

Therapeutic Strategies for Spinocerebellar Ataxia Type 1
 , , and
, , and
Abstract
:1. Introduction
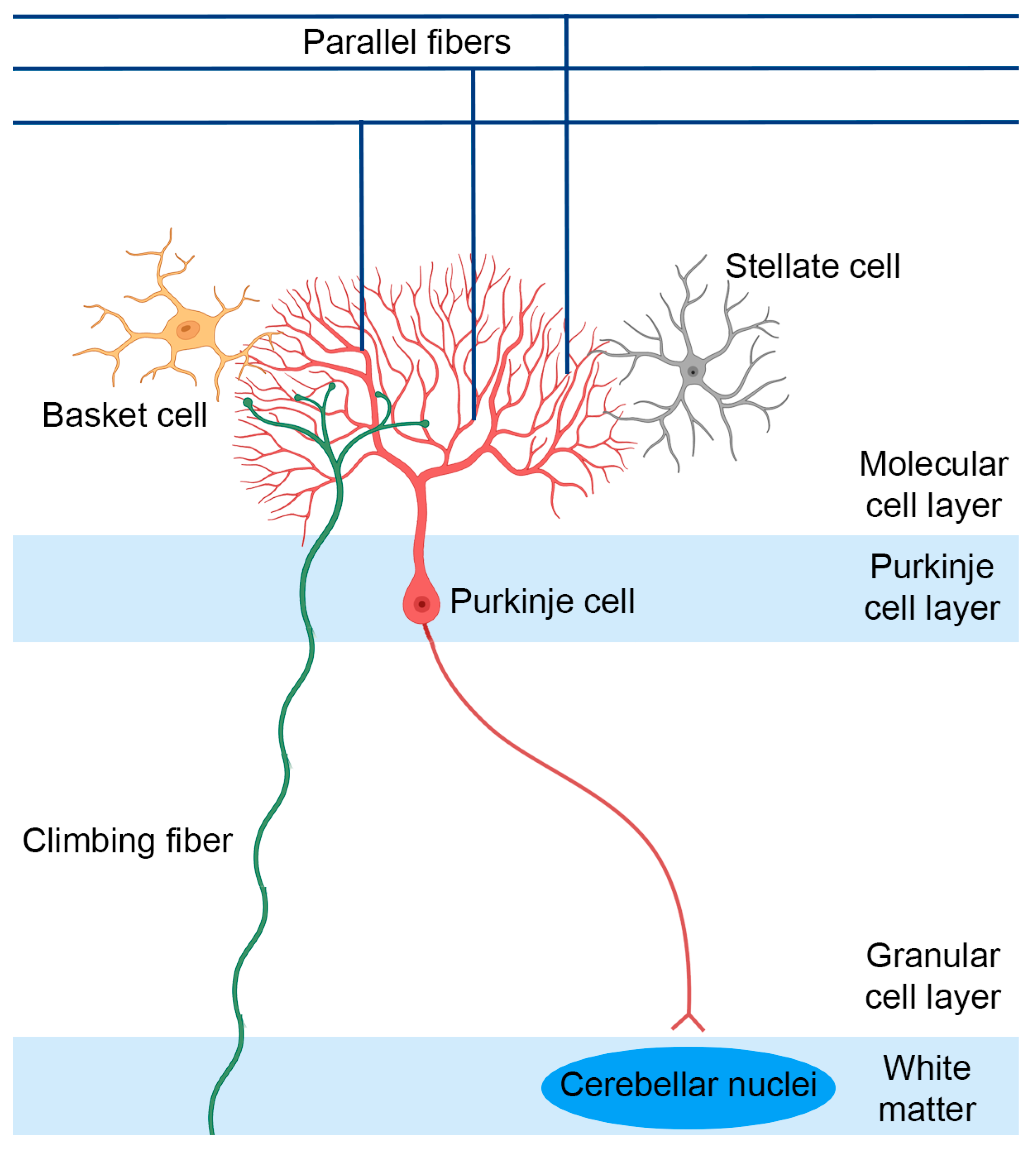
2. SCA1 Pathophysiology
3. Therapeutic Strategies
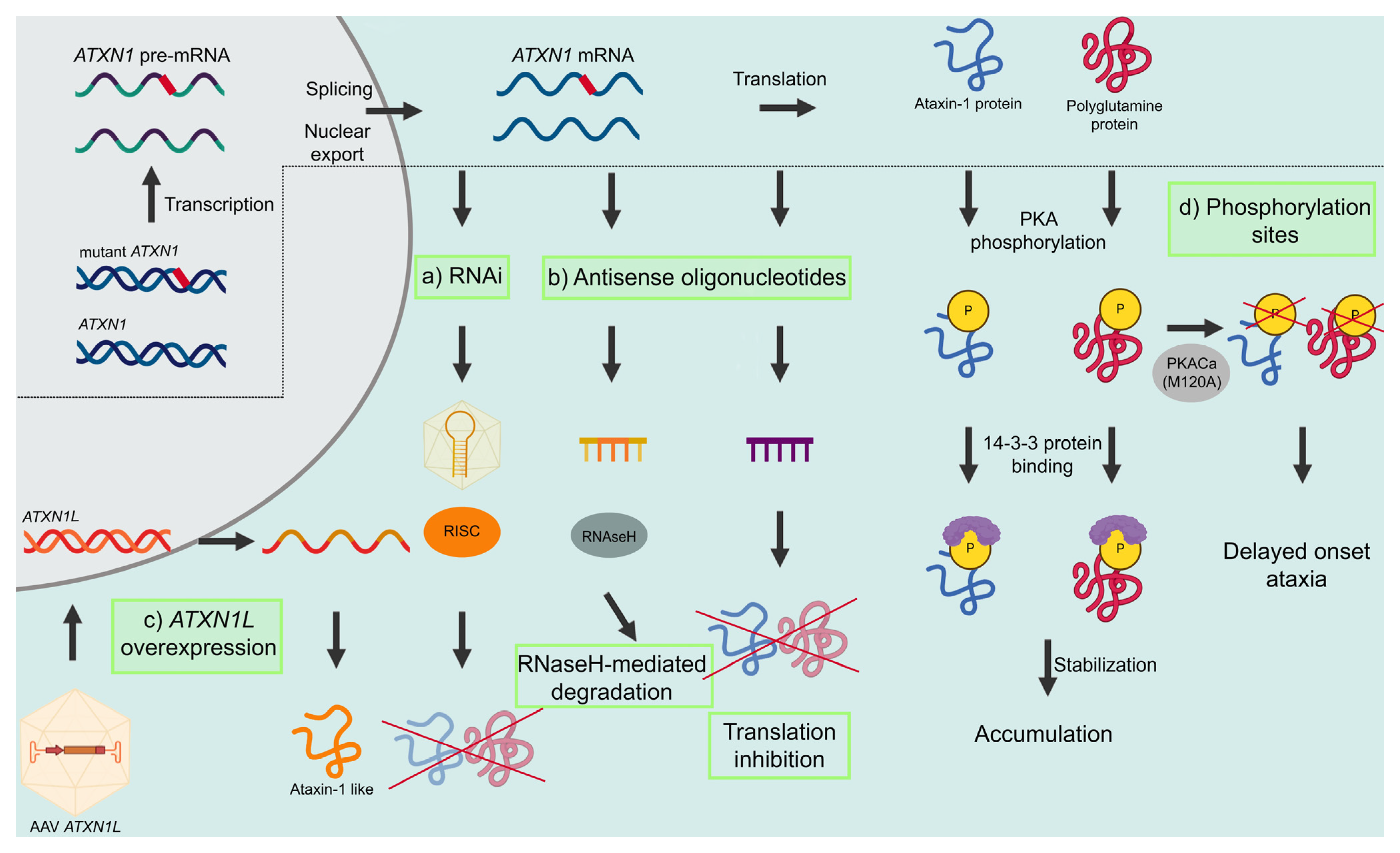
3.1. Modulating Ataxin-1 Levels
3.1.1. RNA Interference
3.1.2. Antisense Oligonucleotides
3.1.3. ATXN1L Overexpression
3.1.4. S776 Phosphorylation
3.2. Pharmacological Targets
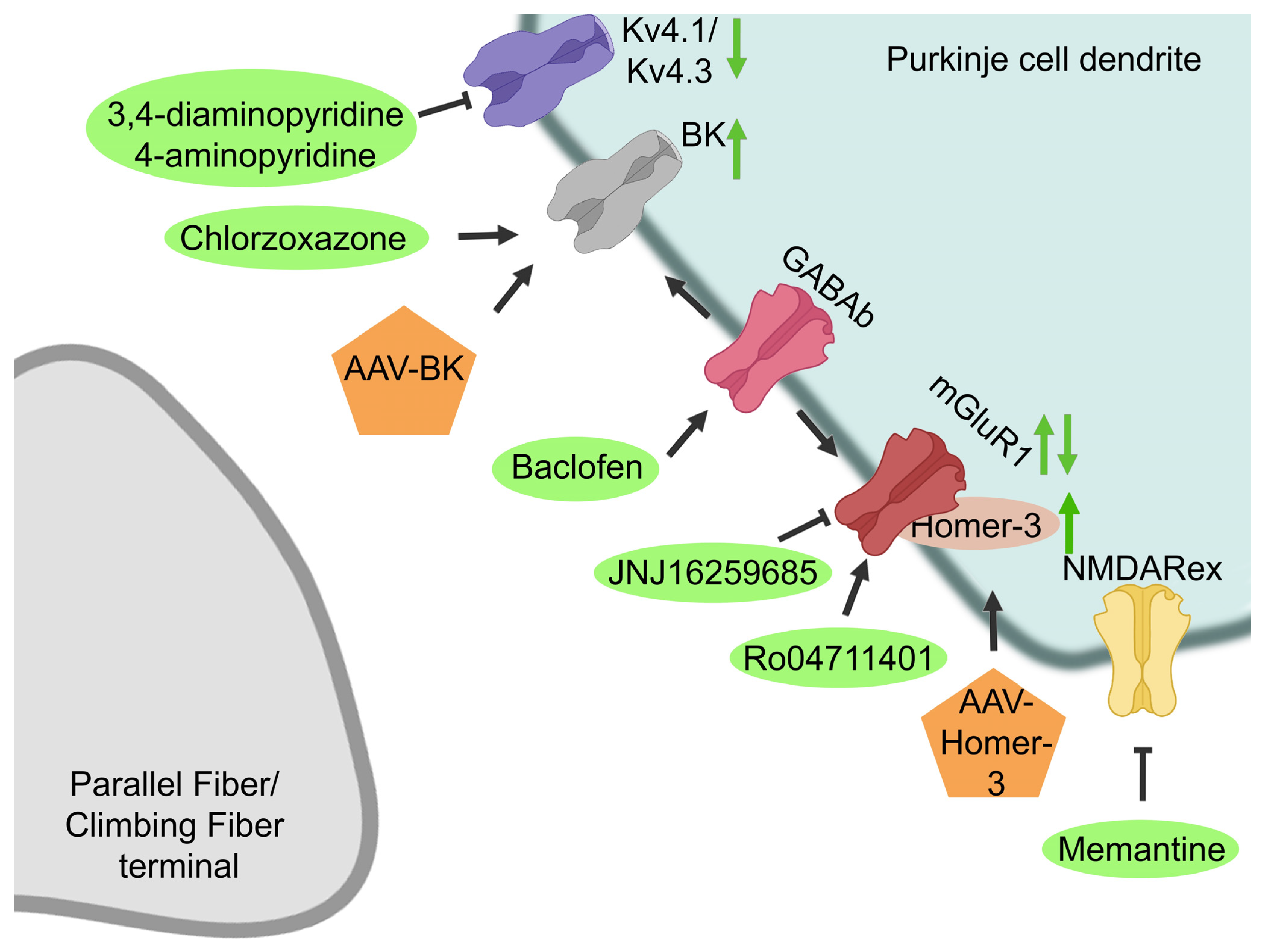
3.2.1. Potassium Channels
3.2.2. Glutamatergic Signaling
3.2.3. NMDA Receptor
3.2.4. Protection of PC Survival and Function
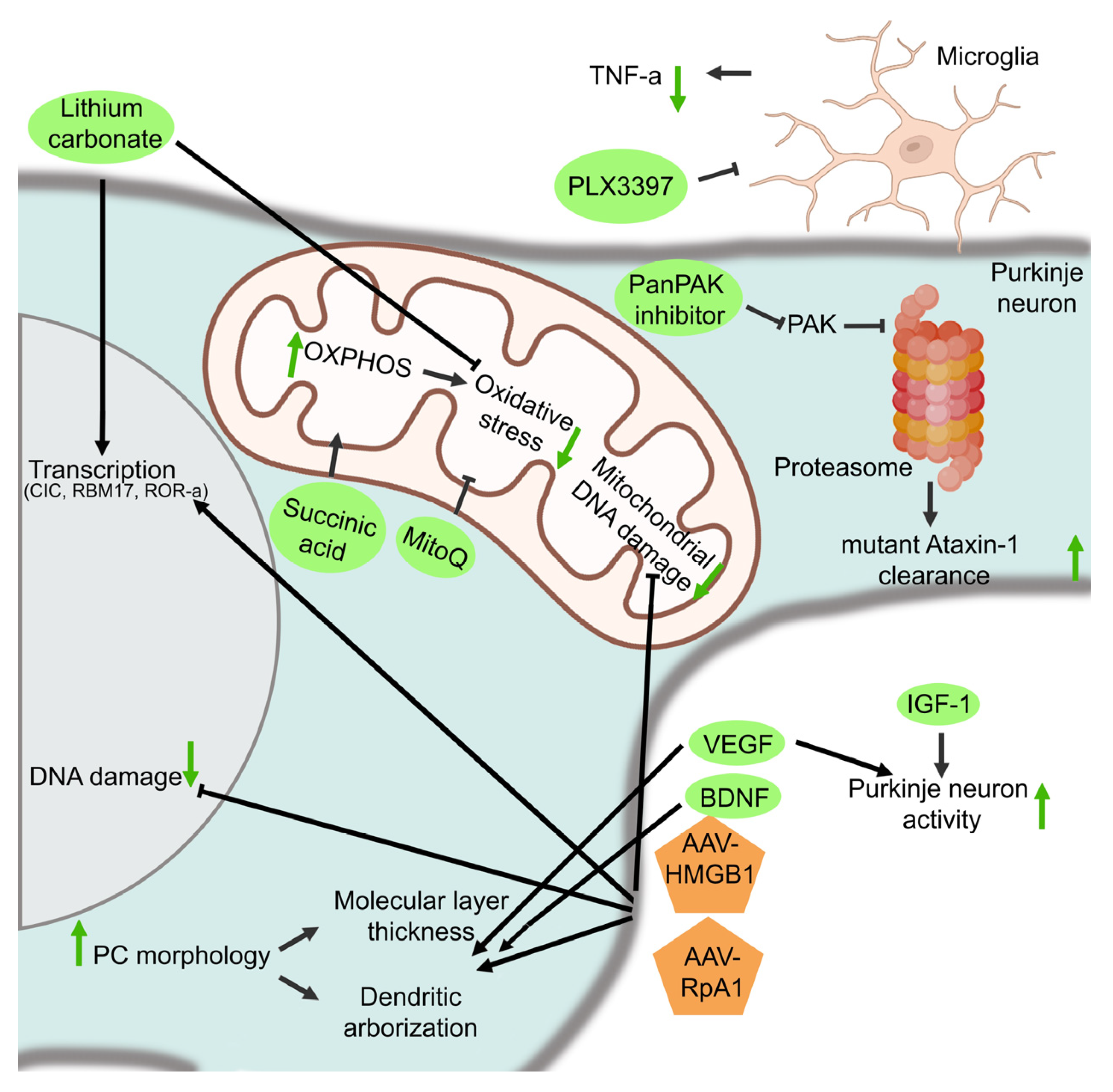
3.2.5. Mitochondrial Functioning
3.2.6. DNA Damage Repair, Transcription, and Replication
3.2.7. The Proteostatic Machinery
3.2.8. Inflammation
3.3. Stem Cell Replacement Therapies
4. Clinical Trials
5. Future Perspectives on SCA1 Therapeutics
Funding
Acknowledgments
Conflicts of Interest
References
- Zoghbi, H.Y.; Orr, H.T. Spinocerebellar ataxia type 1. Semin Cell Biol. 1995, 6, 29–35. [Google Scholar] [CrossRef] [PubMed]
- Banfi, S.; Servadio, A.; Chung, M.-Y.; Kwiatkowski, T.J.; McCall, A.E.; Duvick, L.A.; Shen, Y.; Roth, E.J.; Orr, H.T.; Zoghbi, H. Identification and characterization of the gene causing type 1 spinocerebellar ataxia. Nat. Genet. 1994, 7, 513–520. [Google Scholar] [CrossRef] [PubMed]
- Buijsen, R.A.; Toonen, L.J.; Gardiner, S.L.; van Roon-Mom, W.M. Genetics, Mechanisms, and Therapeutic Progress in Polyglutamine Spinocerebellar Ataxias. Neurotherapeutics 2019, 16, 263–286. [Google Scholar] [CrossRef] [PubMed]
- Jacobi, H.; Bauer, P.; Giunti, P.; Labrum, R.; Sweeney, M.G.; Charles, P.; Durr, A.; Marelli, C.; Globas, C.; Linnemann, C.; et al. The natural history of spinocerebellar ataxia type 1, 2, 3, and 6: A 2-year follow-up study. Neurology 2011, 77, 1035–1041. [Google Scholar] [CrossRef]
- Orengo, J.P.; van der Heijden, M.E.; Hao, S.; Tang, J.; Orr, H.T.; Zoghbi, H.Y. Motor neuron degeneration correlates with respiratory dysfunction in SCA1. Dis. Model. Mech. 2018, 11, dmm032623. [Google Scholar] [CrossRef]
- Schöls, L.; Bauer, P.; Schmidt, T.; Schulte, T.; Riess, O. Autosomal dominant cerebellar ataxias: Clinical features, genetics, and pathogenesis. Lancet Neurol. 2004, 3, 291–304. [Google Scholar] [CrossRef]
- Opal, P.; Ashizawa, T. Spinocerebellar Ataxia Type 1. In GeneReviews®; Adam, M.P., Everman, D.B., Mirzaa, G.M., Pagon, R.A., Wallace, S.E., Bean, L.J.H., Gripp, K.W., Amemiya, A., Eds.; University of Washington Seattle: Seattle, WA, USA, 1993. [Google Scholar]
- van de Warrenburg, B.; Sinke, R.; Verschuuren-Bemelmans, C.; Scheffer, H.; Brunt, E.; Ippel, P.; Maat-Kievit, J.; Dooijes, D.; Notermans, N.; Lindhout, D.; et al. Spinocerebellar ataxias in the Netherlands: Prevalence and age at onset variance analysis. Neurology 2002, 58, 702–708. [Google Scholar] [CrossRef]
- Tang, B.; Liu, C.; Shen, L.; Dai, H.; Pan, Q.; Jing, L.; Ouyang, S.; Xia, J. Frequency of SCA1, SCA2, SCA3/MJD, SCA6, SCA7, and DRPLA CAG trinucleotide repeat expansion in patients with hereditary spinocerebellar ataxia from Chinese kindreds. Arch. Neurol. 2000, 57, 540–544. [Google Scholar] [CrossRef]
- Bryer, A.; Krause, A.; Bill, P.; Davids, V.; Bryant, D.; Butler, J.; Heckmann, J.; Ramesar, R.; Greenberg, J. The hereditary adult-onset ataxias in South Africa. J. Neurol. Sci. 2003, 216, 47–54. [Google Scholar] [CrossRef]
- Orr, H.; Chung, M.-Y.; Banfi, S.; Kwiatkowski, T.J.; Servadio, A.; Beaudet, A.L.; McCall, A.E.; Duvick, L.A.; Ranum, L.P.W.; Zoghbi, H. Expansion of an unstable trinucleotide CAG repeat in spinocerebellar ataxia type 1. Nat. Genet. 1993, 4, 221–226. [Google Scholar] [CrossRef]
- Zühlke, C.; Dalski, A.; Hellenbroich, Y.; Bubel, S.; Schwinger, E.; Bürk, K. Spinocerebellar ataxia type 1 (SCA1): Phenotype-genotype correlation studies in intermediate alleles. Eur. J. Hum. Genet. 2002, 10, 204–209. [Google Scholar] [CrossRef]
- Menon, R.P.; Nethisinghe, S.; Faggiano, S.; Vannocci, T.; Rezaei, H.; Pemble, S.; Sweeney, M.G.; Wood, N.W.; Davis, M.B.; Pastore, A.; et al. The Role of Interruptions in polyQ in the Pathology of SCA1. PLoS Genet. 2013, 9, e1003648. [Google Scholar] [CrossRef]
- Gardiner, S.L.; Boogaard, M.W.; Trompet, S.; De Mutsert, R.; Rosendaal, F.R.; Gussekloo, J.; Jukema, J.W.; Roos, R.A.C.; Aziz, N.A. Prevalence of Carriers of Intermediate and Pathological Polyglutamine Disease–Associated Alleles Among Large Population-Based Cohorts. JAMA Neurol. 2019, 76, 650–656. [Google Scholar] [CrossRef]
- Kraus-Perrotta, C.; Lagalwar, S. Expansion, mosaicism and interruption: Mechanisms of the CAG repeat mutation in spinocerebellar ataxia type 1. Cerebellum Ataxias 2016, 3, 20. [Google Scholar] [CrossRef]
- Servadio, A.; Koshy, B.; Armstrong, D.D.; Antalffy, B.; Orr, H.; Zoghbi, H. Expression analysis of the ataxin–1 protein in tissues from normal and spinocerebellar ataxia type 1 individuals. Nat. Genet. 1995, 10, 94–98. [Google Scholar] [CrossRef]
- Manek, R.; Nelson, T.; Tseng, E.; Rodriguez-Lebron, E. 5′UTR-mediated regulation of Ataxin-1 expression. Neurobiol. Dis. 2020, 134, 104564. [Google Scholar] [CrossRef]
- Paulson, H.L.; Shakkottai, V.G.; Clark, H.B.; Orr, H. Polyglutamine spinocerebellar ataxias—From genes to potential treatments. Nat. Rev. Neurosci. 2017, 18, 613–626. [Google Scholar] [CrossRef]
- Ilg, W.; Bastian, A.J.; Boesch, S.; Burciu, R.G.; Celnik, P.; Claaßen, J.; Feil, K.; Kalla, R.; Miyai, I.; Nachbauer, W.; et al. Consensus Paper: Management of Degenerative Cerebellar Disorders. Cerebellum 2013, 13, 248–268. [Google Scholar] [CrossRef]
- Rüb, U.; Bürk, K.; Timmann, D.; Dunnen, W.D.; Seidel, K.; Farrag, K.; Brunt, E.; Heinsen, H.; Egensperger, R.; Bornemann, A.; et al. Spinocerebellar ataxia type 1 (SCA1): New pathoanatomical and clinico-pathological insights. Neuropathol. Appl. Neurobiol. 2012, 38, 665–680. [Google Scholar] [CrossRef]
- Mieda, T.; Suto, N.; Iizuka, A.; Matsuura, S.; Iizuka, H.; Takagishi, K.; Nakamura, K.; Hirai, H. Mesenchymal stem cells attenuate peripheral neuronal degeneration in spinocerebellar ataxia type 1 knockin mice. J. Neurosci. Res. 2015, 94, 246–252. [Google Scholar] [CrossRef]
- Rüb, U.; Schöls, L.; Paulson, H.; Auburger, G.; Kermer, P.; Jen, J.C.; Seidel, K.; Korf, H.; Deller, T. Clinical features, neurogenetics and neuropathology of the polyglutamine spinocerebellar ataxias type 1, 2, 3, 6 and 7. Prog. Neurobiol. 2013, 104, 38–66. [Google Scholar] [CrossRef] [PubMed]
- Cvetanovic, M.; Ingram, M.; Orr, H.; Opal, P. Early activation of microglia and astrocytes in mouse models of spinocerebellar ataxia type 1. Neuroscience 2015, 289, 289–299. [Google Scholar] [CrossRef] [PubMed]
- Zoghbi, H.; Orr, H. Pathogenic Mechanisms of a Polyglutamine-mediated Neurodegenerative Disease, Spinocerebellar Ataxia Type 1. J. Biol. Chem. 2009, 284, 7425–7429. [Google Scholar] [CrossRef] [PubMed]
- Tejwani, L.; Lim, J. Pathogenic mechanisms underlying spinocerebellar ataxia type 1. Cell. Mol. Life Sci. 2020, 77, 4015–4029. [Google Scholar] [CrossRef]
- Matilla, A.; Roberson, E.D.; Banfi, S.; Morales, J.; Armstrong, D.L.; Burright, E.N.; Orr, H.T.; Sweatt, J.D.; Zoghbi, H.Y.; Matzuk, M.M. Mice Lacking Ataxin-1 Display Learning Deficits and Decreased Hippocampal Paired-Pulse Facilitation. J. Neurosci. 1998, 18, 5508–5516. [Google Scholar] [CrossRef]
- Burright, E.N.; Clark, H.B.; Servadio, A.; Matilla, T.; Feddersen, R.M.; Yunis, W.S.; Duvick, L.A.; Zoghbi, H.Y.; Orr, H.T. SCA1 transgenic mice: A model for neurodegeneration caused by an expanded CAG trinucleotide repeat. Cell 1995, 82, 937–948. [Google Scholar] [CrossRef]
- Watase, K.; Weeber, E.J.; Xu, B.; Antalffy, B.; Yuva-Paylor, L.; Hashimoto, K.; Kano, M.; Atkinson, R.; Sun, Y.; Armstrong, D.L.; et al. A Long CAG Repeat in the Mouse Sca1 Locus Replicates SCA1 Features and Reveals the Impact of Protein Solubility on Selective Neurodegeneration. Neuron 2002, 34, 905–919. [Google Scholar] [CrossRef]
- Fernandez-Funez, P.; Nino-Rosales, M.L.; De Gouyon, B.; She, W.-C.; Luchak, J.M.; Martinez, P.; Turiegano, E.; Benito, J.; Capovilla, M.; Skinner, P.J.; et al. Identification of genes that modify ataxin-1-induced neurodegeneration. Nature 2000, 408, 101–106. [Google Scholar] [CrossRef]
- Klement, I.A.; Skinner, P.J.; Kaytor, M.D.; Yi, H.; Hersch, S.M.; Clark, H.; Zoghbi, H.Y.; Orr, H.T. Ataxin-1 Nuclear Localization and Aggregation: Role in Polyglutamine-Induced Disease in SCA1 Transgenic Mice. Cell 1998, 95, 41–53. [Google Scholar] [CrossRef]
- Chen, H.-K.; Fernandez-Funez, P.; Acevedo, S.F.; Lam, Y.C.; Kaytor, M.D.; Fernandez, M.H.; Aitken, A.; Skoulakis, E.M.; Orr, H.T.; Botas, J.; et al. Interaction of Akt-Phosphorylated Ataxin-1 with 14-3-3 Mediates Neurodegeneration in Spinocerebellar Ataxia Type 1. Cell 2003, 113, 457–468. [Google Scholar] [CrossRef]
- Cummings, C.J.; Mancini, M.A.; Antalffy, B.; DeFranco, D.B.; Orr, H.; Zoghbi, H. Chaperone suppression of aggregation and altered subcellular proteasome localization imply protein misfolding in SCA1. Nat. Genet. 1998, 19, 148–154. [Google Scholar] [CrossRef]
- Handler, H.P.; Duvick, L.; Mitchell, J.S.; Cvetanovic, M.; Reighard, M.; Soles, A.; Mather, K.B.; Rainwater, O.; Serres, S.; Nichols-Meade, T.; et al. Decreasing mutant ATXN1 nuclear localization improves a spectrum of SCA1-like phenotypes and brain region transcriptomic profiles. Neuron 2022, 111, 493–507.e6. [Google Scholar] [CrossRef]
- Putka, A.F.; McLoughlin, H.S. Diverse regional mechanisms drive spinocerebellar ataxia type 1 phenotypes. Neuron 2023, 111, 447–449. [Google Scholar] [CrossRef]
- Burright, E.N.; Davidson, J.D.; Duvick, L.A.; Koshy, B.; Zoghbi, H.Y.; Orr, H.T. Identification of a self-association region within the SCA1 gene product, ataxin-1. Hum. Mol. Genet. 1997, 6, 513–518. [Google Scholar] [CrossRef]
- Nitschke, L.; Coffin, S.L.; Xhako, E.; El-Najjar, D.B.; Orengo, J.P.; Alcala, E.; Dai, Y.; Wan, Y.-W.; Liu, Z.; Orr, H.T.; et al. Modulation of ATXN1 S776 phosphorylation reveals the importance of allele-specific targeting in SCA1. J. Clin. Investig. 2021, 6, e144955. [Google Scholar] [CrossRef]
- Jorgensen, N.D.; Andresen, J.M.; Lagalwar, S.; Armstrong, B.; Stevens, S.; Byam, C.E.; Duvick, L.A.; Lai, S.; Jafar-Nejad, P.; Zoghbi, H.Y.; et al. Phosphorylation of ATXN1 at Ser776 in the cerebellum. J. Neurochem. 2009, 110, 675–686. [Google Scholar] [CrossRef]
- Ortiz, J.M.P.; Mollema, N.; Toker, N.; Adamski, C.J.; O’Callaghan, B.; Duvick, L.; Friedrich, J.; Walters, M.A.; Strasser, J.; Hawkinson, J.E.; et al. Reduction of protein kinase A-mediated phosphorylation of ATXN1-S776 in Purkinje cells delays onset of Ataxia in a SCA1 mouse model. Neurobiol. Dis. 2018, 116, 93–105. [Google Scholar] [CrossRef]
- Lim, J.; Crespo-Barreto, J.; Jafar-Nejad, P.; Bowman, A.B.; Richman, R.; Hill, D.E.; Orr, H.T.; Zoghbi, H.Y. Opposing effects of polyglutamine expansion on native protein complexes contribute to SCA1. Nature 2008, 452, 713–718. [Google Scholar] [CrossRef]
- Lam, Y.C.; Bowman, A.B.; Jafar-Nejad, P.; Lim, J.; Richman, R.; Fryer, J.D.; Hyun, E.D.; Duvick, L.A.; Orr, H.T.; Botas, J.; et al. ATAXIN-1 Interacts with the Repressor Capicua in Its Native Complex to Cause SCA1 Neuropathology. Cell 2006, 127, 1335–1347. [Google Scholar] [CrossRef]
- Cook, A.A.; Fields, E.; Watt, A.J. Losing the Beat: Contribution of Purkinje Cell Firing Dysfunction to Disease, and Its Reversal. Neuroscience 2021, 462, 247–261. [Google Scholar] [CrossRef]
- Coffin, S.L.; Durham, M.A.; Nitschke, L.; Xhako, E.; Brown, A.M.; Revelli, J.-P.; Gonzalez, E.V.; Lin, T.; Handler, H.P.; Dai, Y.; et al. Disruption of the ATXN1-CIC complex reveals the role of additional nuclear ATXN1 interactors in spinocerebellar ataxia type 1. Neuron 2022, 111, 481–492.e8. [Google Scholar] [CrossRef] [PubMed]
- Serra, H.G.; Duvick, L.; Zu, T.; Carlson, K.; Stevens, S.; Jorgensen, N.; Lysholm, A.; Burright, E.; Zoghbi, H.; Clark, H.B.; et al. RORα-Mediated Purkinje Cell Development Determines Disease Severity in Adult SCA1 Mice. Cell 2006, 127, 697–708. [Google Scholar] [CrossRef] [PubMed]
- Gehrking, K.M.; Andresen, J.M.; Duvick, L.; Lough, J.; Zoghbi, H.Y.; Orr, H.T. Partial loss of Tip60 slows mid-stage neurodegeneration in a spinocerebellar ataxia type 1 (SCA1) mouse model. Hum. Mol. Genet. 2011, 20, 2204–2212. [Google Scholar] [CrossRef] [PubMed]
- Bowman, A.; Lam, Y.C.; Jafar-Nejad, P.; Chen, H.-K.; Richman, R.; Samaco, R.; Fryer, J.D.; Kahle, J.J.; Orr, H.; Zoghbi, H. Duplication of Atxn1l suppresses SCA1 neuropathology by decreasing incorporation of polyglutamine-expanded ataxin-1 into native complexes. Nat. Genet. 2007, 39, 373–379. [Google Scholar] [CrossRef] [PubMed]
- Yue, S.; Serra, H.G.; Zoghbi, H.Y.; Orr, H.T. The spinocerebellar ataxia type 1 protein, ataxin-1, has RNA-binding activity that is inversely affected by the length of its polyglutamine tract. Hum. Mol. Genet. 2001, 10, 25–30. [Google Scholar] [CrossRef]
- Elsaey, M.A.; Namikawa, K.; Köster, R.W. Genetic Modeling of the Neurodegenerative Disease Spinocerebellar Ataxia Type 1 in Zebrafish. Int. J. Mol. Sci. 2021, 22, 7351. [Google Scholar] [CrossRef]
- Clark, H.B.; Burright, E.N.; Yunis, W.S.; Larson, S.; Wilcox, C.; Hartman, B.; Matilla, A.; Zoghbi, H.Y.; Orr, H.T. Purkinje Cell Expression of a Mutant Allele of SCA1 in Transgenic Mice Leads to Disparate Effects on Motor Behaviors, Followed by a Progressive Cerebellar Dysfunction and Histological Alterations. J. Neurosci. 1997, 17, 7385–7395. [Google Scholar] [CrossRef]
- Burk, K.; Globas, C.; Bosch, S.; Klockgether, T.; Zuhlke, C.; Daum, I.; Dichgans, J. Cognitive deficits in spinocerebellar ataxia type 1, 2, and 3. J. Neurol. 2003, 250, 207–211. [Google Scholar] [CrossRef]
- Tichanek, F.; Salomova, M.; Jedlicka, J.; Kuncova, J.; Pitule, P.; Macanova, T.; Petrankova, Z.; Tuma, Z.; Cendelin, J. Hippocampal mitochondrial dysfunction and psychiatric-relevant behavioral deficits in spinocerebellar ataxia 1 mouse model. Sci. Rep. 2020, 10, 5418. [Google Scholar] [CrossRef]
- Asher, M.; Rosa, J.-G.; Rainwater, O.; Duvick, L.; Bennyworth, M.; Lai, R.-Y.; Kuo, S.-H.; Cvetanovic, M.; Sca, C. Cerebellar contribution to the cognitive alterations in SCA1: Evidence from mouse models. Hum. Mol. Genet. 2019, 29, 117–131. [Google Scholar] [CrossRef]
- Fire, A.; Xu, S.; Montgomery, M.K.; Kostas, S.A.; Driver, S.E.; Mello, C.C. Potent and specific genetic interference by double-stranded RNA in Caenorhabditis elegans. Nature 1998, 391, 806–811. [Google Scholar] [CrossRef]
- Elbashir, S.M.; Harborth, J.; Lendeckel, W.; Yalcin, A.; Weber, K.; Tuschl, T. Duplexes of 21-nucleotide RNAs mediate RNA interference in cultured mammalian cells. Nature 2001, 411, 494–498. [Google Scholar] [CrossRef]
- Vázquez-Mojena, Y.; León-Arcia, K.; González-Zaldivar, Y.; Rodríguez-Labrada, R.; Velázquez-Pérez, L. Gene Therapy for Polyglutamine Spinocerebellar Ataxias: Advances, Challenges, and Perspectives. Mov. Disord. 2021, 36, 2731–2744. [Google Scholar] [CrossRef]
- Xia, H.; Mao, Q.; Eliason, S.L.; Harper, S.Q.; Martins, I.H.; Orr, H.T.; Paulson, H.L.; Yang, L.; Kotin, R.M.; Davidson, B.L. RNAi suppresses polyglutamine-induced neurodegeneration in a model of spinocerebellar ataxia. Nat. Med. 2004, 10, 816–820. [Google Scholar] [CrossRef]
- Keiser, M.S.; Kordasiewicz, H.B.; McBride, J.L. Gene suppression strategies for dominantly inherited neurodegenerative diseases: Lessons from Huntington’s disease and spinocerebellar ataxia. Hum. Mol. Genet. 2015, 25, R53–R64. [Google Scholar] [CrossRef]
- Keiser, M.S.; Geoghegan, J.C.; Boudreau, R.L.; Lennox, K.A.; Davidson, B.L. RNAi or overexpression: Alternative therapies for Spinocerebellar Ataxia Type 1. Neurobiol. Dis. 2013, 56, 6–13. [Google Scholar] [CrossRef]
- Keiser, M.S.; Boudreau, R.; Davidson, B.L. Broad Therapeutic Benefit After RNAi Expression Vector Delivery to Deep Cerebellar Nuclei: Implications for Spinocerebellar Ataxia Type 1 Therapy. Mol. Ther. 2014, 22, 588–595. [Google Scholar] [CrossRef]
- Keiser, M.S.; Monteys, A.M.; Corbau, R.; Gonzalez-Alegre, P.; Davidson, B.L. RNAi prevents and reverses phenotypes induced by mutant human ataxin-1. Ann. Neurol. 2016, 80, 754–765. [Google Scholar] [CrossRef]
- Keiser, M.S.; Kordower, J.H.; Gonzalez-Alegre, P.; Davidson, B.L. Broad distribution of ataxin 1 silencing in rhesus cerebella for spinocerebellar ataxia type 1 therapy. Brain 2015, 138, 3555–3566. [Google Scholar] [CrossRef]
- Keiser, M.S.; Ranum, P.T.; Yrigollen, C.M.; Carrell, E.M.; Smith, G.R.; Muehlmatt, A.L.; Chen, Y.H.; Stein, J.M.; Wolf, R.L.; Radaelli, E.; et al. Toxicity after AAV delivery of RNAi expression constructs into nonhuman primate brain. Nat. Med. 2021, 27, 1982–1989. [Google Scholar] [CrossRef]
- Friedrich, J.; Kordasiewicz, H.B.; O’Callaghan, B.L.; Handler, H.P.; Wagener, C.; Duvick, L.; Swayze, E.; Rainwater, O.; Hofstra, B.; Benneyworth, M.; et al. Antisense oligonucleotide–mediated ataxin-1 reduction prolongs survival in SCA1 mice and reveals disease-associated transcriptome profiles. J. Clin. Investig. 2018, 3, e123193. [Google Scholar] [CrossRef] [PubMed]
- O’callaghan, B.; Hofstra, B.; Handler, H.P.; Kordasiewicz, H.B.; Cole, T.; Duvick, L.; Friedrich, J.; Rainwater, O.; Yang, P.; Benneyworth, M.; et al. Antisense Oligonucleotide Therapeutic Approach for Suppression of Ataxin-1 Expression: A Safety Assessment. Mol. Ther.-Nucleic Acids 2020, 21, 1006–1016. [Google Scholar] [CrossRef]
- Kourkouta, E.; Weij, R.; González-Barriga, A.; Mulder, M.; Verheul, R.; Bosgra, S.; Groenendaal, B.; Puoliväli, J.; Toivanen, J.; van Deutekom, J.C.; et al. Suppression of Mutant Protein Expression in SCA3 and SCA1 Mice Using a CAG Repeat-Targeting Antisense Oligonucleotide. Mol. Ther.-Nucleic Acids 2019, 17, 601–614. [Google Scholar] [CrossRef] [PubMed]
- Carrell, E.M.; Keiser, M.S.; Robbins, A.B.; Davidson, B.L. Combined overexpression of ATXN1L and mutant ATXN1 knockdown by AAV rescue motor phenotypes and gene signatures in SCA1 mice. Mol. Ther.-Methods Clin. Dev. 2022, 25, 333–343. [Google Scholar] [CrossRef] [PubMed]
- Hammond, S.M.; Aartsma-Rus, A.; Alves, S.; Borgos, S.E.; Buijsen, R.A.M.; Collin, R.W.J.; Covello, G.; Denti, M.A.; Desviat, L.R.; Echevarría, L.; et al. Delivery of oligonucleotide-based therapeutics: Challenges and opportunities. EMBO Mol. Med. 2021, 13, e13243. [Google Scholar] [CrossRef]
- Quemener, A.M.; Bachelot, L.; Forestier, A.; Donnou-Fournet, E.; Gilot, D.; Galibert, M.-D. The powerful world of antisense oligonucleotides: From bench to bedside. Wiley Interdiscip. Rev. RNA 2020, 11, e1594. [Google Scholar] [CrossRef]
- Silva, A.C.; Lobo, D.D.; Martins, I.; Lopes, S.M.; Henriques, C.; Duarte, S.P.; Dodart, J.-C.; Nobre, R.J.; De Almeida, L.P. Antisense oligonucleotide therapeutics in neurodegenerative diseases: The case of polyglutamine disorders. Brain 2019, 143, 407–429. [Google Scholar] [CrossRef]
- Ashizawa, A.T.; Holt, J.; Faust, K.; Liu, W.; Tiwari, A.; Zhang, N.; Ashizawa, T. Intravenously Administered Novel Liposomes, DCL64, Deliver Oligonucleotides to Cerebellar Purkinje Cells. Cerebellum 2018, 18, 99–108. [Google Scholar] [CrossRef]
- Kozlu, S.; Caban, S.; Yerlikaya, F.; Fernandez-Megia, E.; Novoa-Carballal, R.; Riguera, R.; Yemisci, M.; Gürsoy-Ozdemir, Y.; Dalkara, T.; Couvreur, P.; et al. An aquaporin 4 antisense oligonucleotide loaded, brain targeted nanoparticulate system design. Die Pharm. 2014, 69, 340–345. [Google Scholar]
- Du, L.; Kayali, R.; Bertoni, C.; Fike, F.; Hu, H.; Iversen, P.L.; Gatti, R.A. Arginine-rich cell-penetrating peptide dramatically enhances AMO-mediated ATM aberrant splicing correction and enables delivery to brain and cerebellum. Hum. Mol. Genet. 2011, 20, 3151–3160. [Google Scholar] [CrossRef]
- Zeniya, S.; Kuwahara, H.; Daizo, K.; Watari, A.; Kondoh, M.; Yoshida-Tanaka, K.; Kaburagi, H.; Asada, K.; Nagata, T.; Nagahama, M.; et al. Angubindin-1 opens the blood–brain barrier in vivo for delivery of antisense oligonucleotide to the central nervous system. J. Control. Release 2018, 283, 126–134. [Google Scholar] [CrossRef]
- Miller, T.M.; Cudkowicz, M.E.; Genge, A.; Shaw, P.J.; Sobue, G.; Bucelli, R.C.; Chiò, A.; Van Damme, P.; Ludolph, A.C.; Glass, J.D.; et al. Trial of Antisense Oligonucleotide Tofersen for SOD1 ALS. N. Engl. J. Med. 2022, 387, 1099–1110. [Google Scholar] [CrossRef]
- Acsadi, G.; Crawford, T.O.; Müller-Felber, W.; Shieh, P.B.; Richardson, R.; Natarajan, N.; Castro, D.; Ramirez-Schrempp, D.; Gambino, G.; Sun, P.; et al. Safety and efficacy of nusinersen in spinal muscular atrophy: The EMBRACE study. Muscle Nerve 2021, 63, 668–677. [Google Scholar] [CrossRef]
- Evers, M.M.; Pepers, B.A.; Van Deutekom, J.C.T.; Mulders, S.A.M.; Dunnen, J.T.D.; Aartsma-Rus, A.; Van Ommen, G.-J.B.; Van Roon-Mom, W.M.C. Targeting Several CAG Expansion Diseases by a Single Antisense Oligonucleotide. PLoS ONE 2011, 6, e24308. [Google Scholar] [CrossRef]
- Suh, J.; Romano, D.M.; Nitschke, L.; Herrick, S.P.; DiMarzio, B.A.; Dzhala, V.; Bae, J.-S.; Oram, M.K.; Zheng, Y.; Hooli, B.; et al. Loss of Ataxin-1 Potentiates Alzheimer’s Pathogenesis by Elevating Cerebral BACE1 Transcription. Cell 2019, 178, 1159–1175.e17. [Google Scholar] [CrossRef]
- Alves, S.; Nascimento-Ferreira, I.; Auregan, G.; Hassig, R.; Dufour, N.; Brouillet, E.; de Lima, M.P.; Hantraye, P.; de Almeida, L.P.; Déglon, N. Allele-Specific RNA Silencing of Mutant Ataxin-3 Mediates Neuroprotection in a Rat Model of Machado-Joseph Disease. PLoS ONE 2008, 3, e3341. [Google Scholar] [CrossRef]
- Scholefield, J.; Greenberg, L.J.; Weinberg, M.; Arbuthnot, P.B.; Abdelgany, A.; Wood, M.J.A. Design of RNAi Hairpins for Mutation-Specific Silencing of Ataxin-7 and Correction of a SCA7 Phenotype. PLoS ONE 2009, 4, e7232. [Google Scholar] [CrossRef]
- Kubodera, T.; Yokota, T.; Ishikawa, K.; Mizusawa, H. New RNAi Strategy for Selective Suppression of a Mutant Allele in Polyglutamine Disease. Oligonucleotides 2005, 15, 298–302. [Google Scholar] [CrossRef]
- Lee, W.; Lavery, L.; Rousseaux, M.W.C.; Rutledge, E.B.; Jang, Y.; Wan, Y.; Wu, S.; Kim, W.; Al-Ramahi, I.; Rath, S.; et al. Dual targeting of brain region-specific kinases potentiates neurological rescue in Spinocerebellar ataxia type 1. EMBO J. 2021, 40, e106106. [Google Scholar] [CrossRef]
- Emamian, E.S.; Kaytor, M.D.; Duvick, L.A.; Zu, T.; Tousey, S.K.; Zoghbi, H.; Clark, H.; Orr, H.T. Serine 776 of Ataxin-1 Is Critical for Polyglutamine-Induced Disease in SCA1 Transgenic Mice. Neuron 2003, 38, 375–387. [Google Scholar] [CrossRef]
- Hourez, R.; Servais, L.; Orduz, D.; Gall, D.; Millard, I.; D’Exaerde, A.d.K.; Cheron, G.; Orr, H.T.; Pandolfo, M.; Schiffmann, S.N. Aminopyridines Correct Early Dysfunction and Delay Neurodegeneration in a Mouse Model of Spinocerebellar Ataxia Type 1. J. Neurosci. 2011, 31, 11795–11807. [Google Scholar] [CrossRef] [PubMed]
- Chopra, R.; Bushart, D.D.; Shakkottai, V.G. Dendritic potassium channel dysfunction may contribute to dendrite degeneration in spinocerebellar ataxia type 1. PLoS ONE 2018, 13, e0198040. [Google Scholar] [CrossRef] [PubMed]
- Bushart, D.; Chopra, R.; Singh, V.; Murphy, G.G.; Wulff, H.; Shakkottai, V.G. Targeting potassium channels to treat cerebellar ataxia. Ann. Clin. Transl. Neurol. 2018, 5, 297–314. [Google Scholar] [CrossRef] [PubMed]
- Ruegsegger, C.; Stucki, D.M.; Steiner, S.; Angliker, N.; Radecke, J.; Keller, E.; Zuber, B.; Rüegg, M.A.; Saxena, S. Impaired mTORC1-Dependent Expression of Homer-3 Influences SCA1 Pathophysiology. Neuron 2016, 89, 129–146. [Google Scholar] [CrossRef] [PubMed]
- Lin, X.; Antalffy, B.; Kang, D.; Orr, H.T.; Zoghbi, H.Y. Polyglutamine expansion down-regulates specific neuronal genes before pathologic changes in SCA1. Nat. Neurosci. 2000, 3, 157–163. [Google Scholar] [CrossRef]
- Barnes, J.A.; Ebner, B.A.; Duvick, L.A.; Gao, W.; Chen, G.; Orr, H.T.; Ebner, T.J. Abnormalities in the Climbing Fiber-Purkinje Cell Circuitry Contribute to Neuronal Dysfunction in ATXN1[82Q] Mice. J. Neurosci. 2011, 31, 12778–12789. [Google Scholar] [CrossRef]
- Edamakanti, C.R.; Do, J.; Didonna, A.; Martina, M.; Opal, P. Mutant ataxin1 disrupts cerebellar development in spinocerebellar ataxia type 1. J. Clin. Investig. 2018, 128, 2252–2265. [Google Scholar] [CrossRef]
- Dell’Orco, J.M.; Wasserman, A.; Chopra, R.; Ingram, M.A.C.; Hu, Y.-S.; Singh, V.; Wulff, H.; Opal, P.; Orr, H.; Shakkottai, V.G. Neuronal Atrophy Early in Degenerative Ataxia Is a Compensatory Mechanism to Regulate Membrane Excitability. J. Neurosci. 2015, 35, 11292–11307. [Google Scholar] [CrossRef]
- Bushart, D.D.; Huang, H.; Man, L.J.; Bs, L.M.M.; Shakkottai, V.G. A Chlorzoxazone-Baclofen Combination Improves Cerebellar Impairment in Spinocerebellar Ataxia Type 1. Mov. Disord. 2020, 36, 622–631. [Google Scholar] [CrossRef]
- Notartomaso, S.; Zappulla, C.; Biagioni, F.; Cannella, M.; Bucci, D.; Mascio, G.; Scarselli, P.; Fazio, F.; Weisz, F.; Lionetto, L.; et al. Pharmacological enhancement of mGlu1 metabotropic glutamate receptors causes a prolonged symptomatic benefit in a mouse model of spinocerebellar ataxia type 1. Mol. Brain 2013, 6, 48. [Google Scholar] [CrossRef]
- Mitsumura, K.; Hosoi, N.; Furuya, N.; Hirai, H. Disruption of metabotropic glutamate receptor signalling is a major defect at cerebellar parallel fibre-Purkinje cell synapses in staggerer mutant mice. J. Physiol. 2011, 589, 3191–3209. [Google Scholar] [CrossRef]
- Shuvaev, A.N.; Hosoi, N.; Sato, Y.; Yanagihara, D.; Hirai, H. Progressive impairment of cerebellar mGluR signalling and its therapeutic potential for cerebellar ataxia in spinocerebellar ataxia type 1 model mice. J. Physiol. 2016, 595, 141–164. [Google Scholar] [CrossRef]
- Liberatore, F.; Antenucci, N.; Tortolani, D.; Mascio, G.; Fanti, F.; Sergi, M.; Battaglia, G.; Bruno, V.; Nicoletti, F.; Maccarrone, M.; et al. Targeting mGlu1 Receptors in the Treatment of Motor and Cognitive Dysfunctions in Mice Modeling Type 1 Spinocerebellar Ataxia. Cells 2022, 11, 3916. [Google Scholar] [CrossRef]
- Power, E.M.; Morales, A.; Empson, R.M. Prolonged Type 1 Metabotropic Glutamate Receptor Dependent Synaptic Signaling Contributes to Spino-Cerebellar Ataxia Type 1. J. Neurosci. 2016, 36, 4910–4916. [Google Scholar] [CrossRef]
- Serra, H.G.; Byam, C.E.; Lande, J.D.; Tousey, S.K.; Zoghbi, H.Y.; Orr, H.T. Gene profiling links SCA1 pathophysiology to glutamate signaling in Purkinje cells of transgenic mice. Hum. Mol. Genet. 2004, 13, 2535–2543. [Google Scholar] [CrossRef]
- Nag, N.; Tarlac, V.; Storey, E. Assessing the Efficacy of Specific Cerebellomodulatory Drugs for Use as Therapy for Spinocerebellar Ataxia Type 1. Cerebellum 2012, 12, 74–82. [Google Scholar] [CrossRef]
- Fick, D.M.; Semla, T.P.; Steinman, M.; Beizer, J.; Brandt, N.; Dombrowski, R. American Geriatrics Society 2019 Updated AGS Beers Criteria® for Potentially Inappropriate Medication Use in Older Adults. J. Am. Geriatr. Soc. 2019, 67, 674–694. [Google Scholar] [CrossRef]
- Iizuka, A.; Nakamura, K.; Hirai, H. Long-term oral administration of the NMDA receptor antagonist memantine extends life span in spinocerebellar ataxia type 1 knock-in mice. Neurosci. Lett. 2015, 592, 37–41. [Google Scholar] [CrossRef]
- Vig, P.; Subramony, S.; D’Souza, D.; Wei, J.; Lopez, M. Intranasal administration of IGF-I improves behavior and Purkinje cell pathology in SCA1 mice. Brain Res. Bull. 2006, 69, 573–579. [Google Scholar] [CrossRef]
- Cvetanovic, M.; Patel, J.; Marti, H.; Kini, A.R.; Opal, P. Vascular endothelial growth factor ameliorates the ataxic phenotype in a mouse model of spinocerebellar ataxia type 1. Nat. Med. 2011, 17, 1445–1447. [Google Scholar] [CrossRef]
- Hu, Y.-S.; Do, J.; Edamakanti, C.R.; Kini, A.R.; Martina, M.; Stupp, S.I.; Opal, P. Self-assembling vascular endothelial growth factor nanoparticles improve function in spinocerebellar ataxia type 1. Brain 2019, 142, 312–321. [Google Scholar] [CrossRef] [PubMed]
- Rosa, J.-G.; Hamel, K.; Soles, A.; Sheeler, C.; Borgenheimer, E.; Gilliat, S.; Sbrocco, K.; Ghanoum, F.; Handler, H.P.; Forster, C.; et al. BDNF is altered in a brain-region specific manner and rescues deficits in Spinocerebellar Ataxia Type 1. Neurobiol. Dis. 2023, 178, 106023. [Google Scholar] [CrossRef] [PubMed]
- Watase, K.; Gatchel, J.; Sun, Y.; Emamian, E.; Atkinson, R.; Richman, R.; Mizusawa, H.; Orr, H.; Shaw, C.; Zoghbi, H.Y. Lithium Therapy Improves Neurological Function and Hippocampal Dendritic Arborization in a Spinocerebellar Ataxia Type 1 Mouse Model. PLoS Med. 2007, 4, e182. [Google Scholar] [CrossRef] [PubMed]
- Perroud, B.; Jafar-Nejad, P.; Wikoff, W.R.; Gatchel, J.R.; Wang, L.; Barupal, D.K.; Crespo-Barreto, J.; Fiehn, O.; Zoghbi, H.Y.; Kaddurah-Daouk, R. Pharmacometabolomic Signature of Ataxia SCA1 Mouse Model and Lithium Effects. PLoS ONE 2013, 8, e70610. [Google Scholar] [CrossRef]
- Stucki, D.M.; Ruegsegger, C.; Steiner, S.; Radecke, J.; Murphy, M.P.; Zuber, B.; Saxena, S. Mitochondrial impairments contribute to Spinocerebellar ataxia type 1 progression and can be ameliorated by the mitochondria-targeted antioxidant MitoQ. Free. Radic. Biol. Med. 2016, 97, 427–440. [Google Scholar] [CrossRef]
- Ferro, A.; Carbone, E.; Zhang, J.; Marzouk, E.; Villegas, M.; Siegel, A.; Nguyen, D.; Possidente, T.; Hartman, J.; Polley, K.; et al. Short-term succinic acid treatment mitigates cerebellar mitochondrial OXPHOS dysfunction, neurodegeneration and ataxia in a Purkinje-specific spinocerebellar ataxia type 1 (SCA1) mouse model. PLoS ONE 2017, 12, e0188425. [Google Scholar] [CrossRef]
- Ito, H.; Fujita, K.; Tagawa, K.; Chen, X.; Homma, H.; Sasabe, T.; Shimizu, J.; Shimizu, S.; Tamura, T.; Muramatsu, S.; et al. HMGB 1 facilitates repair of mitochondrial DNA damage and extends the lifespan of mutant ataxin-1 knock-in mice. EMBO Mol. Med. 2014, 7, 78–101. [Google Scholar] [CrossRef]
- Taniguchi, J.B.; Kondo, K.; Fujita, K.; Chen, X.; Homma, H.; Sudo, T.; Mao, Y.; Watase, K.; Tanaka, T.; Tagawa, K.; et al. RpA1 ameliorates symptoms of mutant ataxin-1 knock-in mice and enhances DNA damage repair. Hum. Mol. Genet. 2016, 25, 4432–4447. [Google Scholar] [CrossRef]
- Bondar, V.V.; Adamski, C.J.; Onur, T.S.; Tan, Q.; Wang, L.; Diaz-Garcia, J.; Park, J.; Orr, H.T.; Botas, J.; Zoghbi, H.Y. PAK1 regulates ATXN1 levels providing an opportunity to modify its toxicity in spinocerebellar ataxia type 1. Hum. Mol. Genet. 2018, 27, 2863–2873. [Google Scholar] [CrossRef]
- Qu, W.; Johnson, A.; Kim, J.H.; Lukowicz, A.; Svedberg, D.; Cvetanovic, M. Inhibition of colony-stimulating factor 1 receptor early in disease ameliorates motor deficits in SCA1 mice. J. Neuroinflamm. 2017, 14, 107. [Google Scholar] [CrossRef]
- Fernandez, A.; Carro, E.; Lopez-Lopez, C.; Torres-Aleman, I. Insulin-like growth factor I treatment for cerebellar ataxia: Addressing a common pathway in the pathological cascade? Brain Res. Rev. 2005, 50, 134–141. [Google Scholar] [CrossRef]
- Sheeler, C.; Rosa, J.-G.; Borgenheimer, E.; Mellesmoen, A.; Rainwater, O.; Cvetanovic, M. Post-symptomatic Delivery of Brain-Derived Neurotrophic Factor (BDNF) Ameliorates Spinocerebellar Ataxia Type 1 (SCA1) Pathogenesis. Cerebellum 2021, 20, 420–429. [Google Scholar] [CrossRef]
- Mellesmoen, A.; Sheeler, C.; Ferro, A.; Rainwater, O.; Cvetanovic, M. Brain Derived Neurotrophic Factor (BDNF) Delays Onset of Pathogenesis in Transgenic Mouse Model of Spinocerebellar Ataxia Type 1 (SCA1). Front. Cell. Neurosci. 2019, 12, 509. [Google Scholar] [CrossRef]
- Chen, G.; Zeng, W.-Z.; Yuan, P.-X.; Huang, L.-D.; Jiang, Y.-M.; Zhao, Z.-H.; Manji, H.K. The Mood-Stabilizing Agents Lithium and Valproate RobustlIncrease the Levels of the Neuroprotective Protein bcl-2 in the CNS. J. Neurochem. 1999, 72, 879–882. [Google Scholar] [CrossRef]
- Jope, R.S. Lithium and GSK-3: One inhibitor, two inhibitory actions, multiple outcomes. Trends Pharmacol. Sci. 2003, 24, 441–443. [Google Scholar] [CrossRef]
- Barclay, S.S.; Tamura, T.; Ito, H.; Fujita, K.; Tagawa, K.; Shimamura, T.; Katsuta, A.; Shiwaku, H.; Sone, M.; Imoto, S.; et al. Systems biology analysis of Drosophila in vivo screen data elucidates core networks for DNA damage repair in SCA1. Hum. Mol. Genet. 2013, 23, 1345–1364. [Google Scholar] [CrossRef]
- Öz, G.; Hutter, D.; Tkáč, I.; Clark, H.B.; Gross, M.D.; Jiang, H.; Eberly, L.E.; Bushara, K.O.; Gomez, C.M. Neurochemical alterations in spinocerebellar ataxia type 1 and their correlations with clinical status. Mov. Disord. 2010, 25, 1253–1261. [Google Scholar] [CrossRef]
- Maciel, P.; Correia, J.; Duarte-Silva, S.; Salgado, A. Cell-based therapeutic strategies for treatment of spinocerebellar ataxias: An update. Neural Regen. Res. 2023, 18, 1203. [Google Scholar] [CrossRef]
- Huda, F.; Fan, Y.; Suzuki, M.; Konno, A.; Matsuzaki, Y.; Takahashi, N.; Chan, J.K.Y.; Hirai, H. Fusion of Human Fetal Mesenchymal Stem Cells with “Degenerating” Cerebellar Neurons in Spinocerebellar Ataxia Type 1 Model Mice. PLoS ONE 2016, 11, e0164202. [Google Scholar] [CrossRef]
- Matsuura, S.; Shuvaev, A.N.; Iizuka, A.; Nakamura, K.; Hirai, H. Mesenchymal Stem Cells Ameliorate Cerebellar Pathology in a Mouse Model of Spinocerebellar Ataxia Type 1. Cerebellum 2013, 13, 323–330. [Google Scholar] [CrossRef]
- Tsai, P.-J.; Yeh, C.-C.; Huang, W.-J.; Min, M.-Y.; Huang, T.-H.; Ko, T.-L.; Huang, P.-Y.; Chen, T.-H.; Hsu, S.P.C.; Soong, B.-W.; et al. Xenografting of human umbilical mesenchymal stem cells from Wharton’s jelly ameliorates mouse spinocerebellar ataxia type 1. Transl. Neurodegener. 2019, 8, 29. [Google Scholar] [CrossRef] [PubMed]
- Suto, N.; Mieda, T.; Iizuka, A.; Nakamura, K.; Hirai, H. Morphological and Functional Attenuation of Degeneration of Peripheral Neurons by Mesenchymal Stem Cell-Conditioned Medium in Spinocerebellar Ataxia Type 1-Knock-in Mice. CNS Neurosci. Ther. 2016, 22, 670–676. [Google Scholar] [CrossRef] [PubMed]
- Chintawar, S.; Hourez, R.; Ravella, A.; Gall, D.; Orduz, D.; Rai, M.; Bishop, D.P.; Geuna, S.; Schiffmann, S.N.; Pandolfo, M. Grafting Neural Precursor Cells Promotes Functional Recovery in an SCA1 Mouse Model. J. Neurosci. 2009, 29, 13126–13135. [Google Scholar] [CrossRef] [PubMed]
- Kaemmerer, W.F.; Low, W.C. Cerebellar Allografts Survive and Transiently Alleviate Ataxia in a Transgenic Model of Spinocerebellar Ataxia Type-1. Exp. Neurol. 1999, 158, 301–311. [Google Scholar] [CrossRef] [PubMed]
- Lige, L.; Zengmin, T. Transplantation of neural precursor cells in the treatment for parkinson disease: An efficacy and safety analysis. Turk. Neurosurg. 2015, 26, 378–383. [Google Scholar] [CrossRef]
- Dongmei, H.; Jing, L.; Mei, X.; Ling, Z.; Hongmin, Y.; Zhidong, W.; Li, D.; Zikuan, G.; Hengxiang, W. Clinical analysis of the treatment of spinocerebellar ataxia and multiple system atrophy-cerebellar type with umbilical cord mesenchymal stromal cells. Cytotherapy 2011, 13, 913–917. [Google Scholar] [CrossRef]
- Jin, J.-L.; Liu, Z.; Lu, Z.-J.; Guan, D.-N.; Wang, C.; Chen, Z.-B.; Zhang, J.; Zhang, W.-Y.; Wu, J.-Y.; Xu, Y. Safety and Efficacy of Umbilical Cord Mesenchymal Stem Cell Therapy in Hereditary Spinocerebellar Ataxia. Curr. Neurovascular Res. 2013, 10, 11–20. [Google Scholar] [CrossRef]
- Giordano, I.; Bogdanow, M.; Jacobi, H.; Jahn, K.; Minnerop, M.; Schoels, L.; Synofzik, M.; Teufel, J.; Klockgether, T. Experience in a short-term trial with 4-Aminopyridine in cerebellar ataxia. J. Neurol. 2013, 260, 2175–2176. [Google Scholar] [CrossRef]
- Schniepp, R.; Wuehr, M.; Neuhaeusser, M.; Benecke, A.K.; Adrion, C.; Brandt, T.; Strupp, M.; Jahn, K. 4-Aminopyridine and cerebellar gait: A retrospective case series. J. Neurol. 2012, 259, 2491–2493. [Google Scholar] [CrossRef]
- Wagner, J.L.; O’Connor, D.M.; Donsante, A.; Boulis, N.M. Gene, Stem Cell, and Alternative Therapies for SCA 1. Front. Mol. Neurosci. 2016, 9, 67. [Google Scholar] [CrossRef]
- Gala, D.; Scharf, S.; Kudlak, M.; Green, C.; Khowaja, F.; Shah, M.; Kumar, V.; Ullal, G. A Comprehensive Review of the Neurological Manifestations of Celiac Disease and Its Treatment. Diseases 2022, 10, 111. [Google Scholar] [CrossRef]
- Ristori, G.; Romano, S.; Visconti, A.; Cannoni, S.; Spadaro, M.; Frontali, M.; Pontieri, F.E.; Vanacore, N.; Salvetti, M. Riluzole in cerebellar ataxia: A randomized, double-blind, placebo-controlled pilot trial. Neurology 2010, 74, 839–845. [Google Scholar] [CrossRef]
- Romano, S.; Coarelli, G.; Marcotulli, C.; Leonardi, L.; Piccolo, F.; Spadaro, M.; Frontali, M.; Ferraldeschi, M.; Vulpiani, M.C.; Ponzelli, F.; et al. Riluzole in patients with hereditary cerebellar ataxia: A randomised, double-blind, placebo-controlled trial. Lancet Neurol. 2015, 14, 985–991. [Google Scholar] [CrossRef]
- Coarelli, G.; Heinzmann, A.; Ewenczyk, C.; Fischer, C.; Chupin, M.; Monin, M.-L.; Hurmic, H.; Calvas, F.; Calvas, P.; Goizet, C.; et al. Safety and efficacy of riluzole in spinocerebellar ataxia type 2 in France (ATRIL): A multicentre, randomised, double-blind, placebo-controlled trial. Lancet Neurol. 2022, 21, 225–233. [Google Scholar] [CrossRef]
- Schniepp, R.; Möhwald, K.; Wuehr, M. Gait ataxia in humans: Vestibular and cerebellar control of dynamic stability. J. Neurol. 2017, 264, 87–92. [Google Scholar] [CrossRef]
- Appelt, P.A.; Comella, K.; de Souza, L.A.P.S.; Luvizutto, G.J. Effect of stem cell treatment on functional recovery of spinocerebellar ataxia: Systematic review and meta-analysis. Cerebellum Ataxias 2021, 8, 8. [Google Scholar] [CrossRef]
- Hommersom, M.P.; Buijsen, R.A.M.; van Roon-Mom, W.M.C.; van de Warrenburg, B.P.C.; van Bokhoven, H. Human Induced Pluripotent Stem Cell-Based Modelling of Spinocerebellar Ataxias. Stem Cell Rev. Rep. 2021, 18, 441–456. [Google Scholar] [CrossRef]
- Olmos, V.; Gogia, N.; Luttik, K.; Haidery, F.; Lim, J. The extra-cerebellar effects of spinocerebellar ataxia type 1 (SCA1): Looking beyond the cerebellum. Cell. Mol. Life Sci. 2022, 79, 404. [Google Scholar] [CrossRef]
- Buijsen, R.A.; Gardiner, S.L.; Bouma, M.J.; van der Graaf, L.M.; Boogaard, M.W.; Pepers, B.A.; Eussen, B.; de Klein, A.; Freund, C.; van Roon-Mom, W.M. Generation of 3 spinocerebellar ataxia type 1 (SCA1) patient-derived induced pluripotent stem cell lines LUMCi002-A, B, and C and 2 unaffected sibling control induced pluripotent stem cell lines LUMCi003-A and B. Stem Cell Res. 2018, 29, 125–128. [Google Scholar] [CrossRef]
- Rubinsztein, D.C.; Orr, H. Diminishing return for mechanistic therapeutics with neurodegenerative disease duration? There may be a point in the course of a neurodegenerative condition where therapeutics targeting disease-causing mechanisms are futile. Bioessays 2016, 38, 977–980. [Google Scholar] [CrossRef]
- Zu, T.; Duvick, L.A.; Kaytor, M.D.; Berlinger, M.S.; Zoghbi, H.Y.; Clark, H.B.; Orr, H.T. Recovery from Polyglutamine-Induced Neurodegeneration in Conditional SCA1 Transgenic Mice. J. Neurosci. 2004, 24, 8853–8861. [Google Scholar] [CrossRef] [PubMed]
- Ibrahim, M.F.; Power, E.M.; Potapov, K.; Empson, R.M. Motor and Cerebellar Architectural Abnormalities during the Early Progression of Ataxia in a Mouse Model of SCA1 and How Early Prevention Leads to a Better Outcome Later in Life. Front. Cell. Neurosci. 2017, 11, 292. [Google Scholar] [CrossRef] [PubMed]
- Brooker, S.M.; Edamakanti, C.R.; Akasha, S.M.; Kuo, S.; Opal, P. Spinocerebellar ataxia clinical trials: Opportunities and challenges. Ann. Clin. Transl. Neurol. 2021, 8, 1543–1556. [Google Scholar] [CrossRef] [PubMed]
- van Prooije, T.; Ruigrok, S.; Berkmortel, N.v.D.; Maas, R.P.P.W.M.; Wijn, S.; van Roon-Mom, W.M.C.; van de Warrenburg, B.; Grutters, J.P.C. The potential value of disease-modifying therapy in patients with spinocerebellar ataxia type 1: An early health economic modeling study. J. Neurol. 2023; Online ahead of print. [Google Scholar] [CrossRef]
- Aartsma-Rus, A.; van Roon-Mom, W.; Lauffer, M.; Siezen, C.; Duijndam, B.; Coenen-de Roo, T.; Schüle, R.; Synofzik, M.; Graessner, H. Development of tailored splice switching oligonucleotides for progressive brain disorders in Europe: Development, regulation and implementation considerations. RNA 2023, 29, 446–454. [Google Scholar] [CrossRef]
- Pizzamiglio, C.; Vernon, H.J.; Hanna, M.G.; Pitceathly, R.D.S. Designing clinical trials for rare diseases: Unique challenges and opportunities. Nat. Rev. Methods Prim. 2022, 2, 13. [Google Scholar] [CrossRef]
- Tabrizi, S.J.; Schobel, S.; Gantman, E.C.; Mansbach, A.; Borowsky, B.; Konstantinova, P.; Mestre, T.A.; Panagoulias, J.; Ross, C.A.; Zauderer, M.; et al. A biological classification of Huntington’s disease: The Integrated Staging System. Lancet Neurol. 2022, 21, 632–644. [Google Scholar] [CrossRef]
- Diallo, A.; Jacobi, H.; du Montcel, S.T.; Klockgether, T. Natural history of most common spinocerebellar ataxia: A systematic review and meta-analysis. J. Neurol. 2020, 268, 2749–2756. [Google Scholar] [CrossRef]
- Wilke, C.; Mengel, D.; Schöls, L.; Hengel, H.; Rakowicz, M.; Klockgether, T.; Durr, A.; Filla, A.; Melegh, B.; Schüle, R.; et al. Levels of Neurofilament Light at the Preataxic and Ataxic Stages of Spinocerebellar Ataxia Type 1. Neurology 2022, 98, e1985–e1996. [Google Scholar] [CrossRef]
- Lin, C.-C.; Ashizawa, T.; Kuo, S.-H. Collaborative Efforts for Spinocerebellar Ataxia Research in the United States: CRC-SCA and READISCA. Front. Neurol. 2020, 11, 902. [Google Scholar] [CrossRef]
- Pappadà, M.; Bonuccelli, O.; Buratto, M.; Fontana, R.; Sicurella, M.; Caproni, A.; Fuselli, S.; Benazzo, A.; Bertorelli, R.; De Sanctis, V.; et al. Suppressing gain-of-function proteins via CRISPR/Cas9 system in SCA1 cells. Sci. Rep. 2022, 12, 20285. [Google Scholar] [CrossRef]
- Uddin, F.; Rudin, C.M.; Sen, T. CRISPR Gene Therapy: Applications, Limitations, and Implications for the Future. Front. Oncol. 2020, 10, 1387. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| SCA1 Animal Model | Genetic Background | Advantages | Disadvantages |
|---|---|---|---|
| SCA1 transgenic B05 mice | Human ATXN1 gene containing 82 CAG repeats under control of a pcp2 promoter | Mimics cerebellar component of SCA1, including ataxia and classical neuropathological changes. Contains human ATXN1. | Expression only in the PCs of the cerebellum. Has 50 to 100 times overexpression of ATXN1 compared with endogenous levels [27,48]. |
| SCA1 knock-in mice (SCA1154Q/2Q) | 154 CAG repeats in the mouse Sca1 locus under control of endogenous promoters | Expresses endogenous Atxn1 levels. Shows development of slow progressive neurodegeneration, ataxia, and deficits in memory (reflecting extra-cerebellar pathology). | Nuclear inclusions were found in more brain regions in the mice than were found in humans. Model contains very long CAG repeats, not in the range of SCA1 patients [28]. |
| SCA1 Drosophila melanogaster (fly) model | Expresses human ATXN1 gene containing 82Q in the eye retina using the GAL4/UAS system | Shows nuclear inclusion formation in the eye photoreceptor cells and CNS neurons as well as degeneration of the retina and neurodegeneration. | The model lacks a cerebellum [29]. |
| Mechanism | Model System | Molecular Findings | Pathology | Behavioral/Functional Tests | |
|---|---|---|---|---|---|
| RNA Level | Protein Level | ||||
| shRNA targeting human ATXN1 | SCA1 transgenic B05 mice | - | - | Amelioration of PC pathology and rescue of nuclear inclusions in PCs. | Improvement in motor performance as assessed by the rotarod assay [55]. |
| AAV expressing an artificial miRNA targeting human ataxin-1 (miS1) | SCA1 knock-in mice (SCA1154Q/2Q); pre-symptomatic | Up to 30% reduction | 58 to 72% reduction of ataxin-1 levels | Amelioration of PC pathology and no glial activation. | Improvement in motor performance as assessed by the rotarod assay and improvement in gait analysis [58]. |
| SCA1 transgenic B05 mice; pre-symptomatic | Up to 70% reduction | Amelioration of PC pathology and no glial activation. | Improvement of motor performance as assessed by the rotarod assay [57]. | ||
| SCA1 transgenic B05 mice; symptomatic | Up to 67% reduction | Amelioration of PC pathology. Mice treated with the highest dose showed no ATXN1-positive PCs but showed enhanced immunoreactivity. | Rescue of motor performance to wildtype levels as assessed by the rotarod assay in a dose-dependent manner; rescue in NAA/inositol ratios [59]. | ||
| Rhesus monkey | Up to 30% reduction in the left cerebellar hemisphere | Eight weeks post-injection, microglial activation and astrogliosis were enhanced in the left cerebellar cortex and deep cerebellar nuclei [60]. | |||
| Rhesus monkey | Significant reduction of ATXN1 in medial and lateral cerebellar cortex | Toxicity in deep cerebellar nuclei was observed; necrosis, demyelination, perivascular/leptomeningeal lymphoid infiltrates, increase in lesion severity, PC loss, and enhanced immunoreactivity. | Monkeys developed ataxia, tremor, head-tilt, and dysmetria 3 months after treatment [61]. | ||
| ASO353 | SCA1 knock-in mice (SCA1154Q/2Q) | Significant reduction of total mouse Atxn1 in brainstem and cerebellum | Rescue of analysis of neurochemicals and transcriptional disease signatures. No unwanted side effects involving BACE1, CIC activity or reduction in neuronal precursor cells. | Improved motor performance at 28 weeks, as assessed by the balance beam test. Pre-symptomatic treatment improved motor performance on both the rotarod and balance beam as well as prolonged survival [62,63]. | |
| (CUG)7 VO659 | SCA1 knock-in mice (SCA1154Q/2Q) | An exon 8 skip product of the Atxn1 gene was detected | A significant dose-dependent reduction in polyglutamine ataxin-1 in several brain regions. Reduction up to 45% in cerebellum and up to 56% in brainstem [64] | ||
| Combination of miS1 and vectors expressing human ATXN1L | SCA1 transgenic B05 mice | Significant increase in human ATXN1L expression and a significant reduction in human ATXN1 levels | Improvement in autonomous transcriptional changes and partial restoration of glial activation. | Significant improvement in latency to fall on the rotarod assay [65] | |
| Mechanism | Model System | Pathology | Behavioral/Functional Test |
|---|---|---|---|
| BK-AAV and baclofen (intracerebellar injection) | SCA1 B05 transgenic mice | Mitigation of dendritic degeneration | Improves motor performance [83] |
| Chlorzoxazone and baclofen (oral administration) | SCA1 B05 transgenic mice | Rescue of dendritic excitability in PCs | Improves motor performance [84] |
| SCA1 knock-in mice (SCA1154Q/2Q) | Rescue of dendritic excitability in PCs | Improves motor performance without affecting muscle strength [90] | |
| 4-aminopyridine and 3,4-diaminopyridine (subcutaneous injection) | SCA1 B05 transgenic mice | Normalization of firing rate of PCs; partial protection against cell atrophy | Improves motor performance [82] |
| mGlu1 receptor PAM (Ro0711401; subcutaneous injection) | SCA1 knock-in mice (SCA1154Q/2Q) | Increase in the number of dendritic spines | Significantly improves motor performance; effect lasted for 6 days [91] |
| Baclofen (intrathecal injection) | SCA1 B05 transgenic mice | Enhancement of mGluR1 signaling | Improves motor performance [93] |
| AAV-Homer-3 (ICV) | SCA1 B05 transgenic mice | Restoration of mTORC1 signaling and neuronal activation; improvement in PC morphology | Significantly improves motor performance [85] |
| Memantine (oral administration) | SCA1 B05 transgenic mice | Rescue of PC density; significant reduction in PC nuclear inclusions | Attenuates body weight loss and extends the life span [99] |
| IGF-1 (intranasal administration) | SCA1 B05 transgenic mice | Restoration of PC pathology | Improves motor performance [100] |
| Recombinant VEGF (ICV) | SCA1 knock-in mice (SCA1154Q/2Q) | Restoration of PC dendrite pathology and firing rate; improved microvascular health. | Improves motor performance [101] |
| Nano-VEGF (ICV) | SCA1 knock-in mice (SCA1154Q/2Q) | Restoration of PC dendrite pathology and firing rate; improved microvascular health. | Improves motor performance [102] |
| BDNF | SCA1 knock-in mice (SCA1154Q/2Q) | Improvement in cerebellar and hippocampal pathology | Ameliorates motor and cognitive deficits [103] |
| Lithium carbonate (oral administration) | SCA1 knock-in mice (SCA1154Q/2Q) | Partial rescue of dendrite pathology, restoration of isoprenylcystein carboxyl methyltransferase levels; reduction of GSK3ß levels; restoration of purine, oxidative stress, and energy production metabolic pathways | Improves motor performance, spatial learning, and memory [104,105] |
| MitoQ (oral administration) | SCA1 knock-in mice (SCA1154Q/2Q) | Amelioration of mitochondrial morphology and function; improvement in PC numbers and function | Improves motor performance [106] |
| Succinic acid (oral administration) | SCA1 B05 transgenic mice | Rescue of complex I OXPHOS dysfunction; amelioration of molecular layer and PC layer degeneration | Improves motor performance [107] |
| AAV1-HMGB1 (intrathecal injection) | SCA1 knock-in mice (SCA1154Q/2Q) | Rescue of PC loss; improvement in PC morphology and molecular layer thickness as well as nuclear and mitochondrial DNA damage | Improves motor performance and increased survival ratio [108] |
| AAV-RpA1 (intrathecal injection) | SCA1 knock-in mice (SCA1154Q/2Q) | Rescue of γH2AX and 53BP1 levels; recovery of mitochondrial DNA damage, dendritic pathology; rescue of molecular layer thickness, impaired splicing, transcription, and abnormal cell cycle | Improves motor performance [109] |
| panPAK inhibitor (intraperitoneal injection) | SCA1 knock-in mice (SCA1154Q/2Q) | Significant reduction in both expanded and wildtype ATXN1 RNA levels in the cerebellum [110] | |
| PLX3397 (oral administration) | SCA1 B05 transgenic mice | Reduction in microglial density in the cerebellum; reduction in TNF-α and increase in PSD-95 expression; increase in WT ataxin-1 protein levels | Improves motor performance [111] |
| Cell Type | Delivery Method | Delivery Location | Model | Treatment Age | Pathology Outcome |
|---|---|---|---|---|---|
| MSCs | Single IT injection | Subarachnoid space | SCA1 B05 transgenic mice | 5 weeks, pre-symptomatic stage | Amelioration of PC and PC dendritic spine pathology as well as improved motor coordination [121] |
| Human fetal MSCs | Single intracranial injection | Cerebellar cortex | SCA1 B05 transgenic mice | 4 weeks, pre-symptomatic stage as well as 6–8-month old symptomatic mice | Fusion of MSCs with cerebellar neurons in the symptomatic mice only [120] |
| Human umbilical MSCs | Single bilaterial intracranial injection | Left and right cerebellar lobules IV | SCA1 B05 transgenic mice | 4 weeks, pre-symptomatic stage | Improvement in molecular layer thickness, PC loss, and PC dendritic morphology; improvement in motor coordination [122] |
| MSCs | Single IT injection | Subarachnoid space | SCA1 knock-in mice (SCA1154Q/2Q) | 5 weeks, pre-symptomatic stage | Larger axon and myelin sizes [21] |
| MSC conditioned medium | Single IT injection and/or multiple intravenous injections | Subarachnoid space and/or systemic circulation | SCA1 knock-in mice (SCA1154Q/2Q) | 4 weeks, pre-symptomatic stage | Improvement in axon and myelin degeneration in the spinal neurons as well as improvement in motor coordination in all administration routes tested [123] |
| E13-E15 primordium mouse NPCs | Intracranial injection | Deep cerebellar nuclei | SCA1 B05 transgenic mice | 6–9 weeks, early symptomatic stage | Improved motor performance and amelioration of PC dendritic morphology [125] |
| Mouse NPCs from subventricular zone | Single, bilateral intracranial injection | Cerebellar white matter | SCA1 B05 transgenic mice | 5 or 13 weeks, pre-symptomatic; 24 weeks, symptomatic stage | Improvement in motor coordination, PC survival, PC function, and PC morphology in the symptomatic mice [124] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kerkhof, L.M.C.; van de Warrenburg, B.P.C.; van Roon-Mom, W.M.C.; Buijsen, R.A.M. Therapeutic Strategies for Spinocerebellar Ataxia Type 1. Biomolecules 2023, 13, 788. https://doi.org/10.3390/biom13050788
Kerkhof LMC, van de Warrenburg BPC, van Roon-Mom WMC, Buijsen RAM. Therapeutic Strategies for Spinocerebellar Ataxia Type 1. Biomolecules. 2023; 13(5):788. https://doi.org/10.3390/biom13050788
Chicago/Turabian StyleKerkhof, Laurie M. C., Bart P. C. van de Warrenburg, Willeke M. C. van Roon-Mom, and Ronald A. M. Buijsen. 2023. "Therapeutic Strategies for Spinocerebellar Ataxia Type 1" Biomolecules 13, no. 5: 788. https://doi.org/10.3390/biom13050788