
Role of Angiotensin II in Cardiovascular Diseases: Introducing Bisartans as a Novel Therapy for Coronavirus 2019
 , ,
, ,  ,
,  and
and
Abstract
:1. Introduction
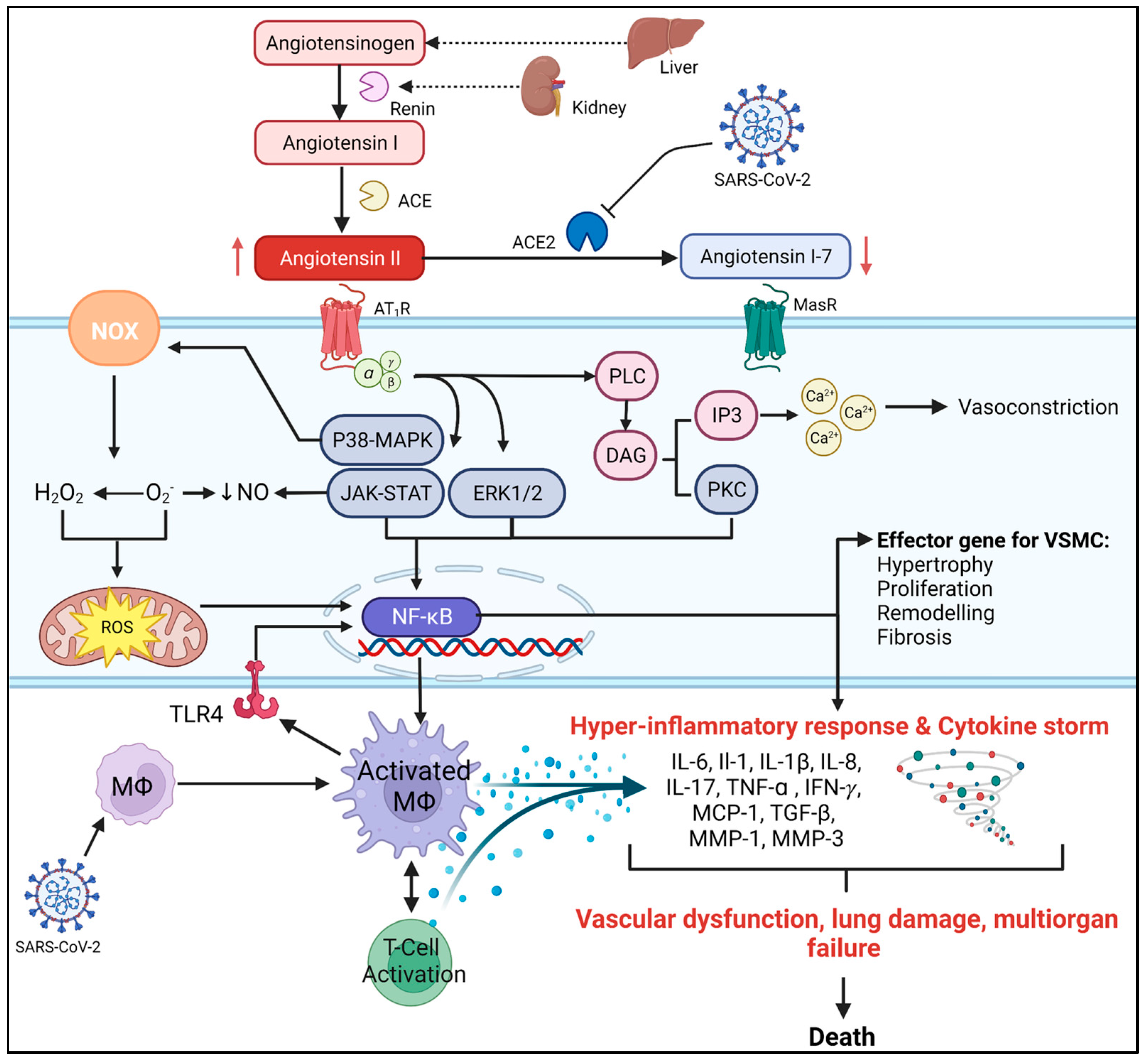
2. The Role of AngII in CVDs
2.1. AngII and Endothelial Dysfunction
2.2. AngII and Oxidative Stress
2.3. AngII and CVD Inflammation
3. The Relationship between CVDs and COVID-19
COVID-19 and RAS
4. ARBs as a Treatment for COVID-19
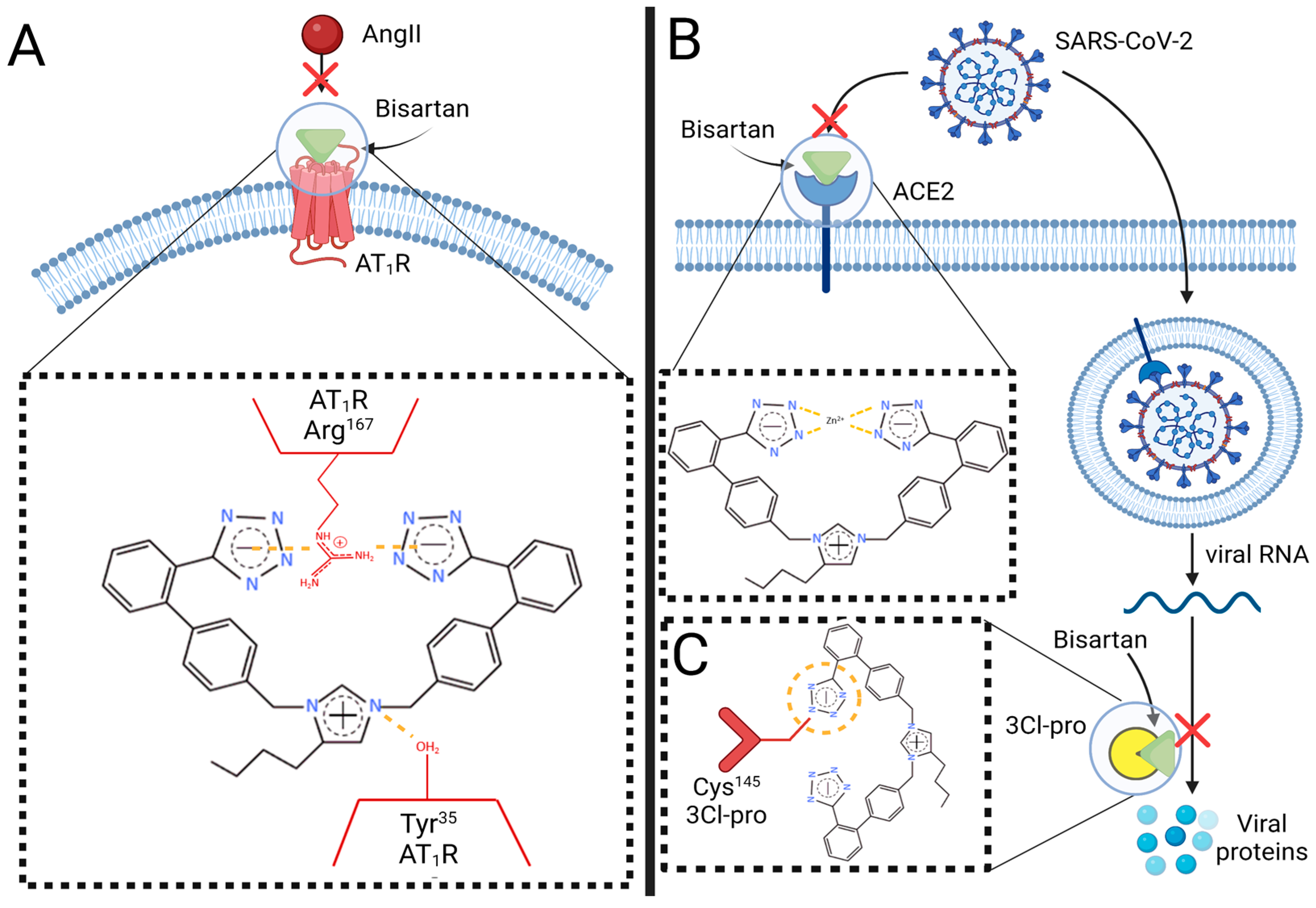
5. Bisartans, a New Generation of ARBs against SARS-CoV-2
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Amini, M.; Zayeri, F.; Salehi, M. Trend analysis of cardiovascular disease mortality, incidence, and mortality-to-incidence ratio: Results from global burden of disease study 2017. BMC Public Health 2021, 21, 401. [Google Scholar] [CrossRef] [PubMed]
- Tsao, C.; Aaron, A.W.; Almarzooq, Z.I.; Alonso, A.; Beaton, A.Z.; Bittencourt, M.S.; Boehme, A.K.; Buxton, A.E.; Carson, A.P.; CommodoreMensah, Y.; et al. Heart Disease and Stroke Statistics—2022 Update: A Report From the American Heart Association. Circulation 2022, 145, e153–e639. [Google Scholar] [CrossRef] [PubMed]
- Roth, G.A.; Mensah, G.A.; Johnson, C.O.; Addolorato, G.; Ammirati, E.; Baddour, L.M.; Barengo, N.C.; Beaton, A.Z.; Benjamin, E.J.; Benziger, C.P.; et al. Global Burden of Cardiovascular Diseases and Risk Factors, 1990-2019: Update From the GBD 2019 Study. J. Am. Coll. Cardiol. 2020, 76, 2982–3021. [Google Scholar] [CrossRef]
- American Heart Association. Cardiovascular Disease: A Costly Burden for America. Projections through 2035; AHA: Dallas, TX, USA, 2017. [Google Scholar]
- Frak, W.; Wojtasinska, A.; Lisinska, W.; Mlynarska, E.; Franczyk, B.; Rysz, J. Pathophysiology of Cardiovascular Diseases: New Insights into Molecular Mechanisms of Atherosclerosis, Arterial Hypertension, and Coronary Artery Disease. Biomedicines 2022, 10, 1938. [Google Scholar] [CrossRef]
- Bitker, L.; Burrell, L.M. Classic and Nonclassic Renin-Angiotensin Systems in the Critically. Crit. Care Clin. 2019, 35, 213–227. [Google Scholar] [CrossRef] [PubMed]
- Abdalla, M.; El-Arabey, A.A.; Gai, Z. Hypertension is still a moving target in the context of COVID-19 and post-acute COVID-19 syndrome. J. Med. Virol. 2023, 95, e28128. [Google Scholar] [CrossRef] [PubMed]
- Muhamad, S.A.; Ugusman, A.; Kumar, J.; Skiba, D.; Hamid, A.A.; Aminuddin, A. COVID-19 and Hypertension: The What, the Why, and the How. Front. Physiol. 2021, 12, 665064. [Google Scholar] [CrossRef] [PubMed]
- Ruiz-Ortega, M.; Lorenzo, O.; Ruperez, M.; Esteban, V.; Suzuki, Y.; Mezzano, S.; Plaza, J.J.; Egido, J. Role of the renin-angiotensin system in vascular diseases: Expanding the field. Hypertension 2001, 38, 1382–1387. [Google Scholar] [CrossRef]
- World Health Organization. WHO Coronavirus (COVD-19) Dashboard. Available online: https://covid19.who.int/ (accessed on 28 February 2023).
- Chung, M.K.; Zidar, D.A.; Bristow, M.R.; Cameron, S.J.; Chan, T.; Harding, C.V.; Kwon, D.H., 3rd; Singh, T.; Tilton, J.C.; Tsai, E.J.; et al. COVID-19 and Cardiovascular Disease: From Bench to Bedside. Circ. Res. 2021, 128, 1214–1236. [Google Scholar] [CrossRef]
- Chavda, V.P.; Kapadia, C.; Soni, S.; Prajapati, R.; Chauhan, S.C.; Yallapu, M.M.; Apostolopoulos, V. A global picture: Therapeutic perspectives for COVID-19. Immunotherapy 2022, 14, 351–371. [Google Scholar] [CrossRef]
- Raisi-Estabragh, Z.; Cooper, J.; Salih, A.; Raman, B.; Lee, A.M.; Neubauer, S.; Harvey, N.C.; Petersen, S.E. Cardiovascular disease and mortality sequelae of COVID-19 in the UK Biobank. Heart 2022, 109, 119–126. [Google Scholar] [CrossRef]
- Chernyak, B.V.; Popova, E.N.; Prikhodko, A.S.; Grebenchikov, O.A.; Zinovkina, L.A.; Zinovkin, R.A. COVID-19 and Oxidative Stress. Biochemistry 2020, 85, 1543–1553. [Google Scholar] [CrossRef] [PubMed]
- Hojyo, S.; Uchida, M.; Tanaka, K.; Hasebe, R.; Tanaka, Y.; Murakami, M.; Hirano, T. How COVID-19 induces cytokine storm with high mortality. Inflamm. Regen. 2020, 40, 37. [Google Scholar] [CrossRef] [PubMed]
- Yuki, K.; Fujiogi, M.; Koutsogiannaki, S. COVID-19 pathophysiology: A review. Clin. Immunol. 2020, 215, 108427. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Petitjean, S.J.L.; Koehler, M.; Zhang, Q.; Dumitru, A.C.; Chen, W.; Derclaye, S.; Vincent, S.P.; Soumillion, P.; Alsteens, D. Molecular interaction and inhibition of SARS-CoV-2 binding to the ACE2 receptor. Nat. Commun. 2020, 11, 4541. [Google Scholar] [CrossRef] [PubMed]
- Dasgupta, C.; Zhang, L. Angiotensin II receptors and drug discovery in cardiovascular disease. Drug Discov. Today 2011, 16, 22–34. [Google Scholar] [CrossRef]
- Wu, C.H.; Mohammadmoradi, S.; Chen, J.Z.; Sawada, H.; Daugherty, A.; Lu, H.S. Renin-Angiotensin System and Cardiovascular Functions. Arterioscler. Thromb. Vasc. Biol. 2018, 38, e108–e116. [Google Scholar] [CrossRef]
- Forrester, S.J.; Booz, G.W.; Sigmund, C.D.; Coffman, T.M.; Kawai, T.; Rizzo, V.; Scalia, R.; Eguchi, S. Angiotensin II Signal Transduction: An Update on Mechanisms of Physiology and Pathophysiology. Physiol. Rev. 2018, 98, 1627–1738. [Google Scholar] [CrossRef]
- Costa, L.B.; Perez, L.G.; Palmeira, V.A.; Macedo, E.C.T.; Ribeiro, V.T.; Lanza, K.; Simoes, E.S.A.C. Insights on SARS-CoV-2 Molecular Interactions With the Renin-Angiotensin System. Front. Cell. Dev. Biol. 2020, 8, 559841. [Google Scholar] [CrossRef]
- Paz Ocaranza, M.; Riquelme, J.A.; Garcia, L.; Jalil, J.E.; Chiong, M.; Santos, R.A.S.; Lavandero, S. Counter-regulatory renin-angiotensin system in cardiovascular disease. Nat. Rev. Cardiol. 2020, 17, 116–129. [Google Scholar] [CrossRef]
- Triposkiadis, F.; Xanthopoulos, A.; Giamouzis, G.; Boudoulas, K.D.; Starling, R.C.; Skoularigis, J.; Boudoulas, H.; Iliodromitis, E. ACE2, the Counter-Regulatory Renin-Angiotensin System Axis and COVID-19 Severity. J. Clin. Med. 2021, 10, 3885. [Google Scholar] [CrossRef] [PubMed]
- Lautner, R.Q.; Villela, D.C.; Fraga-Silva, R.A.; Silva, N.; Verano-Braga, T.; Costa-Fraga, F.; Jankowski, J.; Jankowski, V.; Sousa, F.; Alzamora, A.; et al. Discovery and characterization of alamandine: A novel component of the renin-angiotensin system. Circ. Res. 2013, 112, 1104–1111. [Google Scholar] [CrossRef] [PubMed]
- Villela, D.C.; Passos-Silva, D.G.; Santos, R.A. Alamandine: A new member of the angiotensin family. Curr. Opin. Nephrol. Hypertens. 2014, 23, 130–134. [Google Scholar] [CrossRef] [PubMed]
- Qaradakhi, T.; Apostolopoulos, V.; Zulli, A. Angiotensin (1–7) and Alamandine: Similarities and differences. Pharmacol. Res. 2016, 111, 820–826. [Google Scholar] [CrossRef]
- De Souza-Neto, F.P.; Silva, M.M.E.; Santuchi, M.C.; de Alcantara-Leonidio, T.C.; Motta-Santos, D.; Oliveira, A.C.; Melo, M.B.; Canta, G.N.; de Souza, L.E.; Irigoyen, M.C.C.; et al. Alamandine attenuates arterial remodelling induced by transverse aortic constriction in mice. Clin. Sci. 2019, 133, 629–643. [Google Scholar] [CrossRef]
- Hekmat, A.S.; Navabi, Z.; Alipanah, H.; Javanmardi, K. Alamandine significantly reduces doxorubicin-induced cardiotoxicity in rats. Hum. Exp. Toxicol. 2021, 40, 1781–1795. [Google Scholar] [CrossRef]
- Liu, Q.; Zheng, B.; Zhang, Y.; Huang, W.; Hong, Q.; Meng, Y. Alamandine via MrgD receptor attenuates pulmonary fibrosis via NOX4 and autophagy pathway. Can. J. Physiol. Pharmacol. 2021, 99, 885–893. [Google Scholar] [CrossRef]
- Soltani Hekmat, A.; Chenari, A.; Alipanah, H.; Javanmardi, K. Protective effect of alamandine on doxorubicin-induced nephrotoxicity in rats. BMC Pharmacol. Toxicol. 2021, 22, 31. [Google Scholar] [CrossRef]
- Fernandes, R.S.; Dias, H.B.; de Souza Jaques, W.A.; Becker, T.; Rigatto, K. Assessment of Alamandine in Pulmonary Fibrosis and Respiratory Mechanics in Rodents. J. Renin. Angiotensin. Aldosterone Syst. 2021, 2021, 9975315. [Google Scholar] [CrossRef]
- Ruiz-Ortega, M.; Esteban, V.; Egido, J. The regulation of the inflammatory response through nuclear factor-kappab pathway by angiotensin IV extends the role of the renin angiotensin system in cardiovascular diseases. Trends Cardiovasc. Med. 2007, 17, 19–25. [Google Scholar] [CrossRef]
- Murphy, T.J.; Alexander, R.W.; Griendling, K.K.; Runge, M.S.; Bernstein, K.E. Isolation of a cDNA encoding the vascular type-1 angiotensin II receptor. Nature 1991, 351, 233–236. [Google Scholar] [CrossRef] [PubMed]
- Reaux, A.; Fournie-Zaluski, M.C.; Llorens-Cortes, C. Angiotensin III: A central regulator of vasopressin release and blood pressure. Trends Endocrinol. Metab. 2001, 12, 157–162. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.; Zeng, X.J.; Wang, H.X.; Zhang, L.K.; Dong, X.L.; Guo, S.; Du, J.; Li, H.H.; Tang, C.S. Angiotensin IV protects against angiotensin II-induced cardiac injury via AT4 receptor. Peptides 2011, 32, 2108–2115. [Google Scholar] [CrossRef]
- Wang, Q.G.; Xue, X.; Yang, Y.; Gong, P.Y.; Jiang, T.; Zhang, Y.D. Angiotensin IV suppresses inflammation in the brains of rats with chronic cerebral hypoperfusion. J. Renin. Angiotensin. Aldosterone Syst. 2018, 19, 1470320318799587. [Google Scholar] [CrossRef]
- Coleman, J.K.; Krebs, L.T.; Hamilton, T.A.; Ong, B.; Lawrence, K.A.; Sardinia, M.F.; Harding, J.W.; Wright, J.W. Autoradiographic identification of kidney angiotensin IV binding sites and angiotensin IV-induced renal cortical blood flow changes in rats. Peptides 1998, 19, 269–277. [Google Scholar] [CrossRef]
- Kramar, E.A.; Krishnan, R.; Harding, J.W.; Wright, J.W. Role of nitric oxide in angiotensin IV-induced increases in cerebral blood flow. Regul. Pept. 1998, 74, 185–192. [Google Scholar] [CrossRef] [PubMed]
- Chansel, D.; Vandermeersch, S.; Oko, A.; Curat, C.; Ardaillou, R. Effects of angiotensin IV and angiotensin-(1-7) on basal and angiotensin II-stimulated cytosolic Ca2+ in mesangial cells. Eur. J. Pharmacol. 2001, 414, 165–175. [Google Scholar] [CrossRef] [PubMed]
- Szczepanska-Sadowska, E.; Czarzasta, K.; Cudnoch-Jedrzejewska, A. Dysregulation of the Renin-Angiotensin System and the Vasopressinergic System Interactions in Cardiovascular Disorders. Curr. Hypertens. Rep. 2018, 20, 19. [Google Scholar] [CrossRef]
- Kawai, T.; Forrester, S.J.; O’Brien, S.; Baggett, A.; Rizzo, V.; Eguchi, S. AT1 receptor signaling pathways in the cardiovascular system. Pharmacol. Res. 2017, 125, 4–13. [Google Scholar] [CrossRef]
- Fellner, S.K.; Arendshorst, W.J. Angiotensin II Ca2+ signaling in rat afferent arterioles: Stimulation of cyclic ADP ribose and IP3 pathways. Am. J. Physiol. Renal. Physiol. 2005, 288, F785–F791. [Google Scholar] [CrossRef]
- Alexander, R.W.; Brock, T.A.; Gimbrone, M.A., Jr.; Rittenhouse, S.E. Angiotensin increases inositol trisphosphate and calcium in vascular smooth muscle. Hypertension 1985, 7, 447–451. [Google Scholar] [CrossRef] [PubMed]
- Simoes, S.C.; Balico-Silva, A.L.; Parreiras, E.S.L.T.; Bitencourt, A.L.B.; Bouvier, M.; Costa-Neto, C.M. Signal Transduction Profiling of Angiotensin II Type 1 Receptor With Mutations Associated to Atrial Fibrillation in Humans. Front. Pharmacol. 2020, 11, 600132. [Google Scholar] [CrossRef] [PubMed]
- Spat, A.; Hunyady, L. Control of aldosterone secretion: A model for convergence in cellular signaling pathways. Physiol. Rev. 2004, 84, 489–539. [Google Scholar] [CrossRef] [PubMed]
- Verma, K.; Pant, M.; Paliwal, S.; Dwivedi, J.; Sharma, S. An Insight on Multicentric Signaling of Angiotensin II in Cardiovascular system: A Recent Update. Front. Pharmacol. 2021, 12, 734917. [Google Scholar] [CrossRef]
- Wei, H.; Ahn, S.; Shenoy, S.K.; Karnik, S.S.; Hunyady, L.; Luttrell, L.M.; Lefkowitz, R.J. Independent beta-arrestin 2 and G protein-mediated pathways for angiotensin II activation of extracellular signal-regulated kinases 1 and 2. Proc. Natl. Acad. Sci. USA 2003, 100, 10782–10787. [Google Scholar] [CrossRef]
- Kim, S.-M.; Huh, J.-W.; Kim, E.-Y.; Shin, M.-K.; Park, J.-E.; Kim, S.W.; Lee, W.; Choi, B.; Chang, E.-J. Endothelial dysfunction induces atherosclerosis: Increased aggrecan expression promotes apoptosis in vascular smooth muscle cells. BMB Rep. 2019, 52, 145. [Google Scholar] [CrossRef]
- Widmer, R.J.; Lerman, A. Endothelial dysfunction and cardiovascular disease. Glob. Cardiol. Sci. Pract. 2014, 2014, 291–308. [Google Scholar] [CrossRef]
- Ruhl, L.; Pink, I.; Kuhne, J.F.; Beushausen, K.; Keil, J.; Christoph, S.; Sauer, A.; Boblitz, L.; Schmidt, J.; David, S.; et al. Endothelial dysfunction contributes to severe COVID-19 in combination with dysregulated lymphocyte responses and cytokine networks. Signal. Transduct. Target Ther. 2021, 6, 418. [Google Scholar] [CrossRef]
- Vukelic, S.; Griendling, K.K. Angiotensin II, from vasoconstrictor to growth factor: A paradigm shift. Circ. Res. 2014, 114, 754–757. [Google Scholar] [CrossRef]
- Welsh, D.G.; Tran, C.H.T.; Hald, B.O.; Sancho, M. The Conducted Vasomotor Response: Function, Biophysical Basis, and Pharmacological Control. Annu. Rev. Pharmacol. Toxicol. 2018, 58, 391–410. [Google Scholar] [CrossRef]
- Touyz, R.M.; Alves-Lopes, R.; Rios, F.J.; Camargo, L.L.; Anagnostopoulou, A.; Arner, A.; Montezano, A.C. Vascular smooth muscle contraction in hypertension. Cardiovasc. Res. 2018, 114, 529–539. [Google Scholar] [CrossRef] [PubMed]
- Shatanawi, A.; Romero, M.J.; Iddings, J.A.; Chandra, S.; Umapathy, N.S.; Verin, A.D.; Caldwell, R.B.; Caldwell, R.W. Angiotensin II-induced vascular endothelial dysfunction through RhoA/Rho kinase/p38 mitogen-activated protein kinase/arginase pathway. Am. J. Physiol. Cell Physiol. 2011, 300, C1181–C1192. [Google Scholar] [CrossRef] [PubMed]
- Satoh, M.; Fujimoto, S.; Arakawa, S.; Yada, T.; Namikoshi, T.; Haruna, Y.; Horike, H.; Sasaki, T.; Kashihara, N. Angiotensin II type 1 receptor blocker ameliorates uncoupled endothelial nitric oxide synthase in rats with experimental diabetic nephropathy. Nephrol. Dial. Transplant. 2008, 23, 3806–3813. [Google Scholar] [CrossRef]
- Prathapan, A.; Vineetha, V.P.; Abhilash, P.A.; Raghu, K.G. Boerhaavia diffusa L. attenuates angiotensin II-induced hypertrophy in H9c2 cardiac myoblast cells via modulating oxidative stress and down-regulating NF-kappabeta and transforming growth factor beta1. Br. J. Nutr. 2013, 110, 1201–1210. [Google Scholar] [CrossRef] [PubMed]
- Chen, K.; Chen, J.; Li, D.; Zhang, X.; Mehta, J.L. Angiotensin II regulation of collagen type I expression in cardiac fibroblasts: Modulation by PPAR-gamma ligand pioglitazone. Hypertension 2004, 44, 655–661. [Google Scholar] [CrossRef]
- Lee, C.Y.; Park, H.K.; Lee, B.S.; Jeong, S.; Hyun, S.A.; Choi, J.W.; Kim, S.W.; Lee, S.; Lim, S.; Hwang, K.C. Novel Therapeutic Effects of Pterosin B on Ang II-Induced Cardiomyocyte Hypertrophy. Molecules 2020, 25, 5279. [Google Scholar] [CrossRef]
- Dubois-Deruy, E.; Peugnet, V.; Turkieh, A.; Pinet, F. Oxidative Stress in Cardiovascular Diseases. Antioxidants 2020, 9, 864C. [Google Scholar] [CrossRef]
- Wen, H.; Gwathmey, J.K.; Xie, L.H. Oxidative stress-mediated effects of angiotensin II in the cardiovascular system. World J. Hypertens. 2012, 2, 34–44. [Google Scholar] [CrossRef]
- Rajagopalan, S.; Kurz, S.; Munzel, T.; Tarpey, M.; Freeman, B.A.; Griendling, K.K.; Harrison, D.G. Angiotensin II-mediated hypertension in the rat increases vascular superoxide production via membrane NADH/NADPH oxidase activation. Contribution to alterations of vasomotor tone. J. Clin. Investig. 1996, 97, 1916–1923. [Google Scholar] [CrossRef]
- Sena, C.M.; Leandro, A.; Azul, L.; Seica, R.; Perry, G. Vascular Oxidative Stress: Impact and Therapeutic Approaches. Front. Physiol. 2018, 9, 1668. [Google Scholar] [CrossRef]
- Sugden, P.H.; Clerk, A. Oxidative stress and growth-regulating intracellular signaling pathways in cardiac myocytes. Antioxid. Redox Signal. 2006, 8, 2111–2124. [Google Scholar] [CrossRef] [PubMed]
- Hirotani, S.; Otsu, K.; Nishida, K.; Higuchi, Y.; Morita, T.; Nakayama, H.; Yamaguchi, O.; Mano, T.; Matsumura, Y.; Ueno, H.; et al. Involvement of nuclear factor-kappaB and apoptosis signal-regulating kinase 1 in G-protein-coupled receptor agonist-induced cardiomyocyte hypertrophy. Circulation 2002, 105, 509–515. [Google Scholar] [CrossRef] [PubMed]
- St. Paul, A.; Corbett, C.B.; Okune, R.; Autieri, M.V. Angiotensin II, Hypercholesterolemia, and Vascular Smooth Muscle Cells: A Perfect Trio for Vascular Pathology. Int. J. Mol. Sci. 2020, 21, 4525. [Google Scholar] [CrossRef] [PubMed]
- Yang, W.; Liu, Z.; Xu, Q.; Peng, H.; Chen, L.; Huang, X.; Yang, T.; Yu, Z.; Cheng, G.; Zhang, G.; et al. Involvement of vascular peroxidase 1 in angiotensin II-induced hypertrophy of H9c2 cells. J. Am. Soc. Hypertens. 2017, 11, 519–529.e511. [Google Scholar] [CrossRef] [PubMed]
- Canty, T.G., Jr.; Boyle, E.M., Jr.; Farr, A.; Morgan, E.N.; Verrier, E.D.; Pohlman, T.H. Oxidative stress induces NF-kappaB nuclear translocation without degradation of IkappaBalpha. Circulation 1999, 100, II361–II364. [Google Scholar] [CrossRef]
- Alfaddagh, A.; Martin, S.S.; Leucker, T.M.; Michos, E.D.; Blaha, M.J.; Lowenstein, C.J.; Jones, S.R.; Toth, P.P. Inflammation and cardiovascular disease: From mechanisms to therapeutics. Am. J. Prev. Cardiol. 2020, 4, 100130. [Google Scholar] [CrossRef]
- Amin, M.N.; Siddiqui, S.A.; Ibrahim, M.; Hakim, M.L.; Ahammed, M.S.; Kabir, A.; Sultana, F. Inflammatory cytokines in the pathogenesis of cardiovascular disease and cancer. SAGE Open Med. 2020, 8, 2050312120965752. [Google Scholar] [CrossRef]
- Tian, R.; Hou, G.; Li, D.; Yuan, T.F. A possible change process of inflammatory cytokines in the prolonged chronic stress and its ultimate implications for health. Sci. World J. 2014, 2014, 780616. [Google Scholar] [CrossRef]
- Ekholm, M.; Kahan, T. The Impact of the Renin-Angiotensin-Aldosterone System on Inflammation, Coagulation, and Atherothrombotic Complications, and to Aggravated COVID-19. Front. Pharmacol. 2021, 12, 640185. [Google Scholar] [CrossRef]
- Yang, D.; Elner, S.G.; Bian, Z.M.; Till, G.O.; Petty, H.R.; Elner, V.M. Pro-inflammatory cytokines increase reactive oxygen species through mitochondria and NADPH oxidase in cultured RPE cells. Exp. Eye Res. 2007, 85, 462–472. [Google Scholar] [CrossRef]
- Senoner, T.; Dichtl, W. Oxidative Stress in Cardiovascular Diseases: Still a Therapeutic Target? Nutrients 2019, 11, 2090. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Du, J.; Hu, Z.; Han, G.; Delafontaine, P.; Garcia, G.; Mitch, W.E. IL-6 and serum amyloid A synergy mediates angiotensin II-induced muscle wasting. J. Am. Soc. Nephrol. 2009, 20, 604–612. [Google Scholar] [CrossRef]
- Guo, F.; Chen, X.L.; Wang, F.; Liang, X.; Sun, Y.X.; Wang, Y.J. Role of angiotensin II type 1 receptor in angiotensin II-induced cytokine production in macrophages. J. Interferon. Cytokine Res. 2011, 31, 351–361. [Google Scholar] [CrossRef] [PubMed]
- Dinarello, C.A. Overview of the IL-1 family in innate inflammation and acquired immunity. Immunol. Rev. 2018, 281, 8–27. [Google Scholar] [CrossRef] [PubMed]
- Gullestad, L.; Ueland, T.; Vinge, L.E.; Finsen, A.; Yndestad, A.; Aukrust, P. Inflammatory cytokines in heart failure: Mediators and markers. Cardiology 2012, 122, 23–35. [Google Scholar] [CrossRef] [PubMed]
- Meng, X.M.; Nikolic-Paterson, D.J.; Lan, H.Y. TGF-beta: The master regulator of fibrosis. Nat. Rev. Nephrol. 2016, 12, 325–338. [Google Scholar] [CrossRef]
- Porter, K.E.; Turner, N.A. Cardiac fibroblasts: At the heart of myocardial remodeling. Pharmacol. Ther. 2009, 123, 255–278. [Google Scholar] [CrossRef]
- Santos, A.G.; da Rocha, G.O.; de Andrade, J.B. Occurrence of the potent mutagens 2-nitrobenzanthrone and 3-nitrobenzanthrone in fine airborne particles. Sci. Rep. 2019, 9, 1. [Google Scholar] [CrossRef]
- Gomolak, J.R.; Didion, S.P. Angiotensin II-induced endothelial dysfunction is temporally linked with increases in interleukin-6 and vascular macrophage accumulation. Front. Physiol. 2014, 5, 396. [Google Scholar] [CrossRef]
- Li, Y.; Kinzenbaw, D.A.; Modrick, M.L.; Pewe, L.L.; Faraci, F.M. Context-dependent effects of SOCS3 in angiotensin II-induced vascular dysfunction and hypertension in mice: Mechanisms and role of bone marrow-derived cells. Am. J. Physiol. Heart Circ. Physiol. 2016, 311, H146–H156. [Google Scholar] [CrossRef]
- Satou, R.; Miyata, K.; Gonzalez-Villalobos, R.A.; Ingelfinger, J.R.; Navar, L.G.; Kobori, H. Interferon-gamma biphasically regulates angiotensinogen expression via a JAK-STAT pathway and suppressor of cytokine signaling 1 (SOCS1) in renal proximal tubular cells. FASEB J. 2012, 26, 1821–1830. [Google Scholar] [CrossRef]
- Hessami, A.; Shamshirian, A.; Heydari, K.; Pourali, F.; Alizadeh-Navaei, R.; Moosazadeh, M.; Abrotan, S.; Shojaie, L.; Sedighi, S.; Shamshirian, D.; et al. Cardiovascular diseases burden in COVID-19: Systematic review and meta-analysis. Am. J. Emerg. Med. 2021, 46, 382–391. [Google Scholar] [CrossRef] [PubMed]
- Ahmad Malik, J.; Ahmed, S.; Shinde, M.; Almermesh, M.H.S.; Alghamdi, S.; Hussain, A.; Anwar, S. The Impact of COVID-19 on Comorbidities: A Review of Recent Updates for Combating It. Saudi J. Biol. Sci. 2022, 29, 3586–3599. [Google Scholar] [CrossRef] [PubMed]
- Wei, N.; Xu, Y.; Wang, H.; Jia, Q.; Shou, X.; Zhang, X.; Zhang, N.; Li, Y.; Zhai, H.; Hu, Y. Bibliometric and visual analysis of cardiovascular diseases and COVID-19 research. Front. Public Health 2022, 10, 1022810. [Google Scholar] [CrossRef]
- Roshdy, A.; Zaher, S.; Fayed, H.; Coghlan, J.G. COVID-19 and the Heart: A Systematic Review of Cardiac Autopsies. Front. Cardiovasc. Med. 2020, 7, 626975. [Google Scholar] [CrossRef]
- Salazar, M.R. Is hypertension without any other comorbidities an independent predictor for COVID-19 severity and mortality? J. Clin. Hypertens. 2021, 23, 232–234. [Google Scholar] [CrossRef]
- Healy, E.F.; Lilic, M. A model for COVID-19-induced dysregulation of ACE2 shedding by ADAM17. Biochem. Biophys. Res. Commun. 2021, 573, 158–163. [Google Scholar] [CrossRef]
- Kuba, K.; Imai, Y.; Ohto-Nakanishi, T.; Penninger, J.M. Trilogy of ACE2: A peptidase in the renin-angiotensin system, a SARS receptor, and a partner for amino acid transporters. Pharmacol. Ther. 2010, 128, 119–128. [Google Scholar] [CrossRef]
- Reddy, R.; Asante, I.; Liu, S.; Parikh, P.; Liebler, J.; Borok, Z.; Rodgers, K.; Baydur, A.; Louie, S.G. Circulating angiotensin peptides levels in Acute Respiratory Distress Syndrome correlate with clinical outcomes: A pilot study. PLoS ONE 2019, 14, e0213096. [Google Scholar] [CrossRef]
- Oliveira, A.C.; Melo, M.B.; Motta-Santos, D.; Peluso, A.A.; Souza-Neto, F.; da Silva, R.F.; Almeida, J.F.Q.; Canta, G.; Reis, A.M.; Goncalves, G.; et al. Genetic deletion of the alamandine receptor MRGD leads to dilated cardiomyopathy in mice. Am. J. Physiol. Heart Circ. Physiol. 2019, 316, H123–H133. [Google Scholar] [CrossRef]
- Qi, Y.F.; Zhang, J.; Wang, L.; Shenoy, V.; Krause, E.; Oh, S.P.; Pepine, C.J.; Katovich, M.J.; Raizada, M.K. Angiotensin-converting enzyme 2 inhibits high-mobility group box 1 and attenuates cardiac dysfunction post-myocardial ischemia. J. Mol. Med. 2016, 94, 37–49. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Qian, C.; Roks, A.J.; Westermann, D.; Schumacher, S.M.; Escher, F.; Schoemaker, R.G.; Reudelhuber, T.L.; van Gilst, W.H.; Schultheiss, H.P.; et al. Circulating rather than cardiac angiotensin-(1-7) stimulates cardioprotection after myocardial infarction. Circ. Heart Fail. 2010, 3, 286–293. [Google Scholar] [CrossRef]
- Zhao, K.; Xu, T.; Mao, Y.; Wu, X.; Hua, D.; Sheng, Y.; Li, P. Alamandine alleviated heart failure and fibrosis in myocardial infarction mice. Biol. Direct 2022, 17, 25. [Google Scholar] [CrossRef] [PubMed]
- Zangrillo, A.; Landoni, G.; Beretta, L.; Morselli, F.; Serpa Neto, A.; Bellomo, R.; the COVID-BioB Study Group. Angiotensin II infusion in COVID-19-associated vasodilatory shock: A case series. Crit. Care 2020, 24, 227. [Google Scholar] [CrossRef] [PubMed]
- Alhenc-Gelas, F.; Drueke, T.B. Blockade of SARS-CoV-2 infection by recombinant soluble ACE2. Kidney Int. 2020, 97, 1091–1093. [Google Scholar] [CrossRef]
- Pang, X.; Cui, Y.; Zhu, Y. Recombinant human ACE2: Potential therapeutics of SARS-CoV-2 infection and its complication. Acta Pharmacol. Sin. 2020, 41, 1255–1257. [Google Scholar] [CrossRef]
- Soy, M.; Keser, G.; Atagunduz, P.; Tabak, F.; Atagunduz, I.; Kayhan, S. Cytokine storm in COVID-19: Pathogenesis and overview of anti-inflammatory agents used in treatment. Clin. Rheumatol. 2020, 39, 2085–2094. [Google Scholar] [CrossRef]
- Ragab, D.; Salah Eldin, H.; Taeimah, M.; Khattab, R.; Salem, R. The COVID-19 Cytokine Storm; What We Know So Far. Front. Immunol. 2020, 11, 1446. [Google Scholar] [CrossRef]
- Gudowska-Sawczuk, M.; Mroczko, B. The Role of Nuclear Factor Kappa B (NF-kappaB) in Development and Treatment of COVID-19: Review. Int. J. Mol. Sci. 2022, 23, 5283. [Google Scholar] [CrossRef]
- Gadanec, L.K.; McSweeney, K.R.; Qaradakhi, T.; Ali, B.; Zulli, A.; Apostolopoulos, V. Can SARS-CoV-2 virus use multiple receptors to enter host cells? Int. J. Mol. Sci. 2021, 22, 992. [Google Scholar] [CrossRef]
- Kosyreva, A.; Dzhalilova, D.; Lokhonina, A.; Vishnyakova, P.; Fatkhudinov, T. The Role of Macrophages in the Pathogenesis of SARS-CoV-2-Associated Acute Respiratory Distress Syndrome. Front. Immunol. 2021, 12, 682871. [Google Scholar] [CrossRef] [PubMed]
- Silva-Filho, J.L.; Caruso-Neves, C.; Pinheiro, A.A. Angiotensin II type-1 receptor (AT(1)R) regulates expansion, differentiation, and functional capacity of antigen-specific CD8(+) T cells. Sci. Rep. 2016, 6, 35997. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, S.; Yancey, P.G.; Zuo, Y.; Ma, L.J.; Kaseda, R.; Fogo, A.B.; Ichikawa, I.; Linton, M.F.; Fazio, S.; Kon, V. Macrophage polarization by angiotensin II-type 1 receptor aggravates renal injury-acceleration of atherosclerosis. Arterioscler. Thromb. Vasc. Biol. 2011, 31, 2856–2864. [Google Scholar] [CrossRef] [PubMed]
- Kate Gadanec, L.; Qaradakhi, T.; Renee McSweeney, K.; Ashiana Ali, B.; Zulli, A.; Apostolopoulos, V. Dual targeting of Toll-like receptor 4 and angiotensin-converting enzyme 2: A proposed approach to SARS-CoV-2 treatment. Future Microbiol. 2021, 16, 205–209. [Google Scholar] [CrossRef]
- Chen, X.S.; Wang, S.H.; Liu, C.Y.; Gao, Y.L.; Meng, X.L.; Wei, W.; Shou, S.T.; Liu, Y.C.; Chai, Y.F. Losartan attenuates sepsis-induced cardiomyopathy by regulating macrophage polarization via TLR4-mediated NF-kappaB and MAPK signaling. Pharmacol. Res. 2022, 185, 106473. [Google Scholar] [CrossRef]
- Mehrabadi, M.E.; Hemmati, R.; Tashakor, A.; Homaei, A.; Yousefzadeh, M.; Hemati, K.; Hosseinkhani, S. Induced dysregulation of ACE2 by SARS-CoV-2 plays a key role in COVID-19 severity. Biomed. Pharmacother. 2021, 137, 111363. [Google Scholar] [CrossRef]
- Liu, F.; Li, L.; Xu, M.; Wu, J.; Luo, D.; Zhu, Y.; Li, B.; Song, X.; Zhou, X. Prognostic value of interleukin-6, C-reactive protein, and procalcitonin in patients with COVID-19. J. Clin. Virol. 2020, 127, 104370. [Google Scholar] [CrossRef]
- Sallusto, F.; Lanzavecchia, A. Heterogeneity of CD4+ memory T cells: Functional modules for tailored immunity. Eur. J. Immunol. 2009, 39, 2076–2082. [Google Scholar] [CrossRef]
- Moss, P. The T cell immune response against SARS-CoV-2. Nat. Immunol. 2022, 23, 186–193. [Google Scholar] [CrossRef]
- Lane, N.; Robins, R.A.; Corne, J.; Fairclough, L. Regulation in chronic obstructive pulmonary disease: The role of regulatory T-cells and Th17 cells. Clin. Sci. 2010, 119, 75–86. [Google Scholar] [CrossRef]
- Almutlaq, M.; Mansour, F.A.; Alghamdi, J.; Alhendi, Y.; Alamro, A.A.; Alghamdi, A.A.; Alamri, H.S.; Alroqi, F.; Barhoumi, T. Angiotensin II Exaggerates SARS-CoV-2 Specific T-Cell Response in Convalescent Individuals following COVID-19. Int. J. Mol. Sci. 2022, 23, 8669. [Google Scholar] [CrossRef] [PubMed]
- Parackova, Z.; Bloomfield, M.; Klocperk, A.; Sediva, A. Neutrophils mediate Th17 promotion in COVID-19 patients. J. Leukoc. Biol. 2021, 109, 73–76. [Google Scholar] [CrossRef] [PubMed]
- Itani, H.A.; McMaster, W.G., Jr.; Saleh, M.A.; Nazarewicz, R.R.; Mikolajczyk, T.P.; Kaszuba, A.M.; Konior, A.; Prejbisz, A.; Januszewicz, A.; Norlander, A.E.; et al. Activation of Human T Cells in Hypertension: Studies of Humanized Mice and Hypertensive Humans. Hypertension 2016, 68, 123–132. [Google Scholar] [CrossRef]
- Schwartz, D.M.; Burma, A.M.; Kitakule, M.M.; Luo, Y.; Mehta, N.N. T Cells in Autoimmunity-Associated Cardiovascular Diseases. Front. Immunol. 2020, 11, 588776. [Google Scholar] [CrossRef] [PubMed]
- Ju, H.; Behm, D.J.; Nerurkar, S.; Eybye, M.E.; Haimbach, R.E.; Olzinski, A.R.; Douglas, S.A.; Willette, R.N. p38 MAPK inhibitors ameliorate target organ damage in hypertension: Part 1. p38 MAPK-dependent endothelial dysfunction and hypertension. J. Pharmacol. Exp. Ther. 2003, 307, 932–938. [Google Scholar] [CrossRef]
- Wen, A.Y.; Sakamoto, K.M.; Miller, L.S. The role of the transcription factor CREB in immune function. J. Immunol. 2010, 185, 6413–6419. [Google Scholar] [CrossRef]
- Kuba, K.; Imai, Y.; Penninger, J.M. Angiotensin-converting enzyme 2 in lung diseases. Curr. Opin. Pharmacol. 2006, 6, 271–276. [Google Scholar] [CrossRef]
- Tan, W.S.D.; Liao, W.; Zhou, S.; Mei, D.; Wong, W.F. Targeting the renin-angiotensin system as novel therapeutic strategy for pulmonary diseases. Curr. Opin. Pharmacol. 2018, 40, 9–17. [Google Scholar] [CrossRef]
- Rossi, G.A.; Sacco, O.; Mancino, E.; Cristiani, L.; Midulla, F. Differences and similarities between SARS-CoV and SARS-CoV-2: Spike receptor-binding domain recognition and host cell infection with support of cellular serine proteases. Infection 2020, 48, 665–669. [Google Scholar] [CrossRef]
- Kuba, K.; Imai, Y.; Rao, S.; Gao, H.; Guo, F.; Guan, B.; Huan, Y.; Yang, P.; Zhang, Y.; Deng, W.; et al. A crucial role of angiotensin converting enzyme 2 (ACE2) in SARS coronavirus-induced lung injury. Nat. Med. 2005, 11, 875–879. [Google Scholar] [CrossRef]
- Imai, Y.; Kuba, K.; Rao, S.; Huan, Y.; Guo, F.; Guan, B.; Yang, P.; Sarao, R.; Wada, T.; Leong-Poi, H.; et al. Angiotensin-converting enzyme 2 protects from severe acute lung failure. Nature 2005, 436, 112–116. [Google Scholar] [CrossRef] [PubMed]
- Patra, T.; Meyer, K.; Geerling, L.; Isbell, T.S.; Hoft, D.F.; Brien, J.; Pinto, A.K.; Ray, R.B.; Ray, R. SARS-CoV-2 spike protein promotes IL-6 trans-signaling by activation of angiotensin II receptor signaling in epithelial cells. PLoS Pathog. 2020, 16, e1009128. [Google Scholar] [CrossRef]
- Santa Cruz, A.; Mendes-Frias, A.; Oliveira, A.I.; Dias, L.; Matos, A.R.; Carvalho, A.; Capela, C.; Pedrosa, J.; Castro, A.G.; Silvestre, R. Interleukin-6 Is a Biomarker for the Development of Fatal Severe Acute Respiratory Syndrome Coronavirus 2 Pneumonia. Front. Immunol. 2021, 12, 613422. [Google Scholar] [CrossRef]
- Wu, Z.; Hu, R.; Zhang, C.; Ren, W.; Yu, A.; Zhou, X. Elevation of plasma angiotensin II level is a potential pathogenesis for the critically ill COVID-19 patients. Crit. Care 2020, 24, 290. [Google Scholar] [CrossRef] [PubMed]
- Osman, I.O.; Melenotte, C.; Brouqui, P.; Million, M.; Lagier, J.C.; Parola, P.; Stein, A.; La Scola, B.; Meddeb, L.; Mege, J.L.; et al. Expression of ACE2, Soluble ACE2, Angiotensin I, Angiotensin II and Angiotensin-(1-7) Is Modulated in COVID-19 Patients. Front. Immunol. 2021, 12, 625732. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Yang, Y.; Zhang, C.; Huang, F.; Wang, F.; Yuan, J.; Wang, Z.; Li, J.; Li, J.; Feng, C.; et al. Clinical and biochemical indexes from 2019-nCoV infected patients linked to viral loads and lung injury. Sci. China Life Sci. 2020, 63, 364–374. [Google Scholar] [CrossRef] [PubMed]
- Camargo, R.L.; Bombassaro, B.; Monfort-Pires, M.; Mansour, E.; Palma, A.C.; Ribeiro, L.C.; Ulaf, R.G.; Bernardes, A.F.; Nunes, T.A.; Agrela, M.V.; et al. Plasma Angiotensin II Is Increased in Critical Coronavirus Disease 2019. Front. Cardiovasc. Med. 2022, 9, 847809. [Google Scholar] [CrossRef] [PubMed]
- Caputo, I.; Caroccia, B.; Frasson, I.; Poggio, E.; Zamberlan, S.; Morpurgo, M.; Seccia, T.M.; Cali, T.; Brini, M.; Richter, S.N.; et al. Angiotensin II Promotes SARS-CoV-2 Infection via Upregulation of ACE2 in Human Bronchial Cells. Int. J. Mol. Sci. 2022, 23, 5125. [Google Scholar] [CrossRef]
- Huertas, A.; Montani, D.; Savale, L.; Pichon, J.; Tu, L.; Parent, F.; Guignabert, C.; Humbert, M. Endothelial cell dysfunction: A major player in SARS-CoV-2 infection (COVID-19)? Eur. Respir. J. 2020, 56, 2001634. [Google Scholar] [CrossRef]
- Ackermann, M.; Verleden, S.E.; Kuehnel, M.; Haverich, A.; Welte, T.; Laenger, F.; Vanstapel, A.; Werlein, C.; Stark, H.; Tzankov, A.; et al. Pulmonary Vascular Endothelialitis, Thrombosis, and Angiogenesis in COVID-19. N. Engl. J. Med. 2020, 383, 120–128. [Google Scholar] [CrossRef]
- Taylor, A.A.; Siragy, H.; Nesbitt, S. Angiotensin receptor blockers: Pharmacology, efficacy, and safety. J. Clin. Hypertens. 2011, 13, 677–686. [Google Scholar] [CrossRef] [PubMed]
- Abraham, H.M.; White, C.M.; White, W.B. The comparative efficacy and safety of the angiotensin receptor blockers in the management of hypertension and other cardiovascular diseases. Drug Saf. 2015, 38, 33–54. [Google Scholar] [CrossRef] [PubMed]
- Siragy, H.M. A current evaluation of the safety of angiotensin receptor blockers and direct renin inhibitors. Vasc. Health Risk Manag. 2011, 7, 297–313. [Google Scholar] [CrossRef] [PubMed]
- Oparil, S.; Williams, D.; Chrysant, S.G.; Marbury, T.C.; Neutel, J. Comparative efficacy of olmesartan, losartan, valsartan, and irbesartan in the control of essential hypertension. J. Clin. Hypertens. 2001, 3, 283–291, 318. [Google Scholar] [CrossRef]
- Kassler-Taub, K.; Littlejohn, T.; Elliott, W.; Ruddy, T.; Adler, E. Comparative efficacy of two angiotensin II receptor antagonists, irbesartan and losartan in mild-to-moderate hypertension. Irbesartan/Losartan Study Investigators. Am. J. Hypertens. 1998, 11, 445–453. [Google Scholar] [CrossRef]
- Giles, T.D.; Oparil, S.; Silfani, T.N.; Wang, A.; Walker, J.F. Comparison of increasing doses of olmesartan medoxomil, losartan potassium, and valsartan in patients with essential hypertension. J. Clin. Hypertens. 2007, 9, 187–195. [Google Scholar] [CrossRef]
- The ONTARGET Investigators; Yusuf, S.; Teo, K.K.; Pogue, J.; Dyal, L.; Copland, I.; Schumacher, H.; Dagenais, G.; Sleight, P.; Anderson, C. Telmisartan, ramipril, or both in patients at high risk for vascular events. N. Engl. J. Med. 2008, 358, 1547–1559. [Google Scholar] [CrossRef]
- Oparil, S.; Melino, M.; Lee, J.; Fernandez, V.; Heyrman, R. Triple therapy with olmesartan medoxomil, amlodipine besylate, and hydrochlorothiazide in adult patients with hypertension: The TRINITY multicenter, randomized, double-blind, 12-week, parallel-group study. Clin. Ther. 2010, 32, 1252–1269. [Google Scholar] [CrossRef] [PubMed]
- Fried, L.F.; Emanuele, N.; Zhang, J.H.; Brophy, M.; Conner, T.A.; Duckworth, W.; Leehey, D.J.; McCullough, P.A.; O’Connor, T.; Palevsky, P.M.; et al. Combined angiotensin inhibition for the treatment of diabetic nephropathy. N. Engl. J. Med. 2013, 369, 1892–1903. [Google Scholar] [CrossRef]
- Matsoukas, J.; Apostolopoulos, V.; Zulli, A.; Moore, G.; Kelaidonis, K.; Moschovou, K.; Mavromoustakos, T. From angiotensin II to cyclic peptides and angiotensin receptor blockers (ARBs): Perspectives of ARBs in COVID-19 therapy. Molecules 2021, 26, 618. [Google Scholar] [CrossRef]
- Ridgway, H.; Ntallis, C.; Chasapis, C.T.; Kelaidonis, K.; Matsoukas, M.-T.; Plotas, P.; Apostolopoulos, V.; Moore, G.; Tsiodras, S.; Paraskevis, D. Molecular Epidemiology of SARS-CoV-2: The Dominant Role of Arginine in Mutations and Infectivity. Viruses 2023, 15, 309. [Google Scholar] [CrossRef]
- Benicky, J.; Sanchez-Lemus, E.; Pavel, J.; Saavedra, J.M. Anti-inflammatory effects of angiotensin receptor blockers in the brain and the periphery. Cell. Mol. Neurobiol. 2009, 29, 781–792. [Google Scholar] [CrossRef]
- Takagi, H.; Mizuno, Y.; Yamamoto, H.; Goto, S.N.; Umemoto, T. All-Literature Investigation of Cardiovascular Evidence G. Effects of telmisartan therapy on interleukin-6 and tumor necrosis factor-alpha levels: A meta-analysis of randomized controlled trials. Hypertens. Res. 2013, 36, 368–373. [Google Scholar] [CrossRef]
- Ararat, E.; Brozovich, F.V. Losartan decreases p42/44 MAPK signaling and preserves LZ+ MYPT1 expression. PLoS ONE 2009, 4, e5144. [Google Scholar] [CrossRef]
- Nejat, R.; Sadr, A.S. Are losartan and imatinib effective against SARS-CoV2 pathogenesis? A pathophysiologic-based in silico study. In Silico Pharmacol. 2021, 9, 1. [Google Scholar] [CrossRef] [PubMed]
- Gurwitz, D. Angiotensin receptor blockers as tentative SARS-CoV-2 therapeutics. Drug Dev. Res. 2020, 81, 537–540. [Google Scholar] [CrossRef]
- Ma, J.; Shi, X.; Yu, J.; Lv, F.; Wu, J.; Sheng, X.; Pan, Q.; Yang, J.; Cao, H.; Li, L. Association of ACEi/ARB Use and Clinical Outcomes of COVID-19 Patients With Hypertension. Front. Cardiovasc. Med. 2021, 8, 577398. [Google Scholar] [CrossRef] [PubMed]
- Zhang, P.; Zhu, L.; Cai, J.; Lei, F.; Qin, J.J.; Xie, J.; Liu, Y.M.; Zhao, Y.C.; Huang, X.; Lin, L.; et al. Association of Inpatient Use of Angiotensin-Converting Enzyme Inhibitors and Angiotensin II Receptor Blockers With Mortality Among Patients With Hypertension Hospitalized With COVID-19. Circ. Res. 2020, 126, 1671–1681. [Google Scholar] [CrossRef] [PubMed]
- Genet, B.; Vidal, J.S.; Cohen, A.; Boully, C.; Beunardeau, M.; Marine Harle, L.; Goncalves, A.; Boudali, Y.; Hernandorena, I.; Bailly, H.; et al. COVID-19 In-Hospital Mortality and Use of Renin-Angiotensin System Blockers in Geriatrics Patients. J. Am. Med. Dir. Assoc. 2020, 21, 1539–1545. [Google Scholar] [CrossRef]
- Soleimani, A.; Kazemian, S.; Karbalai Saleh, S.; Aminorroaya, A.; Shajari, Z.; Hadadi, A.; Talebpour, M.; Sadeghian, H.; Payandemehr, P.; Sotoodehnia, M.; et al. Effects of Angiotensin Receptor Blockers (ARBs) on In-Hospital Outcomes of Patients With Hypertension and Confirmed or Clinically Suspected COVID-19. Am. J. Hypertens. 2020, 33, 1102–1111. [Google Scholar] [CrossRef]
- Meng, J.; Xiao, G.; Zhang, J.; He, X.; Ou, M.; Bi, J.; Yang, R.; Di, W.; Wang, Z.; Li, Z.; et al. Renin-angiotensin system inhibitors improve the clinical outcomes of COVID-19 patients with hypertension. Emerg. Microbes Infect. 2020, 9, 757–760. [Google Scholar] [CrossRef]
- Xie, Q.; Tang, S.; Li, Y. The divergent protective effects of angiotensin-converting enzyme inhibitors and angiotensin receptor blockers on clinical outcomes of coronavirus disease 2019 (COVID-19): A systematic review and meta-analysis. Ann. Palliat. Med. 2022, 11, 1253–1263. [Google Scholar] [CrossRef] [PubMed]
- Rothlin, R.P.; Duarte, M.; Pelorosso, F.G.; Nicolosi, L.; Salgado, M.V.; Vetulli, H.M.; Spitzer, E. Angiotensin Receptor Blockers for COVID-19: Pathophysiological and Pharmacological Considerations About Ongoing and Future Prospective Clinical Trials. Front. Pharmacol. 2021, 12, 603736. [Google Scholar] [CrossRef] [PubMed]
- Rothlin, R.P.; Vetulli, H.M.; Duarte, M.; Pelorosso, F.G. Telmisartan as tentative angiotensin receptor blocker therapeutic for COVID-19. Drug Dev. Res. 2020, 81, 768–770. [Google Scholar] [CrossRef]
- Lukito, A.A.; Widysanto, A.; Lemuel, T.A.Y.; Prasetya, I.B.; Massie, B.; Yuniarti, M.; Lumbuun, N.; Pranata, R.; Meidy, C.; Wahjoepramono, E.J.; et al. Candesartan as a tentative treatment for COVID-19: A prospective non-randomized open-label study. Int. J. Infect. Dis. 2021, 108, 159–166. [Google Scholar] [CrossRef]
- Duarte, M.; Pelorosso, F.; Nicolosi, L.N.; Salgado, M.V.; Vetulli, H.; Aquieri, A.; Azzato, F.; Castro, M.; Coyle, J.; Davolos, I.; et al. Telmisartan for treatment of Covid-19 patients: An open multicenter randomized clinical trial. eClinicalMedicine 2021, 37, 100962. [Google Scholar] [CrossRef]
- Kakuta, H.; Sudoh, K.; Sasamata, M.; Yamagishi, S. Telmisartan has the strongest binding affinity to angiotensin II type 1 receptor: Comparison with other angiotensin II type 1 receptor blockers. Int. J. Clin. Pharmacol. Res. 2005, 25, 41–46. [Google Scholar] [PubMed]
- Michel, M.C.; Foster, C.; Brunner, H.R.; Liu, L. A systematic comparison of the properties of clinically used angiotensin II type 1 receptor antagonists. Pharmacol. Rev. 2013, 65, 809–848. [Google Scholar] [CrossRef]
- Elkahloun, A.G.; Saavedra, J.M. Candesartan could ameliorate the COVID-19 cytokine storm. Biomed. Pharmacother. 2020, 131, 110653. [Google Scholar] [CrossRef]
- Liu, D.; Wu, P.; Gu, W.; Yang, C.; Yang, X.; Deng, X.; Xu, J.; Jiang, J.; Jiang, C. Potential of angiotensin II receptor blocker telmisartan in reducing mortality among hospitalized patients with COVID-19 compared with recommended drugs. Cell Discov. 2022, 8, 91. [Google Scholar] [CrossRef]
- Hippisley-Cox, J.; Young, D.; Coupland, C.; Channon, K.M.; Tan, P.S.; Harrison, D.A.; Rowan, K.; Aveyard, P.; Pavord, I.D.; Watkinson, P.J. Risk of severe COVID-19 disease with ACE inhibitors and angiotensin receptor blockers: Cohort study including 8.3 million people. Heart 2020, 106, 1503–1511. [Google Scholar] [CrossRef] [PubMed]
- Angeli, F.; Verdecchia, P.; Balestrino, A.; Bruschi, C.; Ceriana, P.; Chiovato, L.; Dalla Vecchia, L.A.; Fanfulla, F.; La Rovere, M.T.; Perego, F.; et al. Renin Angiotensin System Blockers and Risk of Mortality in Hypertensive Patients Hospitalized for COVID-19: An Italian Registry. J. Cardiovasc. Dev. Dis. 2022, 9, 15. [Google Scholar] [CrossRef]
- Cremer, S.; Pilgram, L.; Berkowitsch, A.; Stecher, M.; Rieg, S.; Shumliakivska, M.; Bojkova, D.; Wagner, J.U.G.; Aslan, G.S.; Spinner, C.; et al. Angiotensin II receptor blocker intake associates with reduced markers of inflammatory activation and decreased mortality in patients with cardiovascular comorbidities and COVID-19 disease. PLoS ONE 2021, 16, e0258684. [Google Scholar] [CrossRef]
- Palazzuoli, A.; Mancone, M.; De Ferrari, G.M.; Forleo, G.; Secco, G.G.; Ruocco, G.M.; D’Ascenzo, F.; Monticone, S.; Paggi, A.; Vicenzi, M.; et al. Antecedent Administration of Angiotensin-Converting Enzyme Inhibitors or Angiotensin II Receptor Antagonists and Survival After Hospitalization for COVID-19 Syndrome. J. Am. Heart Assoc. 2020, 9, e017364. [Google Scholar] [CrossRef] [PubMed]
- Koka, V.; Huang, X.R.; Chung, A.C.; Wang, W.; Truong, L.D.; Lan, H.Y. Angiotensin II up-regulates angiotensin I-converting enzyme (ACE), but down-regulates ACE2 via the AT1-ERK/p38 MAP kinase pathway. Am. J. Pathol. 2008, 172, 1174–1183. [Google Scholar] [CrossRef] [PubMed]
- Burrell, L.M.; Risvanis, J.; Kubota, E.; Dean, R.G.; MacDonald, P.S.; Lu, S.; Tikellis, C.; Grant, S.L.; Lew, R.A.; Smith, A.I.; et al. Myocardial infarction increases ACE2 expression in rat and humans. Eur. Heart J. 2005, 26, 369–375, discussion 322–364. [Google Scholar] [CrossRef]
- Ishiyama, Y.; Gallagher, P.E.; Averill, D.B.; Tallant, E.A.; Brosnihan, K.B.; Ferrario, C.M. Upregulation of angiotensin-converting enzyme 2 after myocardial infarction by blockade of angiotensin II receptors. Hypertension 2004, 43, 970–976. [Google Scholar] [CrossRef]
- Pires de Souza, G.A.; Osman, I.O.; Le Bideau, M.; Baudoin, J.P.; Jaafar, R.; Devaux, C.; La Scola, B. Angiotensin II Receptor Blockers (ARBs Antihypertensive Agents) Increase Replication of SARS-CoV-2 in Vero E6 Cells. Front. Cell. Infect. Microbiol. 2021, 11, 639177. [Google Scholar] [CrossRef]
- Essaidi-Laziosi, M.; Perez Rodriguez, F.J.; Hulo, N.; Jacquerioz, F.; Kaiser, L.; Eckerle, I. Estimating clinical SARS-CoV-2 infectiousness in Vero E6 and primary airway epithelial cells. Lancet Microbe 2021, 2, e571. [Google Scholar] [CrossRef]
- Matsoukas, J.M.; Gadanec, L.K.; Zulli, A.; Apostolopoulos, V.; Kelaidonis, K.; Ligielli, I.; Moschovou, K.; Georgiou, N.; Plotas, P.; Chasapis, C.T. Diminazene aceturate reduces angiotensin II constriction and interacts with the spike protein of severe acute respiratory syndrome coronavirus 2. Biomedicines 2022, 10, 1731. [Google Scholar] [CrossRef]
- Qaradakhi, T.; Gadanec, L.; Matsoukas, J.; Apostolopoulos, V.; Zulli, A. Could DIZE be the answer to COVID-19? Maturitas 2020, 140, 83–84. [Google Scholar] [CrossRef] [PubMed]
- Rico-Mesa, J.S.; White, A.; Anderson, A.S. Outcomes in Patients with COVID-19 Infection Taking ACEI/ARB. Curr. Cardiol. Rep. 2020, 22, 31. [Google Scholar] [CrossRef] [PubMed]
- Ridgway, H.; Moore, G.J.; Mavromoustakos, T.; Tsiodras, S.; Ligielli, I.; Kelaidonis, K.; Chasapis, C.T.; Gadanec, L.K.; Zulli, A.; Apostolopoulos, V.; et al. Discovery of a new generation of angiotensin receptor blocking drugs: Receptor mechanisms and in silico binding to enzymes relevant to SARS-CoV-2. Comput. Struct. Biotechnol. J. 2022, 20, 2091–2111. [Google Scholar] [CrossRef] [PubMed]
- Moore, G.J.; Pires, J.M.; Kelaidonis, K.; Gadanec, L.K.; Zulli, A.; Apostolopoulos, V.; Matsoukas, J.M. Receptor Interactions of Angiotensin II and Angiotensin Receptor Blockers-Relevance to COVID-19. Biomolecules 2021, 11, 979c. [Google Scholar] [CrossRef]
- Takezako, T.; Unal, H.; Karnik, S.S.; Node, K. Current topics in angiotensin II type 1 receptor research: Focus on inverse agonism, receptor dimerization and biased agonism. Pharmacol. Res. 2017, 123, 40–50. [Google Scholar] [CrossRef]
- Moore, G.J. Designing peptide mimetics. Trends Pharmacol. Sci. 1994, 15, 124–129. [Google Scholar] [CrossRef]
- Moore, G.J.; Ridgway, H.; Kelaidonis, K.; Chasapis, C.T.; Ligielli, I.; Mavromoustakos, T.; Bojarska, J.; Matsoukas, J.M. Actions of Novel Angiotensin Receptor Blocking Drugs, Bisartans, Relevant for COVID-19 Therapy: Biased Agonism at Angiotensin Receptors and the Beneficial Effects of Neprilysin in the Renin Angiotensin System. Molecules 2022, 27, 4854. [Google Scholar] [CrossRef]
- Agelis, G.; Resvani, A.; Koukoulitsa, C.; Tumova, T.; Slaninova, J.; Kalavrizioti, D.; Spyridaki, K.; Afantitis, A.; Melagraki, G.; Siafaka, A.; et al. Rational design, efficient syntheses and biological evaluation of N,N′-symmetrically bis-substituted butylimidazole analogs as a new class of potent Angiotensin II receptor blockers. Eur. J. Med. Chem. 2013, 62, 352–370. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| Subject | ARB/Dose | Outcome | Ref |
|---|---|---|---|
| 103 patients with hypertension and COVID-19 receiving ARB therapy | NA | Pre-administered ARBs reduced the need for mechanical ventilation, intensive care admission, and death. | [149] |
| 157 patients diagnosed with COVID-19 and hypertension on ARBs | NA | ARB group is associated with decreased mortality. | [150] |
| 201 hospitalized patients diagnosed with COVID019 | NA | Pre-administered ARB had a lower mortality rate compared to other antihypertensive medications. | [151] |
| 636 COVID-19 patients,1 of which 22 receiving ARBs | NA | Discontinuing ARB therapy during COVID-19 infection resulted in greater mortality, ventilation, and increased risk of acute kidney injury. | [152] |
| 19 586 patients with COVID-19 | NA | ARBs associated with reduced risk of COVID-19 in patients with hypertension. | [163] |
| 566 hypertensive patients with COVID-19, 147 on ARBs. | NA | ARB therapy resulted in a lower risk of mortality in patients with a high prognostic factor, low oxygen saturation, and high lymphocyte count than other RAS inhibitors. | [164] |
| 63,969 hospitalized participants with COVID-19 | Telmisartan | Telmisartan showed a reduction in mortality risks greater than standard care. | [162] |
| 52 COVID-19-diagnosed patients not receiving antihypertensive medication | Telmisartan 160 mg/day, 14 days | Telmisartan reduced morbidity and mortality of COVID-19 patients through anti-inflammatory effects. | [158] |
| 1946 patients with COVID019 and cardiovascular comorbidities of which 493 on ARB | NA | ARBs reduced mortality, leukocyte count, inflammatory markers, and IL-6. No changes in ACE2 expression were observed. | [165] |
| 178 patients with COVID-19 of which 133 used ARBs. | NA | ARBs reduced mortality in patients hospitalized with COVID-19. | [166] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Swiderski, J.; Gadanec, L.K.; Apostolopoulos, V.; Moore, G.J.; Kelaidonis, K.; Matsoukas, J.M.; Zulli, A. Role of Angiotensin II in Cardiovascular Diseases: Introducing Bisartans as a Novel Therapy for Coronavirus 2019. Biomolecules 2023, 13, 787. https://doi.org/10.3390/biom13050787
Swiderski J, Gadanec LK, Apostolopoulos V, Moore GJ, Kelaidonis K, Matsoukas JM, Zulli A. Role of Angiotensin II in Cardiovascular Diseases: Introducing Bisartans as a Novel Therapy for Coronavirus 2019. Biomolecules. 2023; 13(5):787. https://doi.org/10.3390/biom13050787
Chicago/Turabian StyleSwiderski, Jordan, Laura Kate Gadanec, Vasso Apostolopoulos, Graham J. Moore, Konstantinos Kelaidonis, John M. Matsoukas, and Anthony Zulli. 2023. "Role of Angiotensin II in Cardiovascular Diseases: Introducing Bisartans as a Novel Therapy for Coronavirus 2019" Biomolecules 13, no. 5: 787. https://doi.org/10.3390/biom13050787