
Microvascular Contributions to Alzheimer Disease Pathogenesis: Is Alzheimer Disease Primarily an Endotheliopathy?

Abstract
:1. Introduction
2. Evidence to Support Vascular Contributions to AD Pathogenesis
3. Vascular Risk Factors Increase the Risk for AD
4. Concomitant Macrovascular Disease Is Present in Most, and Microvascular Disease in Almost All, AD Brains
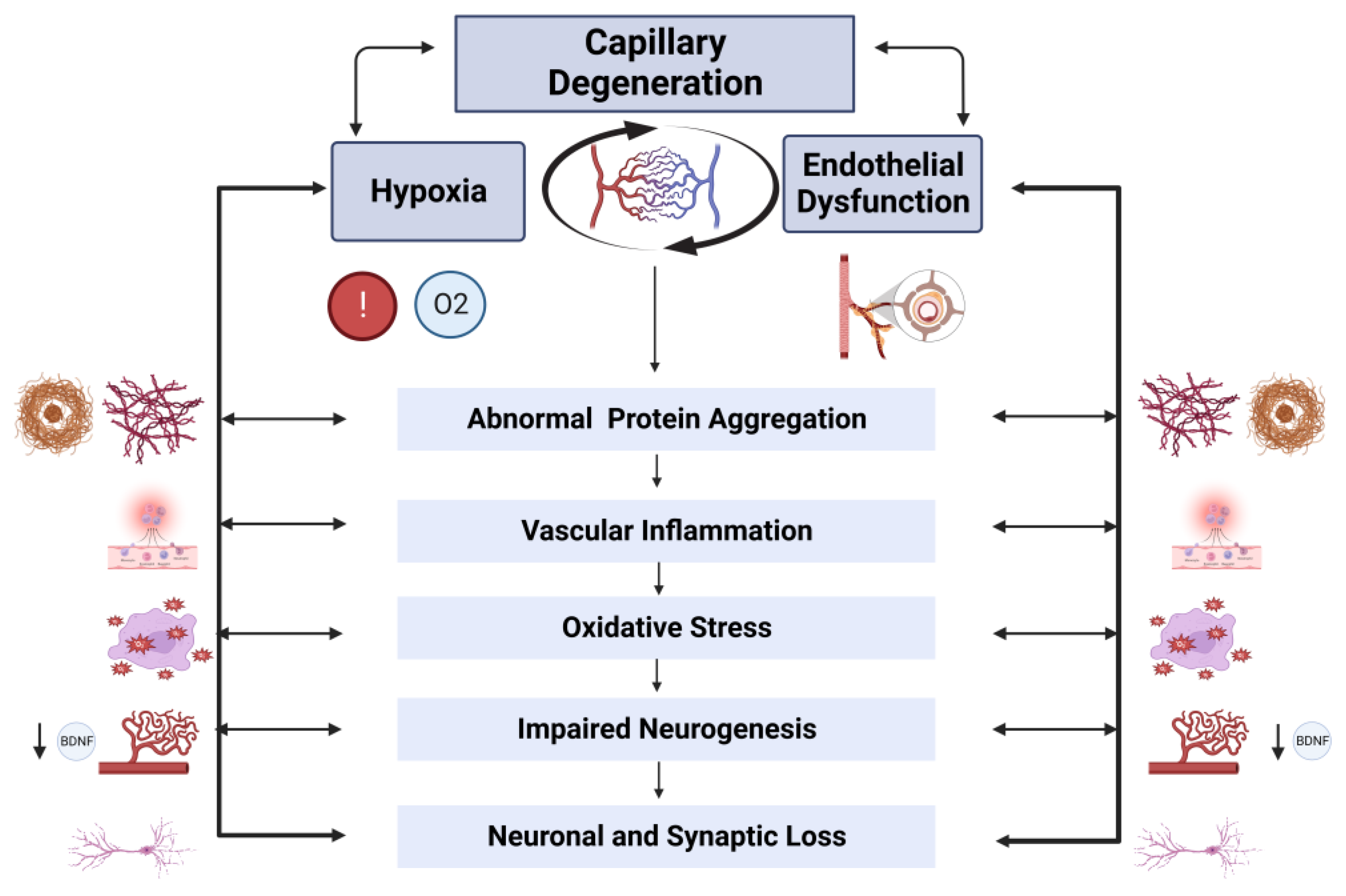
4.1. Brain Capillary Degeneration
4.2. Endothelium
4.3. Pericytes
4.4. Astrocytic End-Foot Processes
4.5. Other Vascular Constituents
5. Impaired Cerebral Perfusion Is a Common and Early Event in AD Which Strongly Predicts Future Cognitive Impairment in Clinical and Animal Studies
6. A Forward-Feedback Loop between Hypoxia and Endothelial Dysfunction Contributes to AD Pathogenesis
6.1. Increased Amyloidogenic APP Processing
6.2. Activation of Tau Kinases
6.3. Endoplasmic Reticulum Stress
6.4. Impaired Endothelial Aβ Clearance
6.5. Dysregulated Endothelial Nitric Oxide Synthase (eNOS)
6.6. Altered Lipid Raft Endocytosis
6.7. Impaired Angiogenesis
6.8. Dysregulated Thrombosis
6.9. Vascular Inflammation and Immune Dysregulation
6.10. Neuronal and Synaptic Dysfunction

6.11. Impaired Neurogenesis
7. Conclusions and Future Directions
Funding
Conflicts of Interest
References
- Tarawneh, R.; Holtzman, D.M. The clinical problem of symptomatic Alzheimer disease and mild cognitive impairment. Cold Spring Harb. Perspect. Med. 2012, 2, a006148. [Google Scholar] [CrossRef] [PubMed]
- Price, J.L.; Davis, P.B.; Morris, J.C.; White, D.L. The distribution of tangles, plaques and related immunohistochemical markers in healthy aging and Alzheimer’s disease. Neurobiol. Aging 1991, 12, 295–312. [Google Scholar] [CrossRef]
- Jack, C.R., Jr.; Knopman, D.S.; Jagust, W.J.; Petersen, R.C.; Weiner, M.W.; Aisen, P.S.; Shaw, L.M.; Vemuri, P.; Wiste, H.J.; Weigand, S.D.; et al. Tracking pathophysiological processes in Alzheimer’s disease: An updated hypothetical model of dynamic biomarkers. Lancet Neurol. 2013, 12, 207–216. [Google Scholar] [CrossRef] [PubMed]
- Esiri, M.M.; Joachim, C.; Sloan, C.; Christie, S.; Agacinski, G.; Bridges, L.R.; Wilcock, G.K.; Smith, A.D. Cerebral Subcortical Small Vessel Disease in Subjects with Pathologically Confirmed Alzheimer Disease: A Clinicopathologic Study in the Oxford Project to Investigate Memory and Ageing (OPTIMA). Alzheimer Dis. Assoc. Disord. 2014, 28, 30–35. [Google Scholar] [CrossRef]
- Attems, J.; Jellinger, K.A. The overlap between vascular disease and Alzheimer’s disease-lessons from pathology. BMC Med. 2014, 12, 206. [Google Scholar] [CrossRef] [PubMed]
- Launer, L.J.; Petrovitch, H.; Ross, G.W.; Markesbery, W.; White, L.R. AD brain pathology: Vascular origins? Results from the HAAS autopsy study. Neurobiol. Aging 2008, 29, 1587–1590. [Google Scholar] [CrossRef]
- Barker, W.W.; Luis, C.A.; Kashuba, A.; Luis, M.; Harwood, D.G.; Loewenstein, D.; Waters, C.; Jimison, P.; Shepherd, E.; Sevush, S.; et al. Relative frequencies of Alzheimer disease, Lewy body, vascular and frontotemporal dementia, and hippocampal sclerosis in the State of Florida Brain Bank. Alzheimer Dis. Assoc. Disord. 2002, 16, 203–212. [Google Scholar] [CrossRef]
- de la Torre, J.C. Alzheimer disease as a vascular disorder: Nosological evidence. Stroke 2002, 33, 1152–1162. [Google Scholar] [CrossRef]
- de la Torre, J.C. Cerebromicrovascular pathology in Alzheimer’s disease compared to normal aging. Gerontology 1997, 43, 26–43. [Google Scholar] [CrossRef]
- Kalaria, R.N.; Hedera, P. Differential degeneration of the cerebral microvasculature in Alzheimer’s disease. Neuroreport 1995, 6, 477–480. [Google Scholar] [CrossRef]
- Kalaria, R.N. Cerebral vessels in ageing and Alzheimer’s disease. Pharmacol. Ther. 1996, 72, 193–214. [Google Scholar] [CrossRef] [PubMed]
- Kelleher, R.J.; Soiza, R.L. Evidence of endothelial dysfunction in the development of Alzheimer’s disease: Is Alzheimer’s a vascular disorder? Am. J. Cardiovasc. Dis. 2013, 3, 197–226. [Google Scholar] [PubMed]
- O’Brien, J.T.; Markus, H.S. Vascular risk factors and Alzheimer’s disease. BMC Med. 2014, 12, 218. [Google Scholar] [CrossRef] [PubMed]
- Grammas, P. Neurovascular dysfunction, inflammation and endothelial activation: Implications for the pathogenesis of Alzheimer’s disease. J. Neuroinflamm. 2011, 8, 26. [Google Scholar] [CrossRef] [PubMed]
- de la Torre, J.C. Is Alzheimer’s disease a neurodegenerative or a vascular disorder? Data, dogma, and dialectics. Lancet. Neurol. 2004, 3, 184–190. [Google Scholar] [CrossRef] [PubMed]
- Claudio, L. Ultrastructural features of the blood-brain barrier in biopsy tissue from Alzheimer’s disease patients. Acta Neuropathol. 1996, 91, 6–14. [Google Scholar] [CrossRef]
- Miyakawa, T.; Kuramoto, R. Ultrastructural study of senile plaques and microvessels in the brain with Alzheimer’s disease and Down’s syndrome. Ann. Med. 1989, 21, 99–102. [Google Scholar] [CrossRef] [PubMed]
- Yamashita, K.; Miyakawa, T.; Katsuragi, S. Vascular changes in the brains with Alzheimer’s disease. Jpn. J. Psychiatry Neurol. 1991, 45, 79–84. [Google Scholar] [CrossRef]
- Iturria-Medina, Y.; Sotero, R.C.; Toussaint, P.J.; Mateos-Pérez, J.M.; Evans, A.C.; Weiner, M.W.; Aisen, P.; Petersen, R.; Jack, C.R.; Jagust, W.; et al. Early role of vascular dysregulation on late-onset Alzheimer’s disease based on multifactorial data-driven analysis. Nat. Commun. 2016, 7, 11934. [Google Scholar] [CrossRef]
- Eisenmenger, L.B.; Peret, A.; Famakin, B.M.; Spahic, A.; Roberts, G.S.; Bockholt, J.H.; Johnson, K.M.; Paulsen, J.S. Vascular contributions to Alzheimer’s disease. Transl. Res. 2022, 254, 41–53. [Google Scholar] [CrossRef]
- Tarawneh, R.; Kasper, R.S.; Sanford, J.; Phuah, C.L.; Hassenstab, J.; Cruchaga, C. Vascular endothelial-cadherin as a marker of endothelial injury in preclinical Alzheimer disease. Ann. Clin. Transl. Neurol. 2022, 9, 1926–1940. [Google Scholar] [CrossRef]
- Jitsuki-Takahashi, A.; Jitsuki, S.; Yamashita, N.; Kawamura, M.; Abe, M.; Sakimura, K.; Sano, A.; Nakamura, F.; Goshima, Y.; Takahashi, T. Activity-induced secretion of semaphorin 3A mediates learning. Eur. J. Neurosci. 2021, 53, 3279–3293. [Google Scholar] [CrossRef]
- de la Torre, J.C.; Mussivand, T. Can disturbed brain microcirculation cause Alzheimer’s disease? Neurol. Res. 1993, 15, 146–153. [Google Scholar] [CrossRef] [PubMed]
- de la Torre, J.C. Impaired brain microcirculation may trigger Alzheimer’s disease. Neurosci. Biobehav. Rev. 1994, 18, 397–401. [Google Scholar] [CrossRef] [PubMed]
- de la Torre, J.C. Critical threshold cerebral hypoperfusion causes Alzheimer’s disease? Acta Neuropathol. 1999, 98, 1–8. [Google Scholar] [CrossRef] [PubMed]
- de la Torre, J.C. Hemodynamic consequences of deformed microvessels in the brain in Alzheimer’s disease. Ann. N. Y. Acad. Sci. 1997, 826, 75–91. [Google Scholar] [CrossRef]
- Stewart, R.; Prince, M.; Mann, A. Vascular risk factors and Alzheimer’s disease. Aust. N. Z. J. Psychiatry 1999, 33, 809–813. [Google Scholar] [CrossRef]
- Breteler, M.M. Vascular risk factors for Alzheimer’s disease: An epidemiologic perspective. Neurobiol. Aging 2000, 21, 153–160. [Google Scholar] [CrossRef]
- Cechetto, D.F.; Hachinski, V.; Whitehead, S.N. Vascular risk factors and Alzheimer’s disease. Expert Rev. Neurother. 2008, 8, 743–750. [Google Scholar] [CrossRef]
- Shi, J.; Perry, G.; Smith, M.A.; Friedland, R.P. Vascular abnormalities: The insidious pathogenesis of Alzheimer’s disease. Neurobiol. Aging 2000, 21, 357–361. [Google Scholar] [CrossRef]
- Sparks, D.L.; Martin, T.A.; Gross, D.R.; Hunsaker, J.C., 3rd. Link between heart disease, cholesterol, and Alzheimer’s disease: A review. Microsc. Res. Tech. 2000, 50, 287–290. [Google Scholar] [CrossRef] [PubMed]
- Rhodin, J.A.; Thomas, T. A vascular connection to Alzheimer’s disease. Microcirculation 2001, 8, 207–220. [Google Scholar] [CrossRef] [PubMed]
- Marx, J. Alzheimer’s disease. Bad for the heart, bad for the mind? Science 2001, 294, 508–509. [Google Scholar] [CrossRef] [PubMed]
- Ronnemaa, E.; Zethelius, B.; Sundelof, J.; Sundstrom, J.; Degerman-Gunnarsson, M.; Berne, C.; Lannfelt, L.; Kilander, L. Impaired insulin secretion increases the risk of Alzheimer disease. Neurology 2008, 71, 1065–1071. [Google Scholar] [CrossRef] [PubMed]
- Irie, F.; Fitzpatrick, A.L.; Lopez, O.L.; Kuller, L.H.; Peila, R.; Newman, A.B.; Launer, L.J. Enhanced risk for Alzheimer disease in persons with type 2 diabetes and APOE epsilon4: The Cardiovascular Health Study Cognition Study. Arch. Neurol. 2008, 65, 89–93. [Google Scholar] [CrossRef]
- Vagelatos, N.T.; Eslick, G.D. Type 2 diabetes as a risk factor for Alzheimer’s disease: The confounders, interactions, and neuropathology associated with this relationship. Epidemiol. Rev. 2013, 35, 152–160. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.J.; Seo, H.I.; Cha, H.Y.; Yang, Y.J.; Kwon, S.H.; Yang, S.J. Diabetes and Alzheimer’s Disease: Mechanisms and Nutritional Aspects. Clin. Nutr. Res. 2018, 7, 229–240. [Google Scholar] [CrossRef]
- Gottesman, R.F.; Albert, M.S.; Alonso, A.; Coker, L.H.; Coresh, J.; Davis, S.M.; Deal, J.A.; McKhann, G.M.; Mosley, T.H.; Sharrett, A.R.; et al. Associations Between Midlife Vascular Risk Factors and 25-Year Incident Dementia in the Atherosclerosis Risk in Communities (ARIC) Cohort. JAMA Neurol. 2017, 74, 1246–1254. [Google Scholar] [CrossRef]
- Liu, C.C.; Liu, C.C.; Kanekiyo, T.; Xu, H.; Bu, G. Apolipoprotein E and Alzheimer disease: Risk, mechanisms and therapy. Nat. Rev. Neurol. 2013, 9, 106–118. [Google Scholar] [CrossRef]
- Corder, E.H.; Saunders, A.M.; Strittmatter, W.J.; Schmechel, D.E.; Gaskell, P.C.; Small, G.W.; Roses, A.D.; Haines, J.L.; Pericak-Vance, M.A. Gene dose of apolipoprotein E type 4 allele and the risk of Alzheimer’s disease in late onset families. Science 1993, 261, 921–923. [Google Scholar] [CrossRef]
- Troutwine, B.R.; Hamid, L.; Lysaker, C.R.; Strope, T.A.; Wilkins, H.M. Apolipoprotein E and Alzheimer’s disease. Acta Pharm. Sinica. B 2022, 12, 496–510. [Google Scholar] [CrossRef] [PubMed]
- Mahley, R.W. Apolipoprotein E: From cardiovascular disease to neurodegenerative disorders. J. Mol. Med. 2016, 94, 739–746. [Google Scholar] [CrossRef] [PubMed]
- Launer, L.J.; Ross, G.W.; Petrovitch, H.; Masaki, K.; Foley, D.; White, L.R.; Havlik, R.J. Midlife blood pressure and dementia: The Honolulu-Asia aging study. Neurobiol. Aging 2000, 21, 49–55. [Google Scholar] [CrossRef] [PubMed]
- DeCarli, C.; Miller, B.L.; Swan, G.E.; Reed, T.; Wolf, P.A.; Carmelli, D. Cerebrovascular and brain morphologic correlates of mild cognitive impairment in the National Heart, Lung, and Blood Institute Twin Study. Arch. Neurol. 2001, 58, 643–647. [Google Scholar] [CrossRef]
- Saunders, A.M.; Roses, A.D. Apolipoprotein E4 allele frequency, ischemic cerebrovascular disease, and Alzheimer’s disease. Stroke 1993, 24, 1416–1417. [Google Scholar] [CrossRef]
- Roses, A.D.; Saunders, A.M. ApoE, Alzheimer’s disease, and recovery from brain stress. Ann. N. Y. Acad. Sci. 1997, 826, 200–212. [Google Scholar] [CrossRef]
- Zhang, X.; Tong, T.; Chang, A.; Ang, T.F.A.; Tao, Q.; Auerbach, S.; Devine, S.; Qiu, W.Q.; Mez, J.; Massaro, J.; et al. Midlife lipid and glucose levels are associated with Alzheimer’s disease. Alzheimer’s Dement. 2023, 19, 181–193. [Google Scholar] [CrossRef]
- Iwagami, M.; Qizilbash, N.; Gregson, J.; Douglas, I.; Johnson, M.; Pearce, N.; Evans, S.; Pocock, S. Blood cholesterol and risk of dementia in more than 1.8 million people over two decades: A retrospective cohort study. Lancet Healthy Longev. 2021, 2, e498–e506. [Google Scholar] [CrossRef]
- Wingo, T.S.; Cutler, D.J.; Wingo, A.P.; Le, N.A.; Rabinovici, G.D.; Miller, B.L.; Lah, J.J.; Levey, A.I. Association of Early-Onset Alzheimer Disease With Elevated Low-Density Lipoprotein Cholesterol Levels and Rare Genetic Coding Variants of APOB. JAMA Neurol. 2019, 76, 809–817. [Google Scholar] [CrossRef]
- Chen, X.; Hui, L.; Geiger, J.D. Role of LDL cholesterol and endolysosomes in amyloidogenesis and Alzheimer’s disease. J. Neurol. Neurophysiol. 2014, 5, 236. [Google Scholar] [CrossRef]
- Feringa, F.M.; van der Kant, R. Cholesterol and Alzheimer’s Disease; From Risk Genes to Pathological Effects. Front. Aging Neurosci. 2021, 13, 690372. [Google Scholar] [CrossRef] [PubMed]
- Di Paolo, G.; Kim, T.-W. Linking lipids to Alzheimer’s disease: Cholesterol and beyond. Nat. Rev. Neurosci. 2011, 12, 284–296. [Google Scholar] [CrossRef] [PubMed]
- Hofman, A.; Ott, A.; Breteler, M.M.; Bots, M.L.; Slooter, A.J.; van Harskamp, F.; van Duijn, C.N.; Van Broeckhoven, C.; Grobbee, D.E. Atherosclerosis, apolipoprotein E, and prevalence of dementia and Alzheimer’s disease in the Rotterdam Study. Lancet 1997, 349, 151–154. [Google Scholar] [CrossRef]
- den Heijer, T.; Vermeer, S.E.; Clarke, R.; Oudkerk, M.; Koudstaal, P.J.; Hofman, A.; Breteler, M.M.B. Homocysteine and brain atrophy on MRI of non-demented elderly. Brain 2002, 126, 170–175. [Google Scholar] [CrossRef] [PubMed]
- Shirafuji, N.; Hamano, T.; Yen, S.H.; Kanaan, N.M.; Yoshida, H.; Hayashi, K.; Ikawa, M.; Yamamura, O.; Kuriyama, M.; Nakamoto, Y. Homocysteine Increases Tau Phosphorylation, Truncation and Oligomerization. Int. J. Mol. Sci. 2018, 19, 891. [Google Scholar] [CrossRef] [PubMed]
- Lominadze, D.; Tyagi, N.; Sen, U.; Ovechkin, A.; Tyagi, S.C. Homocysteine alters cerebral microvascular integrity and causes remodeling by antagonizing GABA-A receptor. Mol. Cell. Biochem. 2012, 371, 89–96. [Google Scholar] [CrossRef]
- Peila, R.; White, L.R.; Petrovich, H.; Masaki, K.; Ross, G.W.; Havlik, R.J.; Launer, L.J. Joint effect of the APOE gene and midlife systolic blood pressure on late-life cognitive impairment: The Honolulu-Asia aging study. Stroke 2001, 32, 2882–2889. [Google Scholar] [CrossRef] [PubMed]
- Kivipelto, M.; Helkala, E.L.; Hanninen, T.; Laakso, M.P.; Hallikainen, M.; Alhainen, K.; Soininen, H.; Tuomilehto, J.; Nissinen, A. Midlife vascular risk factors and late-life mild cognitive impairment: A population-based study. Neurology 2001, 56, 1683–1689. [Google Scholar] [CrossRef]
- Luchsinger, J.A.; Reitz, C.; Honig, L.S.; Tang, M.X.; Shea, S.; Mayeux, R. Aggregation of vascular risk factors and risk of incident Alzheimer disease. Neurology 2005, 65, 545–551. [Google Scholar] [CrossRef]
- Phuah, C.L.; Chen, Y.; Strain, J.F.; Yechoor, N.; Laurido-Soto, O.J.; Ances, B.M.; Lee, J.M.; for the Alzheimer’s Disease Neuroimaging, I. Association of Data-Driven White Matter Hyperintensity Spatial Signatures with Distinct Cerebral Small Vessel Disease Etiologies. Neurology 2022, 99, e2535–e2547. [Google Scholar] [CrossRef]
- Rabin, J.S.; Pruzin, J.; Scott, M.; Yang, H.S.; Hampton, O.; Hsieh, S.; Schultz, A.P.; Buckley, R.F.; Hedden, T.; Rentz, D.; et al. Association of beta-Amyloid and Vascular Risk on Longitudinal Patterns of Brain Atrophy. Neurology 2022, 99, e270–e280. [Google Scholar] [CrossRef] [PubMed]
- Rizvi, B.; Lao, P.J.; Chesebro, A.G.; Dworkin, J.D.; Amarante, E.; Beato, J.M.; Gutierrez, J.; Zahodne, L.B.; Schupf, N.; Manly, J.J.; et al. Association of Regional White Matter Hyperintensities with Longitudinal Alzheimer-like Pattern of Neurodegeneration in Older Adults. JAMA Netw. Open 2021, 4, e2125166. [Google Scholar] [CrossRef] [PubMed]
- Brickman, A.M. Contemplating Alzheimer’s disease and the contribution of white matter hyperintensities. Curr. Neurol. Neurosci. Rep. 2013, 13, 415. [Google Scholar] [CrossRef] [PubMed]
- Tuke, J.B. On the Morbid Histology of the Brain and Spinal Cord as Observed in the Insane. Br. Foreign Med. Chir. Rev. 1873, 51, 450–460. [Google Scholar] [PubMed]
- Buée, L.; Hof, P.R.; Bouras, C.; Delacourte, A.; Perl, D.P.; Morrison, J.H.; Fillit, H.M. Pathological alterations of the cerebral microvasculature in Alzheimer’s disease and related dementing disorders. Acta Neuropathol. 1994, 87, 469–480. [Google Scholar] [CrossRef]
- Miyakawa, T.; Shimoji, A.; Kuramoto, R.; Higuchi, Y. The relationship between senile plaques and cerebral blood vessels in Alzheimer’s disease and senile dementia. Morphological mechanism of senile plaque production. Virchows Archiv. B Cell. Pathol. Incl. Mol. Pathol. 1982, 40, 121–129. [Google Scholar] [CrossRef]
- Araki, K.; Miyakawa, T.; Katsuragi, S. Ultrastructure of senile plaque using thick sections in the brain with Alzheimer’s disease. Jpn. J. Psychiatry Neurol. 1991, 45, 85–89. [Google Scholar] [CrossRef]
- Mancardi, G.L.; Perdelli, F.; Rivano, C.; Leonardi, A.; Bugiani, O. Thickening of the basement membrane of cortical capillaries in Alzheimer’s disease. Acta Neuropathol. 1980, 49, 79–83. [Google Scholar] [CrossRef]
- Meyer, E.P.; Ulmann-Schuler, A.; Staufenbiel, M.; Krucker, T. Altered morphology and 3D architecture of brain vasculature in a mouse model for Alzheimer’s disease. Proc. Natl. Acad. Sci. USA 2008, 105, 3587–3592. [Google Scholar] [CrossRef]
- Bell, R.D.; Zlokovic, B.V. Neurovascular mechanisms and blood-brain barrier disorder in Alzheimer’s disease. Acta Neuropathol. 2009, 118, 103–113. [Google Scholar] [CrossRef]
- De Jong, G.I.; Farkas, E.; Stienstra, C.M.; Plass, J.R.; Keijser, J.N.; de la Torre, J.C.; Luiten, P.G. Cerebral hypoperfusion yields capillary damage in the hippocampal CA1 area that correlates with spatial memory impairment. Neuroscience 1999, 91, 203–210. [Google Scholar] [CrossRef] [PubMed]
- De Jong, G.I.; De Vos, R.A.; Steur, E.N.; Luiten, P.G. Cerebrovascular hypoperfusion: A risk factor for Alzheimer’s disease? Animal model and postmortem human studies. Ann. N. Y. Acad. Sci. 1997, 826, 56–74. [Google Scholar] [CrossRef] [PubMed]
- Christov, A.; Ottman, J.; Hamdheydari, L.; Grammas, P. Structural changes in Alzheimer’s disease brain microvessels. Curr. Alzheimer Res. 2008, 5, 392–395. [Google Scholar] [CrossRef] [PubMed]
- Farkas, E.; Luiten, P.G. Cerebral microvascular pathology in aging and Alzheimer’s disease. Prog. Neurobiol. 2001, 64, 575–611. [Google Scholar] [CrossRef]
- Zuliani, G.; Cavalieri, M.; Galvani, M.; Passaro, A.; Munari, M.R.; Bosi, C.; Zurlo, A.; Fellin, R. Markers of endothelial dysfunction in older subjects with late onset Alzheimer’s disease or vascular dementia. J. Neurol. Sci. 2008, 272, 164–170. [Google Scholar] [CrossRef]
- Lau, S.-F.; Cao, H.; Fu, A.K.Y.; Ip, N.Y. Single-nucleus transcriptome analysis reveals dysregulation of angiogenic endothelial cells and neuroprotective glia in Alzheimer’s disease. Proc. Natl. Acad. Sci. USA 2020, 117, 25800. [Google Scholar] [CrossRef] [PubMed]
- Yang, A.C.; Vest, R.T.; Kern, F.; Lee, D.P.; Agam, M.; Maat, C.A.; Losada, P.M.; Chen, M.B.; Schaum, N.; Khoury, N.; et al. A human brain vascular atlas reveals diverse mediators of Alzheimer’s risk. Nature 2022, 603, 885–892. [Google Scholar] [CrossRef]
- Takechi, R.; Lam, V.; Brook, E.; Giles, C.; Fimognari, N.; Mooranian, A.; Al-Salami, H.; Coulson, S.H.; Nesbit, M.; Mamo, J.C.L. Blood-Brain Barrier Dysfunction Precedes Cognitive Decline and Neurodegeneration in Diabetic Insulin Resistant Mouse Model: An Implication for Causal Link. Front. Aging Neurosci. 2017, 9, 399. [Google Scholar] [CrossRef] [PubMed]
- Shah, G.N.; Morofuji, Y.; Banks, W.A.; Price, T.O. High glucose-induced mitochondrial respiration and reactive oxygen species in mouse cerebral pericytes is reversed by pharmacological inhibition of mitochondrial carbonic anhydrases: Implications for cerebral microvascular disease in diabetes. Biochem. Biophys. Res. Commun. 2013, 440, 354–358. [Google Scholar] [CrossRef]
- Pietronigro, E.; Zenaro, E.; Bianca, V.D.; Dusi, S.; Terrabuio, E.; Iannoto, G.; Slanzi, A.; Ghasemi, S.; Nagarajan, R.; Piacentino, G.; et al. Blockade of α4 integrins reduces leukocyte-endothelial interactions in cerebral vessels and improves memory in a mouse model of Alzheimer’s disease. Sci. Rep. 2019, 9, 12055. [Google Scholar] [CrossRef]
- ElAli, A.; Theriault, P.; Rivest, S. The role of pericytes in neurovascular unit remodeling in brain disorders. Int. J. Mol. Sci. 2014, 15, 6453–6474. [Google Scholar] [CrossRef]
- Alcendor, D.J. Interactions between Amyloid-Beta Proteins and Human Brain Pericytes: Implications for the Pathobiology of Alzheimer’s Disease. J. Clin. Med. 2020, 9, 1490. [Google Scholar] [CrossRef]
- Ma, Q.; Zhao, Z.; Sagare, A.P.; Wu, Y.; Wang, M.; Owens, N.C.; Verghese, P.B.; Herz, J.; Holtzman, D.M.; Zlokovic, B.V. Blood-brain barrier-associated pericytes internalize and clear aggregated amyloid-β42 by LRP1-dependent apolipoprotein E isoform-specific mechanism. Mol. Neurodegener. 2018, 13, 57. [Google Scholar] [CrossRef] [PubMed]
- Verbeek, M.M.; Van Nostrand, W.E.; Otte-Holler, I.; Wesseling, P.; De Waal, R.M. Amyloid-beta-induced degeneration of human brain pericytes is dependent on the apolipoprotein E genotype. Ann. N. Y. Acad. Sci. 2000, 903, 187–199. [Google Scholar] [CrossRef]
- Verbeek, M.M.; de Waal, R.M.; Schipper, J.J.; Van Nostrand, W.E. Rapid degeneration of cultured human brain pericytes by amyloid beta protein. J. Neurochem. 1997, 68, 1135–1141. [Google Scholar] [CrossRef]
- Sengillo, J.D.; Winkler, E.A.; Walker, C.T.; Sullivan, J.S.; Johnson, M.; Zlokovic, B.V. Deficiency in mural vascular cells coincides with blood-brain barrier disruption in Alzheimer’s disease. Brain Pathol. 2013, 23, 303–310. [Google Scholar] [CrossRef]
- Montagne, A.; Nikolakopoulou, A.M.; Zhao, Z.; Sagare, A.P.; Si, G.; Lazic, D.; Barnes, S.R.; Daianu, M.; Ramanathan, A.; Go, A.; et al. Pericyte degeneration causes white matter dysfunction in the mouse central nervous system. Nat. Med. 2018, 24, 326–337. [Google Scholar] [CrossRef] [PubMed]
- Halliday, M.R.; Rege, S.V.; Ma, Q.; Zhao, Z.; Miller, C.A.; Winkler, E.A.; Zlokovic, B.V. Accelerated pericyte degeneration and blood-brain barrier breakdown in apolipoprotein E4 carriers with Alzheimer’s disease. J. Cereb. Blood Flow Metab. 2016, 36, 216–227. [Google Scholar] [CrossRef] [PubMed]
- Montagne, A.; Nation, D.A.; Sagare, A.P.; Barisano, G.; Sweeney, M.D.; Chakhoyan, A.; Pachicano, M.; Joe, E.; Nelson, A.R.; D’Orazio, L.M.; et al. APOE4 leads to blood-brain barrier dysfunction predicting cognitive decline. Nature 2020, 581, 71–76. [Google Scholar] [CrossRef]
- Winkler, E.A.; Sagare, A.P.; Zlokovic, B.V. The pericyte: A forgotten cell type with important implications for Alzheimer’s disease? Brain Pathol. 2014, 24, 371–386. [Google Scholar] [CrossRef] [PubMed]
- Sagare, A.P.; Bell, R.D.; Zhao, Z.; Ma, Q.; Winkler, E.A.; Ramanathan, A.; Zlokovic, B.V. Pericyte loss influences Alzheimer-like neurodegeneration in mice. Nat. Commun. 2013, 4, 2932. [Google Scholar] [CrossRef] [PubMed]
- Castillo-Carranza, D.L.; Nilson, A.N.; Van Skike, C.E.; Jahrling, J.B.; Patel, K.; Garach, P.; Gerson, J.E.; Sengupta, U.; Abisambra, J.; Nelson, P.; et al. Cerebral Microvascular Accumulation of Tau Oligomers in Alzheimer’s Disease and Related Tauopathies. Aging Dis. 2017, 8, 257–266. [Google Scholar] [CrossRef] [PubMed]
- Ojo, J.; Eisenbaum, M.; Shackleton, B.; Lynch, C.; Joshi, U.; Saltiel, N.; Pearson, A.; Ringland, C.; Paris, D.; Mouzon, B.; et al. Mural cell dysfunction leads to altered cerebrovascular tau uptake following repetitive head trauma. Neurobiol. Dis. 2021, 150, 105237. [Google Scholar] [CrossRef]
- González-Reyes, R.E.; Nava-Mesa, M.O.; Vargas-Sánchez, K.; Ariza-Salamanca, D.; Mora-Muñoz, L. Involvement of Astrocytes in Alzheimer’s Disease from a Neuroinflammatory and Oxidative Stress Perspective. Front. Mol. Neurosci. 2017, 10, 427. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez-Giraldo, M.; Gonzalez-Reyes, R.E.; Ramirez-Guerrero, S.; Bonilla-Trilleras, C.E.; Guardo-Maya, S.; Nava-Mesa, M.O. Astrocytes as a Therapeutic Target in Alzheimer’s Disease-Comprehensive Review and Recent Developments. Int. J. Mol. Sci. 2022, 23, 13630. [Google Scholar] [CrossRef] [PubMed]
- Davis, N.; Mota, B.C.; Stead, L.; Palmer, E.O.C.; Lombardero, L.; Rodriguez-Puertas, R.; de Paola, V.; Barnes, S.J.; Sastre, M. Pharmacological ablation of astrocytes reduces Abeta degradation and synaptic connectivity in an ex vivo model of Alzheimer’s disease. J. Neuroinflamm. 2021, 18, 73. [Google Scholar] [CrossRef] [PubMed]
- Silva, I.; Silva, J.; Ferreira, R.; Trigo, D. Glymphatic system, AQP4, and their implications in Alzheimer’s disease. Neurol. Res. Pract. 2021, 3, 5. [Google Scholar] [CrossRef]
- Iliff, J.J.; Wang, M.; Liao, Y.; Plogg, B.A.; Peng, W.; Gundersen, G.A.; Benveniste, H.; Vates, G.E.; Deane, R.; Goldman, S.A.; et al. A paravascular pathway facilitates CSF flow through the brain parenchyma and the clearance of interstitial solutes, including amyloid beta. Sci. Transl. Med. 2012, 4, 147ra111. [Google Scholar] [CrossRef]
- Arnaud, L.; Benech, P.; Greetham, L.; Stephan, D.; Jimenez, A.; Jullien, N.; García-González, L.; Tsvetkov, P.O.; Devred, F.; Sancho-Martinez, I.; et al. APOE4 drives inflammation in human astrocytes via TAGLN3 repression and NF-κB activation. Cell Rep. 2022, 40, 111200. [Google Scholar] [CrossRef]
- Avila-Muñoz, E.; Arias, C. When astrocytes become harmful: Functional and inflammatory responses that contribute to Alzheimer’s disease. Ageing Res. Rev. 2014, 18, 29–40. [Google Scholar] [CrossRef]
- Amro, Z.; Yool, A.J.; Collins-Praino, L.E. The potential role of glial cells in driving the prion-like transcellular propagation of tau in tauopathies. Brain Behav. Immun.-Health 2021, 14, 100242. [Google Scholar] [CrossRef] [PubMed]
- Saroja, S.R.; Gorbachev, K.; Julia, T.; Goate, A.M.; Pereira, A.C. Astrocyte-secreted glypican-4 drives APOE4-dependent tau hyperphosphorylation. Proc. Natl. Acad. Sci. USA 2022, 119, e2108870119. [Google Scholar] [CrossRef]
- Smith, A.M.; Davey, K.; Tsartsalis, S.; Khozoie, C.; Fancy, N.; Tang, S.S.; Liaptsi, E.; Weinert, M.; McGarry, A.; Muirhead, R.C.J.; et al. Diverse human astrocyte and microglial transcriptional responses to Alzheimer’s pathology. Acta Neuropathol. 2022, 143, 75–91. [Google Scholar] [CrossRef] [PubMed]
- Jiang, R.; Smailovic, U.; Haytural, H.; Tijms, B.M.; Li, H.; Haret, R.M.; Shevchenko, G.; Chen, G.; Abelein, A.; Gobom, J.; et al. Increased CSF-decorin predicts brain pathological changes driven by Alzheimer’s Aβ amyloidosis. Acta Neuropathol. Commun. 2022, 10, 96. [Google Scholar] [CrossRef]
- Prohovnik, I.; Mayeux, R.; Sackeim, H.A.; Smith, G.; Stern, Y.; Alderson, P.O. Cerebral perfusion as a diagnostic marker of early Alzheimer’s disease. Neurology 1988, 38, 931–937. [Google Scholar] [CrossRef] [PubMed]
- Mattsson, N.; Tosun, D.; Insel, P.S.; Simonson, A.; Jack, C.R., Jr.; Beckett, L.A.; Donohue, M.; Jagust, W.; Schuff, N.; Weiner, M.W.; et al. Association of brain amyloid-beta with cerebral perfusion and structure in Alzheimer’s disease and mild cognitive impairment. Brain 2014, 137 Pt 5, 1550–1561. [Google Scholar] [CrossRef]
- Korte, N.; Nortley, R.; Attwell, D. Cerebral blood flow decrease as an early pathological mechanism in Alzheimer’s disease. Acta Neuropathol. 2020, 140, 793–810. [Google Scholar] [CrossRef]
- Tanaka, K.; Wada, N.; Ogawa, N. Chronic cerebral hypoperfusion induces transient reversible monoaminergic changes in the rat brain. Neurochem. Res. 2000, 25, 313–320. [Google Scholar] [CrossRef]
- Johnson, K.A.; Albert, M.S. Perfusion abnormalities in prodromal AD. Neurobiol. Aging 2000, 21, 289–292. [Google Scholar] [CrossRef]
- Duan, W.; Zhou, G.D.; Balachandrasekaran, A.; Bhumkar, A.B.; Boraste, P.B.; Becker, J.T.; Kuller, L.H.; Lopez, O.L.; Gach, H.M.; Dai, W. Cerebral Blood Flow Predicts Conversion of Mild Cognitive Impairment into Alzheimer’s Disease and Cognitive Decline: An Arterial Spin Labeling Follow-up Study. J. Alzheimer’s Dis. 2021, 82, 293–305. [Google Scholar] [CrossRef]
- Bracko, O.; Cruz Hernandez, J.C.; Park, L.; Nishimura, N.; Schaffer, C.B. Causes and consequences of baseline cerebral blood flow reductions in Alzheimer’s disease. J. Cereb. Blood Flow Metab. 2021, 41, 1501–1516. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez, G.; Vitali, P.; Calvini, P.; Bordoni, C.; Girtler, N.; Taddei, G.; Mariani, G.; Nobili, F. Hippocampal perfusion in mild Alzheimer’s disease. Psychiatry Res. 2000, 100, 65–74. [Google Scholar] [CrossRef] [PubMed]
- Johnson, K.A.; Jones, K.; Holman, B.L.; Becker, J.A.; Spiers, P.A.; Satlin, A.; Albert, M.S. Preclinical prediction of Alzheimer’s disease using SPECT. Neurology 1998, 50, 1563–1571. [Google Scholar] [CrossRef] [PubMed]
- Ruitenberg, A.; den Heijer, T.; Bakker, S.L.; van Swieten, J.C.; Koudstaal, P.J.; Hofman, A.; Breteler, M.M. Cerebral hypoperfusion and clinical onset of dementia: The Rotterdam Study. Ann. Neurol. 2005, 57, 789–794. [Google Scholar] [CrossRef] [PubMed]
- Berman, S.E.; Rivera-Rivera, L.A.; Clark, L.R.; Racine, A.M.; Keevil, J.G.; Bratzke, L.C.; Carlsson, C.M.; Bendlin, B.B.; Rowley, H.A.; Blennow, K.; et al. Intracranial Arterial 4D-Flow is Associated with Metrics of Brain Health and Alzheimer’s Disease. Alzheimer’s Dement. 2015, 1, 420–428. [Google Scholar] [CrossRef]
- Kogure, D.; Matsuda, H.; Ohnishi, T.; Asada, T.; Uno, M.; Kunihiro, T.; Nakano, S.; Takasaki, M. Longitudinal evaluation of early Alzheimer’s disease using brain perfusion SPECT. J. Nucl. Med. 2000, 41, 1155–1162. [Google Scholar]
- Okamura, N.; Shinkawa, M.; Arai, H.; Matsui, T.; Nakajo, K.; Maruyama, M.; Hu, X.S.; Sasaki, H. Prediction of progression in patients with mild cognitive impairment using IMP-SPECT. Nihon Ronen Igakkai Zasshi 2000, 37, 974–978. [Google Scholar] [CrossRef]
- Tanaka, K.; Wada, N.; Hori, K.; Asanuma, M.; Nomura, M.; Ogawa, N. Chronic cerebral hypoperfusion disrupts discriminative behavior in acquired-learning rats. J. Neurosci. Methods 1998, 84, 63–68. [Google Scholar] [CrossRef]
- Ni, J.; Ohta, H.; Matsumoto, K.; Watanabe, H. Progressive cognitive impairment following chronic cerebral hypoperfusion induced by permanent occlusion of bilateral carotid arteries in rats. Brain Res. 1994, 653, 231–236. [Google Scholar] [CrossRef]
- Abdollahian, N.P.; Cada, A.; Gonzalez-Lima, F.; de la Torre, J.C. Cytochrome oxidase: A predictive marker of neurodegeneration. In Cytochrome Oxidase in Neuronal Metabolism and Alzheimer’s Disease; Gonzalez-Lima, F., Ed.; Plenum Press: New York, NY, USA, 1998; pp. 233–261. [Google Scholar]
- de la Torre, J.C.; Cada, A.; Nelson, N.; Davis, G.; Sutherland, R.J.; Gonzalez-Lima, F. Reduced cytochrome oxidase and memory dysfunction after chronic brain ischemia in aged rats. Neurosci. Lett. 1997, 223, 165–168. [Google Scholar] [CrossRef]
- Otori, T.; Katsumata, T.; Katayama, Y.; Terashi, A. Measurement of regional cerebral blood flow and glucose utilization in rat brain under chronic hypoperfusion conditions following bilateral carotid artery occlusion. Analyzed by autoradiographical methods. Nihon Ika Daigaku Zasshi 1997, 64, 428–439. [Google Scholar] [CrossRef] [PubMed]
- Ouchi, Y.; Tsukada, H.; Kakiuchi, T.; Nishiyama, S.; Futatsubashi, M. Changes in cerebral blood flow and postsynaptic muscarinic cholinergic activity in rats with bilateral carotid artery ligation. J. Nucl. Med. 1998, 39, 198–202. [Google Scholar] [PubMed]
- Pappas, B.A.; Davidson, C.M.; Bennett, S.A.; de la Torre, J.C.; Fortin, T.; Tenniswood, M.P. Chronic ischemia: Memory impairment and neural pathology in the rat. Ann. N. Y. Acad. Sci. 1997, 826, 498–501. [Google Scholar] [CrossRef] [PubMed]
- Ihara, M.; Tomimoto, H.; Kinoshita, M.; Oh, J.; Noda, M.; Wakita, H.; Akiguchi, I.; Shibasaki, H. Chronic cerebral hypoperfusion induces MMP-2 but not MMP-9 expression in the microglia and vascular endothelium of white matter. J. Cereb. Blood Flow Metab. 2001, 21, 828–834. [Google Scholar] [CrossRef]
- Cai, Z.; Liu, Z.; Xiao, M.; Wang, C.; Tian, F. Chronic Cerebral Hypoperfusion Promotes Amyloid-Beta Pathogenesis via Activating beta/gamma-Secretases. Neurochem. Res. 2017, 42, 3446–3455. [Google Scholar] [CrossRef]
- Park, J.H.; Hong, J.H.; Lee, S.W.; Ji, H.D.; Jung, J.A.; Yoon, K.W.; Lee, J.I.; Won, K.S.; Song, B.I.; Kim, H.W. The effect of chronic cerebral hypoperfusion on the pathology of Alzheimer’s disease: A positron emission tomography study in rats. Sci. Rep. 2019, 9, 14102. [Google Scholar] [CrossRef]
- Shang, J.; Yamashita, T.; Tian, F.; Li, X.; Liu, X.; Shi, X.; Nakano, Y.; Tsunoda, K.; Nomura, E.; Sasaki, R.; et al. Chronic cerebral hypoperfusion alters amyloid-beta transport related proteins in the cortical blood vessels of Alzheimer’s disease model mouse. Brain Res. 2019, 1723, 146379. [Google Scholar] [CrossRef]
- Zhu, W.M.; Neuhaus, A.; Beard, D.J.; Sutherland, B.A.; DeLuca, G.C. Neurovascular coupling mechanisms in health and neurovascular uncoupling in Alzheimer’s disease. Brain 2022, 145, 2276–2292. [Google Scholar] [CrossRef]
- Smith, G.S.; de Leon, M.J.; George, A.E.; Kluger, A.; Volkow, N.D.; McRae, T.; Golomb, J.; Ferris, S.H.; Reisberg, B.; Ciaravino, J.; et al. Topography of cross-sectional and longitudinal glucose metabolic deficits in Alzheimer’s disease. Pathophysiologic implications. Arch. Neurol. 1992, 49, 1142–1150. [Google Scholar] [CrossRef]
- Hansra, G.K.; Popov, G.; Banaczek, P.O.; Vogiatzis, M.; Jegathees, T.; Goldsbury, C.S.; Cullen, K.M. The neuritic plaque in Alzheimer’s disease: Perivascular degeneration of neuronal processes. Neurobiol. Aging 2019, 82, 88–101. [Google Scholar] [CrossRef]
- Kimura, T.; Hashimura, T.; Miyakawa, T. Observations of microvessels in the brain with Alzheimer’s disease by the scanning electron microscopy. Jpn. J. Psychiatry Neurol. 1991, 45, 671–676. [Google Scholar] [CrossRef]
- Nortley, R.; Korte, N.; Izquierdo, P.; Hirunpattarasilp, C.; Mishra, A.; Jaunmuktane, Z.; Kyrargyri, V.; Pfeiffer, T.; Khennouf, L.; Madry, C.; et al. Amyloid beta oligomers constrict human capillaries in Alzheimer’s disease via signaling to pericytes. Science 2019, 365, eaav9518. [Google Scholar] [CrossRef]
- Cruz Hernandez, J.C.; Bracko, O.; Kersbergen, C.J.; Muse, V.; Haft-Javaherian, M.; Berg, M.; Park, L.; Vinarcsik, L.K.; Ivasyk, I.; Rivera, D.A.; et al. Neutrophil adhesion in brain capillaries reduces cortical blood flow and impairs memory function in Alzheimer’s disease mouse models. Nat. Neurosci. 2019, 22, 413–420. [Google Scholar] [CrossRef] [PubMed]
- Cortes-Canteli, M.; Kruyer, A.; Fernandez-Nueda, I.; Marcos-Diaz, A.; Ceron, C.; Richards, A.T.; Jno-Charles, O.C.; Rodriguez, I.; Callejas, S.; Norris, E.H.; et al. Long-Term Dabigatran Treatment Delays Alzheimer’s Disease Pathogenesis in the TgCRND8 Mouse Model. J. Am. Coll. Cardiol. 2019, 74, 1910–1923. [Google Scholar] [CrossRef]
- Burmester, T.; Weich, B.; Reinhardt, S.; Hankeln, T. A vertebrate globin expressed in the brain. Nature 2000, 407, 520–523. [Google Scholar] [CrossRef] [PubMed]
- Jones, A.M.; Kennedy, N.; Hanson, J.; Fenton, G.W. A study of dementia in adults with Down’s syndrome using 99Tc(m)-HMPAO SPET. Nucl. Med. Commun. 1997, 18, 662–667. [Google Scholar] [CrossRef] [PubMed]
- Kao, C.H.; Wang, P.Y.; Wang, S.J.; Chou, K.T.; Hsu, C.Y.; Lin, W.Y.; Liao, S.Q.; Yeh, S.H. Regional cerebral blood flow of Alzheimer’s disease-like pattern in young patients with Down’s syndrome detected by 99Tcm-HMPAO brain SPECT. Nucl. Med. Commun. 1993, 14, 47–51. [Google Scholar] [CrossRef]
- Nunomura, A.; Perry, G.; Pappolla, M.A.; Friedland, R.P.; Hirai, K.; Chiba, S.; Smith, M.A. Neuronal oxidative stress precedes amyloid-beta deposition in Down syndrome. J. Neuropathol. Exp. Neurol. 2000, 59, 1011–1017. [Google Scholar] [CrossRef]
- Sumpio, B.E.; Riley, J.T.; Dardik, A. Cells in focus: Endothelial cell. Int. J. Biochem. Cell Biol. 2002, 34, 1508–1512. [Google Scholar] [CrossRef]
- Li, M.M.; Zheng, Y.L.; Wang, W.D.; Lin, S.; Lin, H.L. Neuropeptide Y: An Update on the Mechanism Underlying Chronic Intermittent Hypoxia-Induced Endothelial Dysfunction. Front. Physiol. 2021, 12, 712281. [Google Scholar] [CrossRef]
- Makarenko, V.V.; Usatyuk, P.V.; Yuan, G.; Lee, M.M.; Nanduri, J.; Natarajan, V.; Kumar, G.K.; Prabhakar, N.R. Intermittent hypoxia-induced endothelial barrier dysfunction requires ROS-dependent MAP kinase activation. Am. J. Physiol. Cell Physiol. 2014, 306, C745–C752. [Google Scholar] [CrossRef] [PubMed]
- Prabhakar, N.R.; Kumar, G.K.; Nanduri, J.; Semenza, G.L. ROS signaling in systemic and cellular responses to chronic intermittent hypoxia. Antioxid. Redox Signal. 2007, 9, 1397–1403. [Google Scholar] [CrossRef] [PubMed]
- Claesson-Welsh, L.; Dejana, E.; McDonald, D.M. Permeability of the Endothelial Barrier: Identifying and Reconciling Controversies. Trends Mol. Med. 2021, 27, 314–331. [Google Scholar] [CrossRef]
- Kovacic, J.C.; Dimmeler, S.; Harvey, R.P.; Finkel, T.; Aikawa, E.; Krenning, G.; Baker, A.H. Endothelial to Mesenchymal Transition in Cardiovascular Disease: JACC State-of-the-Art Review. J. Am. Coll. Cardiol. 2019, 73, 190–209. [Google Scholar] [CrossRef]
- Zeng, X.; Guo, R.; Dong, M.; Zheng, J.; Lin, H.; Lu, H. Contribution of TLR4 signaling in intermittent hypoxia-mediated atherosclerosis progression. J. Transl. Med. 2018, 16, 106. [Google Scholar] [CrossRef] [PubMed]
- Meszaros, M.; Kis, A.; Kunos, L.; Tarnoki, A.D.; Tarnoki, D.L.; Lazar, Z.; Bikov, A. The role of hyaluronic acid and hyaluronidase-1 in obstructive sleep apnoea. Sci. Rep. 2020, 10, 19484. [Google Scholar] [CrossRef]
- Hampel, H.; Vassar, R.; De Strooper, B.; Hardy, J.; Willem, M.; Singh, N.; Zhou, J.; Yan, R.; Vanmechelen, E.; De Vos, A.; et al. The beta-Secretase BACE1 in Alzheimer’s Disease. Biol. Psychiatry 2021, 89, 745–756. [Google Scholar] [CrossRef]
- Salminen, A.; Kauppinen, A.; Kaarniranta, K. Hypoxia/ischemia activate processing of Amyloid Precursor Protein: Impact of vascular dysfunction in the pathogenesis of Alzheimer’s disease. J. Neurochem. 2017, 140, 536–549. [Google Scholar] [CrossRef]
- Tesco, G.; Koh, Y.H.; Kang, E.L.; Cameron, A.N.; Das, S.; Sena-Esteves, M.; Hiltunen, M.; Yang, S.H.; Zhong, Z.; Shen, Y.; et al. Depletion of GGA3 stabilizes BACE and enhances beta-secretase activity. Neuron 2007, 54, 721–737. [Google Scholar] [CrossRef]
- Xiong, M.; Zhang, T.; Zhang, L.M.; Lu, S.D.; Huang, Y.L.; Sun, F.Y. Caspase inhibition attenuates accumulation of beta-amyloid by reducing beta-secretase production and activity in rat brains after stroke. Neurobiol. Dis. 2008, 32, 433–441. [Google Scholar] [CrossRef]
- Zhang, Y.; Xiong, M.; Yan, R.Q.; Sun, F.Y. Mutant ubiquitin-mediated beta-secretase stability via activation of caspase-3 is related to beta-amyloid accumulation in ischemic striatum in rats. J. Cereb. Blood Flow Metab. 2010, 30, 566–575. [Google Scholar] [CrossRef]
- Villa, J.C.; Chiu, D.; Brandes, A.H.; Escorcia, F.E.; Villa, C.H.; Maguire, W.F.; Hu, C.J.; de Stanchina, E.; Simon, M.C.; Sisodia, S.S.; et al. Nontranscriptional role of Hif-1α in activation of γ-secretase and notch signaling in breast cancer. Cell Rep. 2014, 8, 1077–1092. [Google Scholar] [CrossRef] [PubMed]
- Kaufmann, M.R.; Barth, S.; Konietzko, U.; Wu, B.; Egger, S.; Kunze, R.; Marti, H.H.; Hick, M.; Muller, U.; Camenisch, G.; et al. Dysregulation of hypoxia-inducible factor by presenilin/gamma-secretase loss-of-function mutations. J. Neurosci. 2013, 33, 1915–1926. [Google Scholar] [CrossRef]
- Cuadrado-Tejedor, M.; Vilariño, M.; Cabodevilla, F.; Del Río, J.; Frechilla, D.; Pérez-Mediavilla, A. Enhanced expression of the voltage-dependent anion channel 1 (VDAC1) in Alzheimer’s disease transgenic mice: An insight into the pathogenic effects of amyloid-β. J. Alzheimer’s Dis. 2011, 23, 195–206. [Google Scholar] [CrossRef]
- Manczak, M.; Sheiko, T.; Craigen, W.J.; Reddy, P.H. Reduced VDAC1 protects against Alzheimer’s disease, mitochondria, and synaptic deficiencies. J. Alzheimer’s Dis. 2013, 37, 679–690. [Google Scholar] [CrossRef]
- Sato, N.; Imaizumi, K.; Manabe, T.; Taniguchi, M.; Hitomi, J.; Katayama, T.; Yoneda, T.; Morihara, T.; Yasuda, Y.; Takagi, T.; et al. Increased production of beta-amyloid and vulnerability to endoplasmic reticulum stress by an aberrant spliced form of presenilin 2. J. Biol. Chem. 2001, 276, 2108–2114. [Google Scholar] [CrossRef] [PubMed]
- Raz, L.; Bhaskar, K.; Weaver, J.; Marini, S.; Zhang, Q.; Thompson, J.F.; Espinoza, C.; Iqbal, S.; Maphis, N.M.; Weston, L.; et al. Hypoxia promotes tau hyperphosphorylation with associated neuropathology in vascular dysfunction. Neurobiol. Dis. 2019, 126, 124–136. [Google Scholar] [CrossRef] [PubMed]
- Qiu, L.; Ng, G.; Tan, E.K.; Liao, P.; Kandiah, N.; Zeng, L. Chronic cerebral hypoperfusion enhances Tau hyperphosphorylation and reduces autophagy in Alzheimer’s disease mice. Sci. Rep. 2016, 6, 23964. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.L.; Wang, C.; Jiang, T.; Tan, L.; Xing, A.; Yu, J.T. The Role of Cdk5 in Alzheimer’s Disease. Mol. Neurobiol. 2016, 53, 4328–4342. [Google Scholar] [CrossRef]
- Seo, J.; Kritskiy, O.; Watson, L.A.; Barker, S.J.; Dey, D.; Raja, W.K.; Lin, Y.T.; Ko, T.; Cho, S.; Penney, J.; et al. Inhibition of p25/Cdk5 Attenuates Tauopathy in Mouse and iPSC Models of Frontotemporal Dementia. J. Neurosci. 2017, 37, 9917–9924. [Google Scholar] [CrossRef]
- Mottet, D.; Dumont, V.; Deccache, Y.; Demazy, C.; Ninane, N.; Raes, M.; Michiels, C. Regulation of hypoxia-inducible factor-1alpha protein level during hypoxic conditions by the phosphatidylinositol 3-kinase/Akt/glycogen synthase kinase 3beta pathway in HepG2 cells. J. Biol. Chem. 2003, 278, 31277–31285. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.Y.; Xie, J.W.; Wang, T.; Xu, Y.; Cai, J.H.; Wang, X.; Zhao, B.L.; An, L.; Wang, Z.Y. Hypoxia-triggered m-calpain activation evokes endoplasmic reticulum stress and neuropathogenesis in a transgenic mouse model of Alzheimer’s disease. CNS Neurosci. Ther. 2013, 19, 820–833. [Google Scholar] [CrossRef] [PubMed]
- Badiola, N.; Penas, C.; Minano-Molina, A.; Barneda-Zahonero, B.; Fado, R.; Sanchez-Opazo, G.; Comella, J.X.; Sabria, J.; Zhu, C.; Blomgren, K.; et al. Induction of ER stress in response to oxygen-glucose deprivation of cortical cultures involves the activation of the PERK and IRE-1 pathways and of caspase-12. Cell Death Dis. 2011, 2, e149. [Google Scholar] [CrossRef] [PubMed]
- Mitsuda, T.; Hayakawa, Y.; Itoh, M.; Ohta, K.; Nakagawa, T. ATF4 regulates gamma-secretase activity during amino acid imbalance. Biochem. Biophys. Res. Commun. 2007, 352, 722–727. [Google Scholar] [CrossRef] [PubMed]
- O’Connor, T.; Sadleir, K.R.; Maus, E.; Velliquette, R.A.; Zhao, J.; Cole, S.L.; Eimer, W.A.; Hitt, B.; Bembinster, L.A.; Lammich, S.; et al. Phosphorylation of the translation initiation factor eIF2alpha increases BACE1 levels and promotes amyloidogenesis. Neuron 2008, 60, 988–1009. [Google Scholar] [CrossRef]
- Rashid, H.O.; Yadav, R.K.; Kim, H.R.; Chae, H.J. ER stress: Autophagy induction, inhibition and selection. Autophagy 2015, 11, 1956–1977. [Google Scholar] [CrossRef]
- Deane, R.; Wu, Z.; Zlokovic, B.V. RAGE (yin) versus LRP (yang) balance regulates alzheimer amyloid beta-peptide clearance through transport across the blood-brain barrier. Stroke 2004, 35 (Suppl. 1), 2628–2631. [Google Scholar] [CrossRef]
- Govindpani, K.; Vinnakota, C.; Waldvogel, H.J.; Faull, R.L.; Kwakowsky, A. Vascular dysfunction in Alzheimer’s disease: A biomarker of disease progression and a potential therapeutic target. Neural Regen. Res. 2020, 15, 1030. [Google Scholar]
- Deane, R.; Wu, Z.; Sagare, A.; Davis, J.; Du Yan, S.; Hamm, K.; Xu, F.; Parisi, M.; LaRue, B.; Hu, H.W.; et al. LRP/Amyloid β-Peptide Interaction Mediates Differential Brain Efflux of Aβ Isoforms. Neuron 2004, 43, 333–344. [Google Scholar] [CrossRef]
- Kanekiyo, T.; Liu, C.C.; Shinohara, M.; Li, J.; Bu, G. LRP1 in brain vascular smooth muscle cells mediates local clearance of Alzheimer’s amyloid-β. J. Neurosci. 2012, 32, 16458–16465. [Google Scholar] [CrossRef]
- Zhao, Z.; Sagare, A.P.; Ma, Q.; Halliday, M.R.; Kong, P.; Kisler, K.; Winkler, E.A.; Ramanathan, A.; Kanekiyo, T.; Bu, G.; et al. Central role for PICALM in amyloid-beta blood-brain barrier transcytosis and clearance. Nat. Neurosci. 2015, 18, 978–987. [Google Scholar] [CrossRef] [PubMed]
- Deane, R.; Du Yan, S.; Submamaryan, R.K.; LaRue, B.; Jovanovic, S.; Hogg, E.; Welch, D.; Manness, L.; Lin, C.; Yu, J.; et al. RAGE mediates amyloid-beta peptide transport across the blood-brain barrier and accumulation in brain. Nat. Med. 2003, 9, 907–913. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Wang, R.; Chen, L.; Bennett, D.A.; Dickson, D.W.; Wang, D.S. Expression and functional profiling of neprilysin, insulin-degrading enzyme, and endothelin-converting enzyme in prospectively studied elderly and Alzheimer’s brain. J. Neurochem. 2010, 115, 47–57. [Google Scholar] [CrossRef] [PubMed]
- Zhang, G.L.; Zhang, X.; Wang, X.M.; Li, J.P. Towards understanding the roles of heparan sulfate proteoglycans in Alzheimer’s disease. BioMed Res. Int. 2014, 2014, 516028. [Google Scholar] [CrossRef]
- Austin, S.A.; Katusic, Z.S. Loss of Endothelial Nitric Oxide Synthase Promotes p25 Generation and Tau Phosphorylation in a Murine Model of Alzheimer’s Disease. Circ. Res. 2016, 119, 1128–1134. [Google Scholar] [CrossRef]
- Iadecola, C. Untangling Neurons With Endothelial Nitric Oxide. Circ. Res. 2016, 119, 1052–1054. [Google Scholar] [CrossRef]
- Jack, C.R., Jr.; Bennett, D.A.; Blennow, K.; Carrillo, M.C.; Dunn, B.; Haeberlein, S.B.; Holtzman, D.M.; Jagust, W.; Jessen, F.; Karlawish, J.; et al. NIA-AA Research Framework: Toward a biological definition of Alzheimer’s disease. Alzheimer’s Dement. 2018, 14, 535–562. [Google Scholar] [CrossRef]
- Ittner, L.M.; Ke, Y.D.; Delerue, F.; Bi, M.; Gladbach, A.; van Eersel, J.; Wolfing, H.; Chieng, B.C.; Christie, M.J.; Napier, I.A.; et al. Dendritic function of tau mediates amyloid-beta toxicity in Alzheimer’s disease mouse models. Cell 2010, 142, 387–397. [Google Scholar] [CrossRef]
- Marin, R.; Fabelo, N.; Fernandez-Echevarria, C.; Canerina-Amaro, A.; Rodriguez-Barreto, D.; Quinto-Alemany, D.; Mesa-Herrera, F.; Diaz, M. Lipid Raft Alterations in Aged-Associated Neuropathologies. Curr. Alzheimer Res. 2016, 13, 973–984. [Google Scholar] [CrossRef]
- Zabroski, I.O.; Nugent, M.A. Lipid Raft Association Stabilizes VEGF Receptor 2 in Endothelial Cells. Int. J. Mol. Sci. 2021, 22, 798. [Google Scholar] [CrossRef]
- Quinn, P.J. A lipid matrix model of membrane raft structure. Prog. Lipid Res. 2010, 49, 390–406. [Google Scholar] [CrossRef] [PubMed]
- Bhattacharyya, R.; Barren, C.; Kovacs, D.M. Palmitoylation of amyloid precursor protein regulates amyloidogenic processing in lipid rafts. J. Neurosci. 2013, 33, 11169–11183. [Google Scholar] [CrossRef] [PubMed]
- Osenkowski, P.; Ye, W.; Wang, R.; Wolfe, M.S.; Selkoe, D.J. Direct and potent regulation of gamma-secretase by its lipid microenvironment. J. Biol. Chem. 2008, 283, 22529–22540. [Google Scholar] [CrossRef] [PubMed]
- Rushworth, J.V.; Hooper, N.M. Lipid Rafts: Linking Alzheimer’s Amyloid-β Production, Aggregation, and Toxicity at Neuronal Membranes. Int. J. Alzheimer’s Dis. 2010, 2011, 603052. [Google Scholar] [CrossRef] [PubMed]
- Danza, G.; Di Serio, C.; Ambrosio, M.R.; Sturli, N.; Lonetto, G.; Rosati, F.; Rocca, B.J.; Ventimiglia, G.; del Vecchio, M.T.; Prudovsky, I.; et al. Notch3 is activated by chronic hypoxia and contributes to the progression of human prostate cancer. Int. J. Cancer 2013, 133, 2577–2586. [Google Scholar] [CrossRef]
- Kawarabayashi, T.; Shoji, M.; Younkin, L.H.; Wen-Lang, L.; Dickson, D.W.; Murakami, T.; Matsubara, E.; Abe, K.; Ashe, K.H.; Younkin, S.G. Dimeric amyloid beta protein rapidly accumulates in lipid rafts followed by apolipoprotein E and phosphorylated tau accumulation in the Tg2576 mouse model of Alzheimer’s disease. J. Neurosci. 2004, 24, 3801–3809. [Google Scholar] [CrossRef]
- Bok, E.; Leem, E.; Lee, B.R.; Lee, J.M.; Yoo, C.J.; Lee, E.M.; Kim, J. Role of the Lipid Membrane and Membrane Proteins in Tau Pathology. Front. Cell. Dev. Biol. 2021, 9, 653815. [Google Scholar] [CrossRef]
- Pilarczyk, M.; Mateuszuk, L.; Rygula, A.; Kepczynski, M.; Chlopicki, S.; Baranska, M.; Kaczor, A. Endothelium in spots--high-content imaging of lipid rafts clusters in db/db mice. PLoS ONE 2014, 9, e106065. [Google Scholar] [CrossRef]
- Touyz, R.M. Lipid rafts take center stage in endothelial cell redox signaling by death receptors. Hypertension 2006, 47, 16–18. [Google Scholar] [CrossRef]
- Thirumangalakudi, L.; Samany, P.G.; Owoso, A.; Wiskar, B.; Grammas, P. Angiogenic proteins are expressed by brain blood vessels in Alzheimer’s disease. J. Alzheimer’s Dis. 2006, 10, 111–118. [Google Scholar] [CrossRef]
- Grammas, P.; Samany, P.G.; Thirumangalakudi, L. Thrombin and inflammatory proteins are elevated in Alzheimer’s disease microvessels: Implications for disease pathogenesis. J. Alzheimer’s Dis. 2006, 9, 51–58. [Google Scholar] [CrossRef]
- Wu, Z.; Guo, H.; Chow, N.; Sallstrom, J.; Bell, R.D.; Deane, R.; Brooks, A.I.; Kanagala, S.; Rubio, A.; Sagare, A.; et al. Role of the MEOX2 homeobox gene in neurovascular dysfunction in Alzheimer disease. Nat. Med. 2005, 11, 959–965. [Google Scholar] [CrossRef] [PubMed]
- Jellinger, K.A. Alzheimer disease and cerebrovascular pathology: An update. J. Neural Transm. 2002, 109, 813–836. [Google Scholar] [CrossRef] [PubMed]
- Buee, L.; Hof, P.R.; Delacourte, A. Brain microvascular changes in Alzheimer’s disease and other dementias. Ann. N. Y. Acad. Sci. 1997, 826, 7–24. [Google Scholar] [CrossRef] [PubMed]
- Kalaria, R.N.; Cohen, D.L.; Premkumar, D.R.; Nag, S.; LaManna, J.C.; Lust, W.D. Vascular endothelial growth factor in Alzheimer’s disease and experimental cerebral ischemia. Brain Res. Mol. Brain Res. 1998, 62, 101–105. [Google Scholar] [CrossRef]
- Guo, H.; Xia, D.; Liao, S.; Niu, B.; Tang, J.; Hu, H.; Qian, H.; Cao, B. Vascular endothelial growth factor improves the cognitive decline of Alzheimer’s disease via concurrently inducing the expression of ADAM10 and reducing the expression of beta-site APP cleaving enzyme 1 in Tg2576 mice. Neurosci. Res. 2019, 142, 49–57. [Google Scholar] [CrossRef]
- Tang, H.; Mao, X.; Xie, L.; Greenberg, D.A.; Jin, K. Expression level of vascular endothelial growth factor in hippocampus is associated with cognitive impairment in patients with Alzheimer’s disease. Neurobiol. Aging 2013, 34, 1412–1415. [Google Scholar] [CrossRef]
- Tarkowski, E.; Issa, R.; Sjögren, M.; Wallin, A.; Blennow, K.; Tarkowski, A.; Kumar, P. Increased intrathecal levels of the angiogenic factors VEGF and TGF-β in Alzheimer’s disease and vascular dementia. Neurobiol. Aging 2002, 23, 237–243. [Google Scholar] [CrossRef]
- Del Bo, R.; Ghezzi, S.; Scarpini, E.; Bresolin, N.; Comi, G.P. VEGF genetic variability is associated with increased risk of developing Alzheimer’s disease. J. Neurol. Sci. 2009, 283, 66–68. [Google Scholar] [CrossRef]
- Paris, D.; Townsend, K.; Quadros, A.; Humphrey, J.; Sun, J.; Brem, S.; Wotoczek-Obadia, M.; DelleDonne, A.; Patel, N.; Obregon, D.F.; et al. Inhibition of angiogenesis by Abeta peptides. Angiogenesis 2004, 7, 75–85. [Google Scholar] [CrossRef]
- Ali, M.; Bracko, O. VEGF Paradoxically Reduces Cerebral Blood Flow in Alzheimer’s Disease Mice. Neurosci. Insights 2022, 17, 1–4. [Google Scholar] [CrossRef] [PubMed]
- Nishino, A.; Suzuki, M.; Ohtani, H.; Motohashi, O.; Umezawa, K.; Nagura, H.; Yoshimoto, T. Thrombin may contribute to the pathophysiology of central nervous system injury. J. Neurotrauma 1993, 10, 167–179. [Google Scholar] [CrossRef] [PubMed]
- Sokolova, E.; Reiser, G. Prothrombin/thrombin and the thrombin receptors PAR-1 and PAR-4 in the brain: Localization, expression and participation in neurodegenerative diseases. Thromb. Haemost. 2008, 100, 576–581. [Google Scholar] [CrossRef] [PubMed]
- Vaughan, P.J.; Su, J.; Cotman, C.W.; Cunningham, D.D. Protease nexin-1, a potent thrombin inhibitor, is reduced around cerebral blood vessels in Alzheimer’s disease. Brain Res. 1994, 668, 160–170. [Google Scholar] [CrossRef]
- Naldini, A.; Carney, D.H.; Pucci, A.; Pasquali, A.; Carraro, F. Thrombin regulates the expression of proangiogenic cytokines via proteolytic activation of protease-activated receptor-1. Gen. Pharm. 2000, 35, 255–259. [Google Scholar] [CrossRef]
- Dupuy, E.; Habib, A.; Lebret, M.; Yang, R.; Levy-Toledano, S.; Tobelem, G. Thrombin induces angiogenesis and vascular endothelial growth factor expression in human endothelial cells: Possible relevance to HIF-1alpha. J. Thromb. Haemost. 2003, 1, 1096–1102. [Google Scholar] [CrossRef]
- Tsopanoglou, N.E.; Andriopoulou, P.; Maragoudakis, M.E. On the mechanism of thrombin-induced angiogenesis: Involvement of alphavbeta3-integrin. Am. J. Physiol. Cell Physiol. 2002, 283, C1501–C1510. [Google Scholar] [CrossRef]
- Smirnova, I.V.; Zhang, S.X.; Citron, B.A.; Arnold, P.M.; Festoff, B.W. Thrombin is an extracellular signal that activates intracellular death protease pathways inducing apoptosis in model motor neurons. J. Neurobiol. 1998, 36, 64–80. [Google Scholar] [CrossRef]
- Mhatre, M.; Nguyen, A.; Kashani, S.; Pham, T.; Adesina, A.; Grammas, P. Thrombin, a mediator of neurotoxicity and memory impairment. Neurobiol. Aging 2004, 25, 783–793. [Google Scholar] [CrossRef]
- Turgeon, V.L.; Milligan, C.E.; Houenou, L.J. Activation of the protease-activated thrombin receptor (PAR)-1 induces motoneuron degeneration in the developing avian embryo. J. Neuropathol. Exp. Neurol. 1999, 58, 499–504. [Google Scholar] [CrossRef]
- Suo, Z.; Wu, M.; Citron, B.A.; Palazzo, R.E.; Festoff, B.W. Rapid tau aggregation and delayed hippocampal neuronal death induced by persistent thrombin signaling. J. Biol. Chem. 2003, 278, 37681–37689. [Google Scholar] [CrossRef] [PubMed]
- Park, K.W.; Jin, B.K. Thrombin-induced oxidative stress contributes to the death of hippocampal neurons: Role of neuronal NADPH oxidase. J. Neurosci. Res. 2008, 86, 1053–1063. [Google Scholar] [CrossRef] [PubMed]
- Lee, D.Y.; Park, K.W.; Jin, B.K. Thrombin induces neurodegeneration and microglial activation in the cortex in vivo and in vitro: Proteolytic and non-proteolytic actions. Biochem. Biophys. Res. Commun. 2006, 346, 727–738. [Google Scholar] [CrossRef]
- Huang, C.F.; Li, G.; Ma, R.; Sun, S.G.; Chen, J.G. Thrombin-induced microglial activation contributes to the degeneration of nigral dopaminergic neurons in vivo. Neurosci. Bull. 2008, 24, 66–72. [Google Scholar] [CrossRef] [PubMed]
- Huang, C.; Ma, R.; Sun, S.; Wei, G.; Fang, Y.; Liu, R.; Li, G. JAK2-STAT3 signaling pathway mediates thrombin-induced proinflammatory actions of microglia in vitro. J. Neuroimmunol. 2008, 204, 118–125. [Google Scholar] [CrossRef] [PubMed]
- Choi, M.S.; Kim, Y.E.; Lee, W.J.; Choi, J.W.; Park, G.H.; Kim, S.D.; Jeon, S.J.; Go, H.S.; Shin, S.M.; Kim, W.K.; et al. Activation of protease-activated receptor1 mediates induction of matrix metalloproteinase-9 by thrombin in rat primary astrocytes. Brain Res. Bull. 2008, 76, 368–375. [Google Scholar] [CrossRef]
- Grammas, P.; Ovase, R. Inflammatory factors are elevated in brain microvessels in Alzheimer’s disease. Neurobiol. Aging 2001, 22, 837–842. [Google Scholar] [CrossRef]
- Grammas, P.; Ovase, R. Cerebrovascular transforming growth factor-beta contributes to inflammation in the Alzheimer’s disease brain. Am. J. Pathol. 2002, 160, 1583–1587. [Google Scholar] [CrossRef]
- Li, M.; Shang, D.S.; Zhao, W.D.; Tian, L.; Li, B.; Fang, W.G.; Zhu, L.; Man, S.M.; Chen, Y.H. Amyloid beta interaction with receptor for advanced glycation end products up-regulates brain endothelial CCR5 expression and promotes T cells crossing the blood-brain barrier. J. Immunol. 2009, 182, 5778–5788. [Google Scholar] [CrossRef]
- Imhof, B.A.; Dunon, D. Leukocyte Migration and Adhesion. In Advances in Immunology; Dixon, F.J., Ed.; Academic Press: Cambridge, MA, USA, 1995; Volume 58, pp. 345–416. [Google Scholar]
- Bevilacqua, M.P.; Pober, J.S.; Mendrick, D.L.; Cotran, R.S.; Gimbrone, M.A., Jr. Identification of an inducible endothelial-leukocyte adhesion molecule. Proc. Natl. Acad. Sci. USA 1987, 84, 9238–9242. [Google Scholar] [CrossRef]
- McCoy, M.G.; Nascimento, D.W.; Veleeparambil, M.; Murtazina, R.; Gao, D.; Tkachenko, S.; Podrez, E.; Byzova, T.V. Endothelial TLR2 promotes proangiogenic immune cell recruitment and tumor angiogenesis. Sci. Signal. 2021, 14, eabc5371. [Google Scholar] [CrossRef] [PubMed]
- Li, G.; Xiong, K.; Korff, A.; Pan, C.; Quinn, J.F.; Galasko, D.R.; Liu, C.; Montine, T.J.; Peskind, E.R.; Zhang, J. Increased CSF E-Selectin in Clinical Alzheimer’s Disease without Altered CSF Abeta42 and Tau. J. Alzheimer’s Dis. 2015, 47, 883–887. [Google Scholar] [CrossRef]
- Govindpani, K.; McNamara, L.G.; Smith, N.R.; Vinnakota, C.; Waldvogel, H.J.; Faull, R.L.; Kwakowsky, A. Vascular Dysfunction in Alzheimer’s Disease: A Prelude to the Pathological Process or a Consequence of It? J. Clin. Med. 2019, 8, 651. [Google Scholar] [CrossRef] [PubMed]
- Howland, S.W.; Gun, S.Y.; Claser, C.; Poh, C.M.; Renia, L. Measuring antigen presentation in mouse brain endothelial cells ex vivo and in vitro. Nat. Protoc. 2015, 10, 2016–2026. [Google Scholar] [CrossRef] [PubMed]
- Park, L.; Wang, G.; Zhou, P.; Zhou, J.; Pitstick, R.; Previti, M.L.; Younkin, L.; Younkin, S.G.; Van Nostrand, W.E.; Cho, S.; et al. Scavenger receptor CD36 is essential for the cerebrovascular oxidative stress and neurovascular dysfunction induced by amyloid-beta. Proc. Natl. Acad. Sci. USA 2011, 108, 5063–5068. [Google Scholar] [CrossRef]
- Amersfoort, J.; Eelen, G.; Carmeliet, P. Immunomodulation by endothelial cells-partnering up with the immune system? Nat. Rev. Immunol. 2022, 22, 576–588. [Google Scholar] [CrossRef]
- Winkler, E.A.; Nishida, Y.; Sagare, A.P.; Rege, S.V.; Bell, R.D.; Perlmutter, D.; Sengillo, J.D.; Hillman, S.; Kong, P.; Nelson, A.R.; et al. GLUT1 reductions exacerbate Alzheimer’s disease vasculo-neuronal dysfunction and degeneration. Nat. Neurosci. 2015, 18, 521–530. [Google Scholar] [CrossRef]
- VanGuilder, H.D.; Farley, J.A.; Yan, H.; Van Kirk, C.A.; Mitschelen, M.; Sonntag, W.E.; Freeman, W.M. Hippocampal dysregulation of synaptic plasticity-associated proteins with age-related cognitive decline. Neurobiol. Dis. 2011, 43, 201–212. [Google Scholar] [CrossRef]
- Yang, J.; Yao, Y.; Wang, L.; Yang, C.; Wang, F.; Guo, J.; Wang, Z.; Yang, Z.; Ming, D. Gastrin-releasing peptide facilitates glutamatergic transmission in the hippocampus and effectively prevents vascular dementia induced cognitive and synaptic plasticity deficits. Exp. Neurol. 2017, 287 Pt 1, 75–83. [Google Scholar] [CrossRef]
- Wang, F.; Cao, Y.; Ma, L.; Pei, H.; Rausch, W.D.; Li, H. Dysfunction of Cerebrovascular Endothelial Cells: Prelude to Vascular Dementia. Front. Aging Neurosci. 2018, 10, 376. [Google Scholar] [CrossRef]
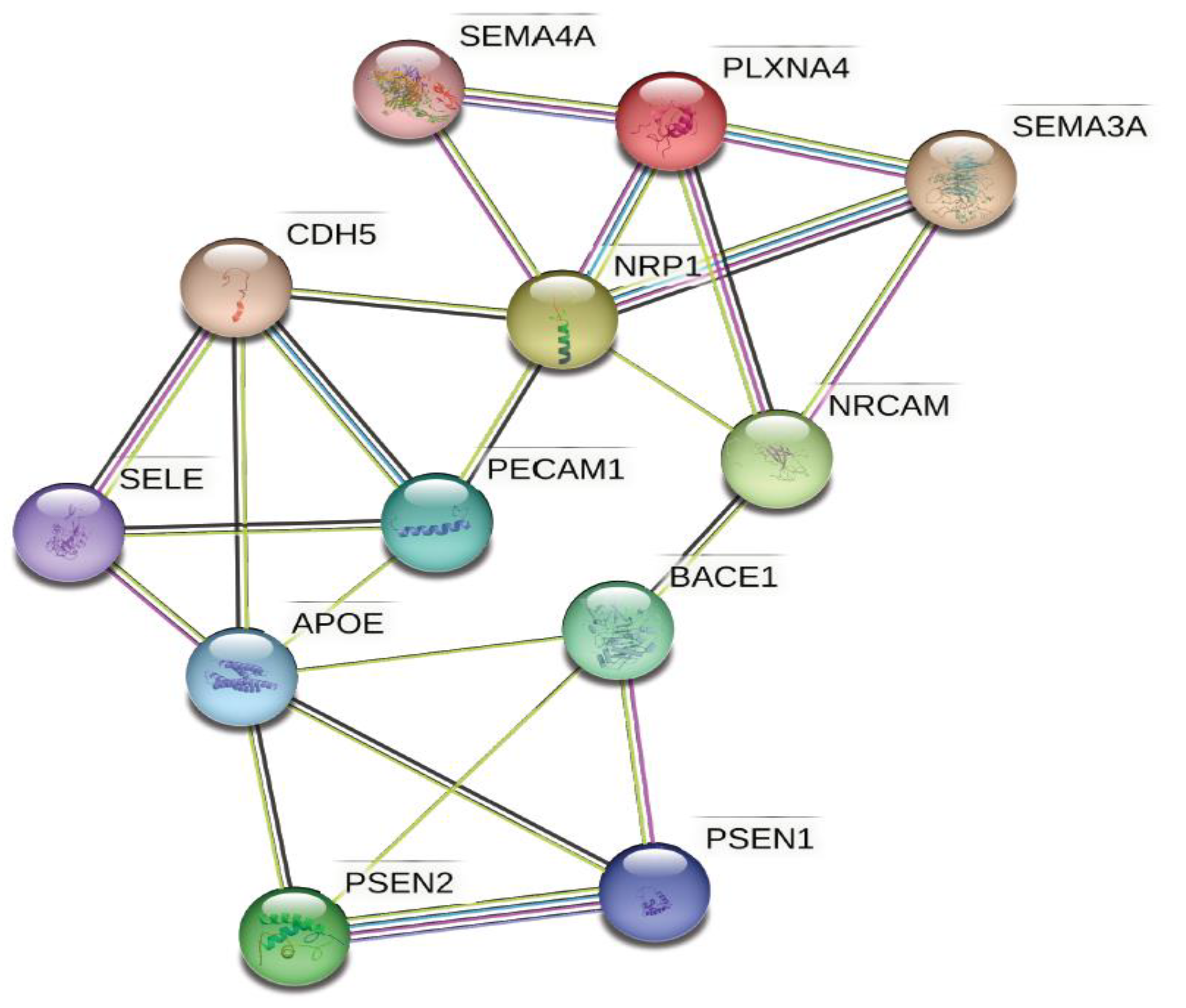
- Demyanenko, G.P.; Mohan, V.; Zhang, X.; Brennaman, L.H.; Dharbal, K.E.S.; Tran, T.S.; Manis, P.B.; Maness, P.F. Neural Cell Adhesion Molecule NrCAM Regulates Semaphorin 3F-Induced Dendritic Spine Remodeling. J. Neurosci. 2014, 34, 11274. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Mandeville, E.T.; Duran-Laforet, V.; Fukuda, N.; Yu, Z.; Zheng, Y.; Held, A.; Park, J.H.; Nakano, T.; Tanaka, M.; et al. Endothelial cells regulate astrocyte to neural progenitor cell trans-differentiation in a mouse model of stroke. Nat. Commun. 2022, 13, 7812. [Google Scholar] [CrossRef] [PubMed]
- Park, L.; Anrather, J.; Girouard, H.; Zhou, P.; Iadecola, C. Nox2-Derived Reactive Oxygen Species Mediate Neurovascular Dysregulation in the Aging Mouse Brain. J. Cereb. Blood Flow Metab. 2007, 27, 1908–1918. [Google Scholar] [CrossRef] [PubMed]
- Toth, P.; Tarantini, S.; Tucsek, Z.; Ashpole, N.M.; Sosnowska, D.; Gautam, T.; Ballabh, P.; Koller, A.; Sonntag, W.E.; Csiszar, A.; et al. Resveratrol treatment rescues neurovascular coupling in aged mice: Role of improved cerebromicrovascular endothelial function and downregulation of NADPH oxidase. Am. J. Physiol. Heart Circ. Physiol. 2014, 306, H299–H308. [Google Scholar] [CrossRef]
- Csiszar, A.; Ungvari, Z.; Edwards, J.G.; Kaminski, P.; Wolin, M.S.; Koller, A.; Kaley, G. Aging-induced phenotypic changes and oxidative stress impair coronary arteriolar function. Circ. Res. 2002, 90, 1159–1166. [Google Scholar] [CrossRef]
- Toth, P.; Tarantini, S.; Ashpole, N.M.; Tucsek, Z.; Milne, G.L.; Valcarcel-Ares, N.M.; Menyhart, A.; Farkas, E.; Sonntag, W.E.; Csiszar, A.; et al. IGF-1 deficiency impairs neurovascular coupling in mice: Implications for cerebromicrovascular aging. Aging Cell 2015, 14, 1034–1044. [Google Scholar] [CrossRef]
- Szklarczyk, D.; Gable, A.L.; Nastou, K.C.; Lyon, D.; Kirsch, R.; Pyysalo, S.; Doncheva, N.T.; Legeay, M.; Fang, T.; Bork, P.; et al. The STRING database in 2021: Customizable protein-protein networks, and functional characterization of user-uploaded gene/measurement sets. Nucleic Acids Res. 2021, 49, D605–D612. [Google Scholar] [CrossRef]
- Szklarczyk, D.; Gable, A.L.; Lyon, D.; Junge, A.; Wyder, S.; Huerta-Cepas, J.; Simonovic, M.; Doncheva, N.T.; Morris, J.H.; Bork, P.; et al. STRING v11: Protein–protein association networks with increased coverage, supporting functional discovery in genome-wide experimental datasets. Nucleic Acids Res. 2018, 47, D607–D613. [Google Scholar] [CrossRef]
- Szklarczyk, D.; Morris, J.H.; Cook, H.; Kuhn, M.; Wyder, S.; Simonovic, M.; Santos, A.; Doncheva, N.T.; Roth, A.; Bork, P.; et al. The STRING database in 2017: Quality-controlled protein-protein association networks, made broadly accessible. Nucleic Acids Res. 2017, 45, D362–D368. [Google Scholar] [CrossRef]
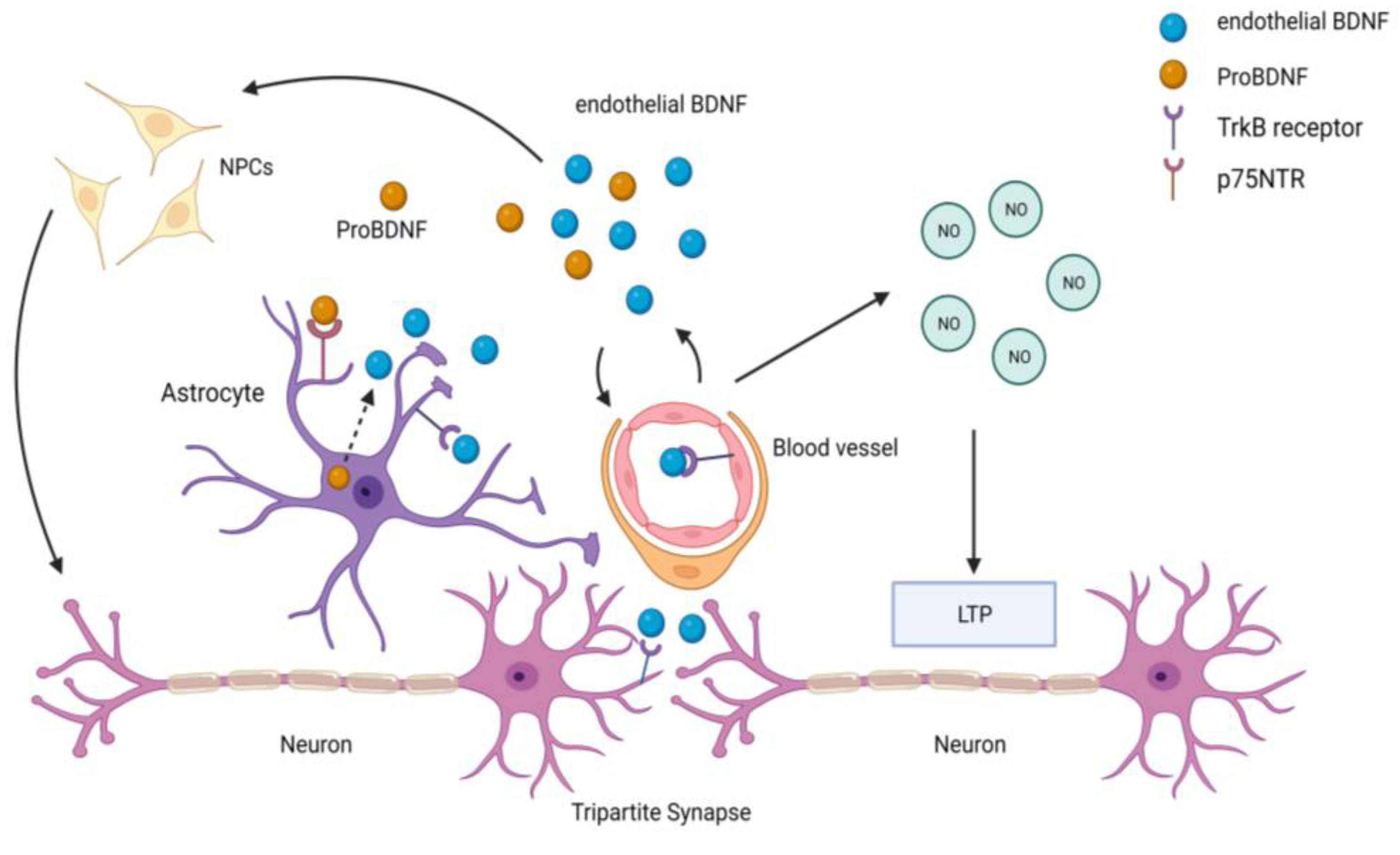
- Marie, C.; Pedard, M.; Quirie, A.; Tessier, A.; Garnier, P.; Totoson, P.; Demougeot, C. Brain-derived neurotrophic factor secreted by the cerebral endothelium: A new actor of brain function? J. Cereb. Blood Flow Metab. 2018, 38, 935–949. [Google Scholar] [CrossRef]
- Amenta, F.; Di Tullio, M.A.; Tomassoni, D. Arterial Hypertension and Brain Damage—Evidence from Animal Models (Review). Clin. Exp. Hypertens. 2003, 25, 359–380. [Google Scholar] [CrossRef] [PubMed]
- Tomiga, Y.; Higaki, Y.; Anzai, K.; Takahashi, H. Behavioral defects and downregulation of hippocampal BDNF and nNOS expression in db/db mice did not improved by chronic TGF-β2 treatment. Front. Physiol. 2022, 13, 969480. [Google Scholar] [CrossRef] [PubMed]
- Guo, S.; Kim, W.J.; Lok, J.; Lee, S.R.; Besancon, E.; Luo, B.H.; Stins, M.F.; Wang, X.; Dedhar, S.; Lo, E.H. Neuroprotection via matrix-trophic coupling between cerebral endothelial cells and neurons. Proc. Natl. Acad. Sci. USA 2008, 105, 7582–7587. [Google Scholar] [CrossRef]
- Navaratna, D.; Guo, S.Z.; Hayakawa, K.; Wang, X.; Gerhardinger, C.; Lo, E.H. Decreased cerebrovascular brain-derived neurotrophic factor-mediated neuroprotection in the diabetic brain. Diabetes 2011, 60, 1789–1796. [Google Scholar] [CrossRef] [PubMed]
- Numakawa, T.; Suzuki, S.; Kumamaru, E.; Adachi, N.; Richards, M.; Kunugi, H. BDNF function and intracellular signaling in neurons. Histol. Histopathol. 2010, 25, 237–258. [Google Scholar] [CrossRef] [PubMed]
- Meuchel, L.W.; Thompson, M.A.; Cassivi, S.D.; Pabelick, C.M.; Prakash, Y.S. Neurotrophins induce nitric oxide generation in human pulmonary artery endothelial cells. Cardiovasc. Res. 2011, 91, 668–676. [Google Scholar] [CrossRef] [PubMed]
- Rudge, J.S.; Li, Y.; Pasnikowski, E.M.; Mattsson, K.; Pan, L.; Yancopoulos, G.D.; Wiegand, S.J.; Lindsay, R.M.; Ip, N.Y. Neurotrophic factor receptors and their signal transduction capabilities in rat astrocytes. Eur. J. Neurosci. 1994, 6, 693–705. [Google Scholar] [CrossRef]
- Vignoli, B.; Battistini, G.; Melani, R.; Blum, R.; Santi, S.; Berardi, N.; Canossa, M. Peri-Synaptic Glia Recycles Brain-Derived Neurotrophic Factor for LTP Stabilization and Memory Retention. Neuron 2016, 92, 873–887. [Google Scholar] [CrossRef]
- Grammas, P.; Moore, P.; Weigel, P.H. Microvessels from Alzheimer’s disease brains kill neurons in vitro. Am. J. Pathol. 1999, 154, 337–342. [Google Scholar] [CrossRef]
- Yoder, M.C. Human endothelial progenitor cells. Cold Spring Harb. Perspect. Med. 2012, 2, a006692. [Google Scholar] [CrossRef]
- Lee, S.T.; Chu, K.; Jung, K.H.; Park, H.K.; Kim, D.H.; Bahn, J.J.; Kim, J.H.; Oh, M.J.; Lee, S.K.; Kim, M.; et al. Reduced circulating angiogenic cells in Alzheimer disease. Neurology 2009, 72, 1858–1863. [Google Scholar] [CrossRef] [PubMed]
- Custodia, A.; Ouro, A.; Romaus-Sanjurjo, D.; Pias-Peleteiro, J.M.; de Vries, H.E.; Castillo, J.; Sobrino, T. Endothelial Progenitor Cells and Vascular Alterations in Alzheimer’s Disease. Front. Aging Neurosci. 2021, 13, 811210. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Zhi, Y.; Li, F.; Huang, S.; Gao, H.; Han, Z.; Ge, X.; Li, D.; Chen, F.; Kong, X.; et al. Transplantation of in vitro cultured endothelial progenitor cells repairs the blood-brain barrier and improves cognitive function of APP/PS1 transgenic AD mice. J. Neurol. Sci. 2018, 387, 6–15. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
| AD Risk Gene | Vascular Cell Types |
|---|---|
| APOE | Smooth muscle cells, Meningeal fibroblasts |
| PICALM * | Brain endothelium (arterial, capillary, and venous) |
| CLU | Meningeal fibroblasts, Ependymal cells |
| ABCA7 | T cells |
| PTK2B | T cells |
| PLCG2 * | Brain endothelium (arterial) |
| HLA-DRB1 * | Brain endothelium (arterial) |
| CD2AP * | Brain endothelium (arterial, capillary, and venous) |
| SLC24A4 | Ependymal cells |
| RIN3 | T cells |
| ADAMTS1 * | Smooth muscle cells, Pericytes, Brain endothelium (arterial) |
| ADAMTS4 | Smooth muscle cells, Pericytes |
| FERMT2 | Smooth muscle cells, Pericytes |
| SCIMP | Ependymal cells |
| CLNK | T cell |
| ECHDC3 | Perivascular fibroblasts |
| TNIP1 * | Brain endothelium (capillary and venous), T cells |
| ABCA1 * | Brain endothelium (venous), Perivascular fibroblasts |
| USP6NL * | Brain endothelium (capillary and venous) |
| INPP5D * | Brain endothelium (capillary), T cells |
| ACE * | Brain endothelium (arterial and capillary) |
| IQCK | Ependymal cells |
| ABI3 | T cells |
| HESX1 | Meningeal fibroblasts |
| FHL2 | Perivascular fibroblasts |
| CHRNE | Perivascular fibroblasts, T cells |
| AGRN | Pericytes |
| IL34 | Smooth muscle cells, Pericytes, Meningeal fibroblasts |
| NYAP1 | Ependymal cells |
| CASS4 * | Brain endothelium (capillary and venous) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tarawneh, R. Microvascular Contributions to Alzheimer Disease Pathogenesis: Is Alzheimer Disease Primarily an Endotheliopathy? Biomolecules 2023, 13, 830. https://doi.org/10.3390/biom13050830
Tarawneh R. Microvascular Contributions to Alzheimer Disease Pathogenesis: Is Alzheimer Disease Primarily an Endotheliopathy? Biomolecules. 2023; 13(5):830. https://doi.org/10.3390/biom13050830
Chicago/Turabian StyleTarawneh, Rawan. 2023. "Microvascular Contributions to Alzheimer Disease Pathogenesis: Is Alzheimer Disease Primarily an Endotheliopathy?" Biomolecules 13, no. 5: 830. https://doi.org/10.3390/biom13050830