis patients: A 2-year follow-up. Am. J. Med. Sci. 2012, 343, 440–445. [Google Scholar] [CrossRef] [PubMed]
- Schaffer, A.; Verdoia, M.; Cassetti, E.; Marino, P.; Suryapranata, H.; De Luca, G.; Novara Atherosclerosis Study Group (NAS). Relationship between homocysteine and coronary artery disease. Results from a large prospective cohort study. Thromb. Res. 2014, 134, 288–293. [Google Scholar] [CrossRef]
- Zylberstein, D.E.; Bengtsson, C.; Björkelund, C.; Landaas, S.; Sundh, V.; Thelle, D.; Lissner, L. Serum homocysteine in relation to mortality and morbidity from coronary heart disease: A 24-year follow-up of the population study of women in Gothenburg. Circulation 2004, 109, 601–606. [Google Scholar] [CrossRef] [Green Version]
- Homocysteine Studies Collaboration. Homocysteine and risk of ischemic heart disease and stroke: A meta-analysis. JAMA 2002, 288, 2015–2022. [Google Scholar] [CrossRef]
- Zylberstein, D.E.; Skoog, I.; Björkelund, C.; Guo, X.; Hultén, B.; Andreasson, L.-A.; Palmertz, B.; Thelle, D.S.; Lissner, L. Homocysteine levels and lacunar brain infarcts in elderly women: The prospective population study of women in Gothenburg. J. Am. Geriatr. Soc. 2008, 56, 1087–1091. [Google Scholar] [CrossRef]
- Casas, J.P.; Bautista, L.E.; Smeeth, L.; Sharma, P.; Hingorani, A.D. Homocysteine and stroke: Evidence on a causal link from mendelian randomisation. Lancet 2005, 365, 224–232. [Google Scholar] [CrossRef]
- Kuan, Y.M.; Dear, A.E.; Grigg, M.J. Homocysteine: An aetiological contributor to peripheral vascular arterial disease. ANZ J. Surg. 2002, 72, 668–671. [Google Scholar] [CrossRef]
- Den Heijer, M.; Lewington, S.; Clarke, R. Homocysteine, MTHFR and risk of venous thrombosis: A meta-analysis of published epidemiological studies. J. Thromb. Haemost. 2005, 3, 292–299. [Google Scholar] [CrossRef]
- Loscalzo, J. Homocysteine and dementias. N. Engl. J. Med. 2002, 346, 466–468. [Google Scholar] [CrossRef]
- Rozycka, A.; Jagodzinski, P.P.; Kozubski, W.; Lianeri, M.; Dorszewska, J. Homocysteine Level and Mechanisms of Injury in Parkinson’s Disease as Related to MTHFR, MTR, and MTHFD1 Genes Polymorphisms and L-Dopa Treatment. Curr. Genom. 2013, 14, 534–542. [Google Scholar] [CrossRef] [Green Version]
- Elias, A.N.; Eng, S. Homocysteine concentrations in patients with diabetes mellitus-relationship to microvascular and macrovascular disease. Diabetes Obes. Metab. 2005, 7, 117–121. [Google Scholar] [CrossRef]
- Van Meurs, J.B.; Dhonukshe-Rutten, R.A.; Pluijm, S.M.; Van Der Klift, M.; De Jonge, R.; Lindemans, J.; De Groot, L.C.; Hofman, A.; Witteman, J.C.; Van Leeuwen, J.P.; et al. Homocysteine levels and the risk of osteoporotic fracture. N. Engl. J. Med. 2004, 350, 2033–2041. [Google Scholar] [CrossRef] [Green Version]
- Yi, F.; Li, P.L. Mechanisms of homocysteine-induced glomerular injury and sclerosis. Am. J. Nephrol. 2008, 28, 254–264. [Google Scholar] [CrossRef] [Green Version]
- Mills, J.L.; Lee, Y.J.; Conley, M.R.; Kirke, P.N.; McPartlin, J.M.; Weir, D.G.; Scott, J.M. Homocysteine metabolism in pregnancies complicated by neural-tube defects. Lancet 1995, 345, 149–151. [Google Scholar] [CrossRef]
- Adinolfi, L.E.; Ingrosso, D.; Cesaro, G.; Cimmino, A.; D’Antò, M.; Capasso, R.; Zappia, V.; Ruggiero, G. Hyperhomocysteinemia and the MTHFR C677T polymorphism promote steatosis and fibrosis in chronic hepatitis C patients. Hepatology 2005, 41, 995–1003. [Google Scholar] [CrossRef]
- Wang, L.; Chen, X.; Tang, B.; Hua, X.; Klein-Szanto, A.; Kruger, W.D. Expression of mutant human cystathionine {beta}-synthase rescues neonatal lethality but not homocystinuria in a mouse model. Hum. Mol. Genet. 2005, 14, 2201–2208. [Google Scholar] [CrossRef]
- Austin, R.C.; Lentz, S.R.; Werstuck, G.H. Role of hyperhomocysteinemia in endothelial dysfunction and atherothrombotic disease. Cell Death Differ. 2004, 11 (Suppl. S1), S56–S64. [Google Scholar] [CrossRef] [Green Version]
- Becker, J.S.; Adler, A.; Schneeberger, A.; Huang, H.; Wang, Z.; Walsh, E.; Koller, A.; Hintze, T.H. Hyperhomocysteinemia, a cardiac metabolic disease: Role of nitric oxide and the p22phox subunit of NADPH oxidase. Circulation 2005, 111, 2112–2118. [Google Scholar] [CrossRef] [Green Version]
- Toole, J.; Malinow, M.; Chambless, L. Lowering homocysteine in patients with ischemic stroke to prevent recurrent stroke, myocardial infarction, and death: The Vitamin Intervention for Stroke Prevention (VISP) randomized controlled trial. JAMA 2004, 291, 565–575. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Spence, J.D.; Stampfer, M.J. Understanding the complexity of homocysteine lowering with vitamins: The potential role of subgroup analyses. JAMA 2011, 306, 2610–2611. [Google Scholar] [CrossRef] [PubMed]
- Mujumdar, V.S.; Hayden, M.R.; Tyagi, S.C. Homocyst(e)ine induces calcium second messenger in vascular smooth muscle cells. J. Cell Physiol. 2000, 183, 28–36. [Google Scholar] [CrossRef]
- Abushik, P.A.; Niittykoski, M.; Giniatullina, R.; Shakirzyanova, A.; Bart, G.; Fayuk, D.; Sibarov, D.A.; Antonov, S.M.; Giniatullin, R. The role of NMDA and mGluR5 receptors in calcium mobilization and neurotoxicity of homocysteine in trigeminal and cortical neurons and glial cells. J. Neurochem. 2014, 129, 264–274. [Google Scholar] [CrossRef]
- Ganapathy, P.S.; White, R.E.; Ha, Y.; Bozard, B.R.; McNeil, P.L.; Caldwell, R.W.; Kumar, S.; Black, S.M.; Smith, S.B. The role of N-methyl-D-aspartate receptor activation in homocysteine-induced death of retinal ganglion cells. Investig. Ophthalmol. Vis. Sci. 2011, 52, 5515–5524. [Google Scholar] [CrossRef]
- Xu, S.-Z.; Muraki, K.; Zeng, F.; Li, J.; Sukumar, P.; Shah, S.; Dedman, A.M.; Flemming, P.K.; McHugh, D.; Naylor, J.; et al. A sphingosine-1-phosphate-activated calcium channel controlling vascular smooth muscle cell motility. Circ. Res. 2006, 98, 1381–1389. [Google Scholar] [CrossRef]
- Kumar, B.; Dreja, K.; Shah, S.S.; Cheong, A.; Xu, S.Z.; Sukumar, P.; Naylor, J.; Forte, A.; Cipollaro, M.; McHugh, D.; et al. Upregulated TRPC1 channel in vascular injury in vivo and its role in human neointimal hyperplasia. Circ. Res. 2006, 98, 557–563. [Google Scholar] [CrossRef] [Green Version]
- Xu, S.Z.; Beech, D.J. TrpC1 is a membrane-spanning subunit of store-operated Ca2+ channels in native vascular smooth muscle cells. Circ. Res. 2001, 88, 84–87. [Google Scholar] [CrossRef] [Green Version]
- Beech, D.J.; Muraki, K.; Flemming, R. Non-selective cationic channels of smooth muscle and the mammalian homologues of Drosophila TRP. J. Physiol. 2004, 559 Pt 3, 685–706. [Google Scholar] [CrossRef]
- Kang, Y.J. Copper and homocysteine in cardiovascular diseases. Pharmacol. Ther. 2010, 129, 321–331. [Google Scholar] [CrossRef]
- Mansoor, M.A.; Bergmark, C.; Haswell, S.J.; Savage, I.F.; Evans, P.H.; Berge, R.K.; Svardal, A.M.; Kristensen, O. Correlation between plasma total homocysteine and copper in patients with peripheral vascular disease. Clin. Chem. 2000, 46, 385–391. [Google Scholar] [CrossRef]
- Dudman, N.P.; Wilcken, D.E. Increased plasma copper in patients with homocystinuria due to cystathionine beta-synthase deficiency. Clin. Chim. Acta 1983, 127, 105–113. [Google Scholar] [CrossRef]
- Gromadzka, G.; Rudnicka, M.; Chabik, G.; Przybyłkowski, A.; Członkowska, A. Genetic variability in the methylenetetrahydrofolate reductase gene (MTHFR) affects clinical expression of Wilson’s disease. J. Hepatol. 2011, 55, 913–919. [Google Scholar] [CrossRef]
- Zhang, L.; Ward, M.-L.; Phillips, A.R.; Zhang, S.; Kennedy, J.; Barry, B.; Cannell, M.B.; Cooper, G.J. Protection of the heart by treatment with a divalent-copper-selective chelator reveals a novel mechanism underlying cardiomyopathy in diabetic rats. Cardiovasc. Diabetol. 2013, 12, 123. [Google Scholar] [CrossRef] [Green Version]
- Xu, S.Z.; Zhong, W.; Watson, N.M.; Dickerson, E.; Wake, J.D.; Lindow, S.W.; Newton, C.J.; Atkin, S.L. Fluvastatin reduces oxidative damage in human vascular endothelial cells by upregulating Bcl-2. J. Thromb. Haemost. 2008, 6, 692–700. [Google Scholar] [CrossRef]
- Daskoulidou, N.; Zeng, B.; Berglund, L.M.; Jiang, H.; Chen, G.-L.; Kotova, O.; Bhandari, S.; Ayoola, J.; Griffin, S.; Atkin, S.L.; et al. High glucose enhances store-operated calcium entry by upregulating ORAI/STIM via calcineurin-NFAT signalling. J. Mol. Med. 2015, 93, 511–521. [Google Scholar] [CrossRef]
- Xu, S.-Z.; Sukumar, P.; Zeng, F.; Li, J.; Jairaman, A.; English, A.; Naylor, J.; Ciurtin, C.; Majeed, Y.; Milligan, C.J.; et al. TRPC channel activation by extracellular thioredoxin. Nature 2008, 451, 69–72. [Google Scholar] [CrossRef] [Green Version]
- Jiang, H.; Zeng, B.; Chen, G.-L.; Bot, D.; Eastmond, S.; Elsenussi, S.E.; Atkin, S.L.; Boa, A.N.; Xu, S.-Z. Effect of non-steroidal anti-inflammatory drugs and new fenamate analogues on TRPC4 and TRPC5 channels. Biochem. Pharmacol. 2012, 83, 923–931. [Google Scholar] [CrossRef]
- Chen, G.-L.; Zeng, B.; Eastmond, S.; Elsenussi, S.E.; Boa, A.; Xu, S.-Z. Pharmacological comparison of novel synthetic fenamate analogues with econazole and 2-APB on the inhibition of TRPM2 channels. Br. J. Pharmacol. 2012, 167, 1232–1243. [Google Scholar] [CrossRef] [Green Version]
- Li, P.; Rubaiy, H.N.; Chen, G.L.; Hallett, T.; Zaibi, N.; Zeng, B.; Saurabh, R.; Xu, S.Z. Mibefradil, a T-type Ca(2+) channel blocker also blocks Orai channels by action at the extracellular surface. Br. J. Pharmacol. 2019, 176, 3845–3856. [Google Scholar] [CrossRef]
- Xu, S.-Z.; Zeng, B.; Daskoulidou, N.; Chen, G.-L.; Atkin, S.L.; Lukhele, B. Activation of TRPC cationic channels by mercurial compounds confers the cytotoxicity of mercury exposure. Toxicol. Sci. 2012, 125, 56–68. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zeng, B.; Yuan, C.; Yang, X.; Atkin, S.L.; Xu, S.-Z. TRPC channels and their splice variants are essential for promoting human ovarian cancer cell proliferation and tumorigenesis. Curr. Cancer Drug Targets 2013, 13, 103–116. [Google Scholar] [CrossRef] [PubMed]
- Zaibi, N.; Li, P.; Xu, S.Z. Protective effects of dapagliflozin against oxidative stress-induced cell injury in human proximal tubular cells. PLoS ONE 2021, 16, e0247234. [Google Scholar] [CrossRef] [PubMed]
- Aranda, E.; Owen, G.I. A semi-quantitative assay to screen for angiogenic compounds and compounds with angiogenic potential using the EA.hy926 endothelial cell line. Biol. Res. 2009, 42, 377–389. [Google Scholar] [CrossRef] [Green Version]
- Xu, S.-Z.; Zeng, F.; Boulay, G.; Grimm, C.; Harteneck, C.; Beech, D.J. Block of TRPC5 channels by 2-aminoethoxydiphenyl borate: A differential, extracellular and voltage-dependent effect. Br. J. Pharmacol. 2005, 145, 405–414. [Google Scholar] [CrossRef]
- Zeng, B.; Chen, G.L.; Xu, S.Z. Divalent copper is a potent extracellular blocker for TRPM2 channel. Biochem. Biophys. Res. Commun. 2012, 424, 279–284. [Google Scholar] [CrossRef]
- Kuang, C.-Y.; Yü, Y.; Wang, K.; Qian, D.-H.; Den, M.-Y.; Huang, L. Knockdown of transient receptor potential canonical-1 reduces the proliferation and migration of endothelial progenitor cells. Stem. Cells Dev. 2012, 21, 487–496. [Google Scholar] [CrossRef]
- Xu, S.-Z.; Zeng, F.; Lei, M.; Li, J.; Gao, B.; Xiong, C.; Sivaprasadarao, A.; Beech, D. Generation of functional ion-channel tools by E3 targeting. Nat. Biotechnol. 2005, 23, 1289–1293. [Google Scholar] [CrossRef]
- Zeng, B.; Chen, G.-L.; Garcia-Vaz, E.; Bhandari, S.; Daskoulidou, N.; Berglund, L.M.; Jiang, H.; Hallett, T.; Zhou, L.-P.; Huang, L.; et al. ORAI channels are critical for receptor-mediated endocytosis of albumin. Nat. Commun. 2017, 8, 1920. [Google Scholar] [CrossRef] [Green Version]
- Yu, P.-C.; Gu, S.-Y.; Bu, J.-W.; Du, J.-L. TRPC1 is essential for in vivo angiogenesis in zebrafish. Circ. Res. 2010, 106, 1221–1232. [Google Scholar] [CrossRef] [Green Version]
- Antigny, F.; Girardin, N.; Frieden, M. Transient receptor potential canonical channels are required for in vitro endothelial tube formation. J. Biol. Chem. 2012, 287, 5917–5927. [Google Scholar] [CrossRef] [Green Version]
- Alexandru, N.; Jardín, I.; Popov, D.; Simionescu, M.; García-Estañ, J.; Salido, G.M.; Rosado, J.A. Effect of homocysteine on calcium mobilization and platelet function in type 2 diabetes mellitus. J. Cell Mol. Med. 2008, 12, 2586–2597. [Google Scholar] [CrossRef] [Green Version]
- Han, H.; Wang, Y.; Li, X.; Wang, P.A.; Wei, X.; Liang, W.; Ding, G.; Yu, X.; Bao, C.; Zhang, Y.; et al. Novel role of NOD2 in mediating Ca2+ signaling: Evidence from NOD2-regulated podocyte TRPC6 channels in hyperhomocysteinemia. Hypertension 2013, 62, 506–511. [Google Scholar] [CrossRef] [Green Version]
- Ovey, I.S.; Naziroglu, M. Homocysteine and cytosolic GSH depletion induce apoptosis and oxidative toxicity through cytosolic calcium overload in the hippocampus of aged mice: Involvement of TRPM2 and TRPV1 channels. Neuroscience 2015, 284, 225–233. [Google Scholar] [CrossRef]
- Chigurupati, S.; Wei, Z.; Belal, C.; Vandermey, M.; Kyriazis, G.; Arumugam, T.; Chan, S.L. The homocysteine-inducible endoplasmic reticulum stress protein counteracts calcium store depletion and induction of CCAAT enhancer-binding protein homologous protein in a neurotoxin model of Parkinson disease. J. Biol. Chem. 2009, 284, 18323–18333. [Google Scholar] [CrossRef] [Green Version]
- Zhang, H.-S.; Xiao, J.-H.; Cao, E.-H.; Qin, J.-F. Homocysteine inhibits store-mediated calcium entry in human endothelial cells: Evidence for involvement of membrane potential and actin cytoskeleton. Mol. Cell Biochem. 2005, 269, 37–47. [Google Scholar] [CrossRef]
- Cai, B.; Gong, D.; Pan, Z.; Liu, Y.; Qian, H.; Zhang, Y.; Jiao, J.; Lu, Y.; Yang, B. Large-conductance Ca2+-activated K+ currents blocked and impaired by homocysteine in human and rat mesenteric artery smooth muscle cells. Life Sci. 2007, 80, 2060–2066. [Google Scholar] [CrossRef]
- Cortés, M.P.; Becerra, J.P.; Vinet, R.; Álvarez, R.; Quintana, I. Inhibition of ATP-induced calcium influx by homocysteine in human umbilical vein endothelial cells. Cell Biol. Int. 2013, 37, 600–607. [Google Scholar] [CrossRef]
- Cai, B.; Gong, D.; Chen, N.; Li, J.; Wang, G.; Lu, Y.; Yang, B. The negative inotropic effects of homocysteine were prevented by matrine via the regulating intracellular calcium level. Int. J. Cardiol. 2011, 150, 113–115. [Google Scholar] [CrossRef]
- Thilo, F.; Liu, Y.; Krueger, K.; Förste, N.; Wittstock, A.; Scholze, A.; Tepel, M. Do cysteine residues regulate transient receptor potential canonical type 6 channel protein expression? Antioxid. Redox Signal 2012, 16, 452–457. [Google Scholar] [CrossRef]
- Lucock, M.; Yates, Z. Folic acid-vitamin and panacea or genetic time bomb? Nat. Rev. Genet. 2005, 6, 235–240. [Google Scholar] [CrossRef] [PubMed]








Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chen, G.-L.; Zeng, B.; Jiang, H.; Daskoulidou, N.; Saurabh, R.; Chitando, R.J.; Xu, S.-Z. Ca2+ Influx through TRPC Channels Is Regulated by Homocysteine–Copper Complexes. Biomolecules 2023, 13, 952. https://doi.org/10.3390/biom13060952
Chen G-L, Zeng B, Jiang H, Daskoulidou N, Saurabh R, Chitando RJ, Xu S-Z. Ca2+ Influx through TRPC Channels Is Regulated by Homocysteine–Copper Complexes. Biomolecules. 2023; 13(6):952. https://doi.org/10.3390/biom13060952
Chicago/Turabian StyleChen, Gui-Lan, Bo Zeng, Hongni Jiang, Nikoleta Daskoulidou, Rahul Saurabh, Rumbidzai J. Chitando, and Shang-Zhong Xu. 2023. "Ca2+ Influx through TRPC Channels Is Regulated by Homocysteine–Copper Complexes" Biomolecules 13, no. 6: 952. https://doi.org/10.3390/biom13060952