
Schizophrenia Animal Modeling with Epidermal Growth Factor and Its Homologs: Their Connections to the Inflammatory Pathway and the Dopamine System

Abstract
:1. Introduction
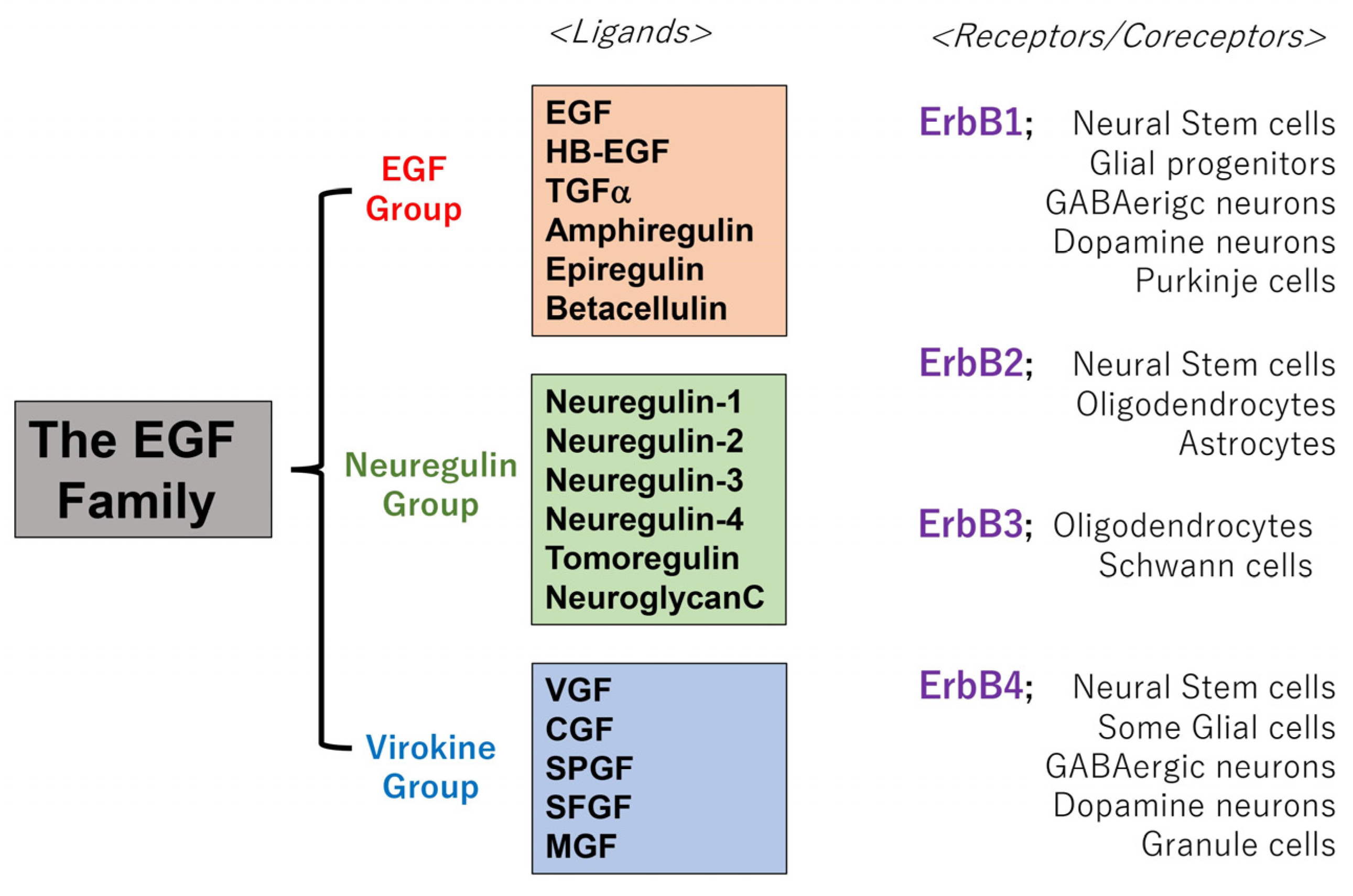
1.1. Biological Overviews of the Epidermal Growth Factor (EGF) Family
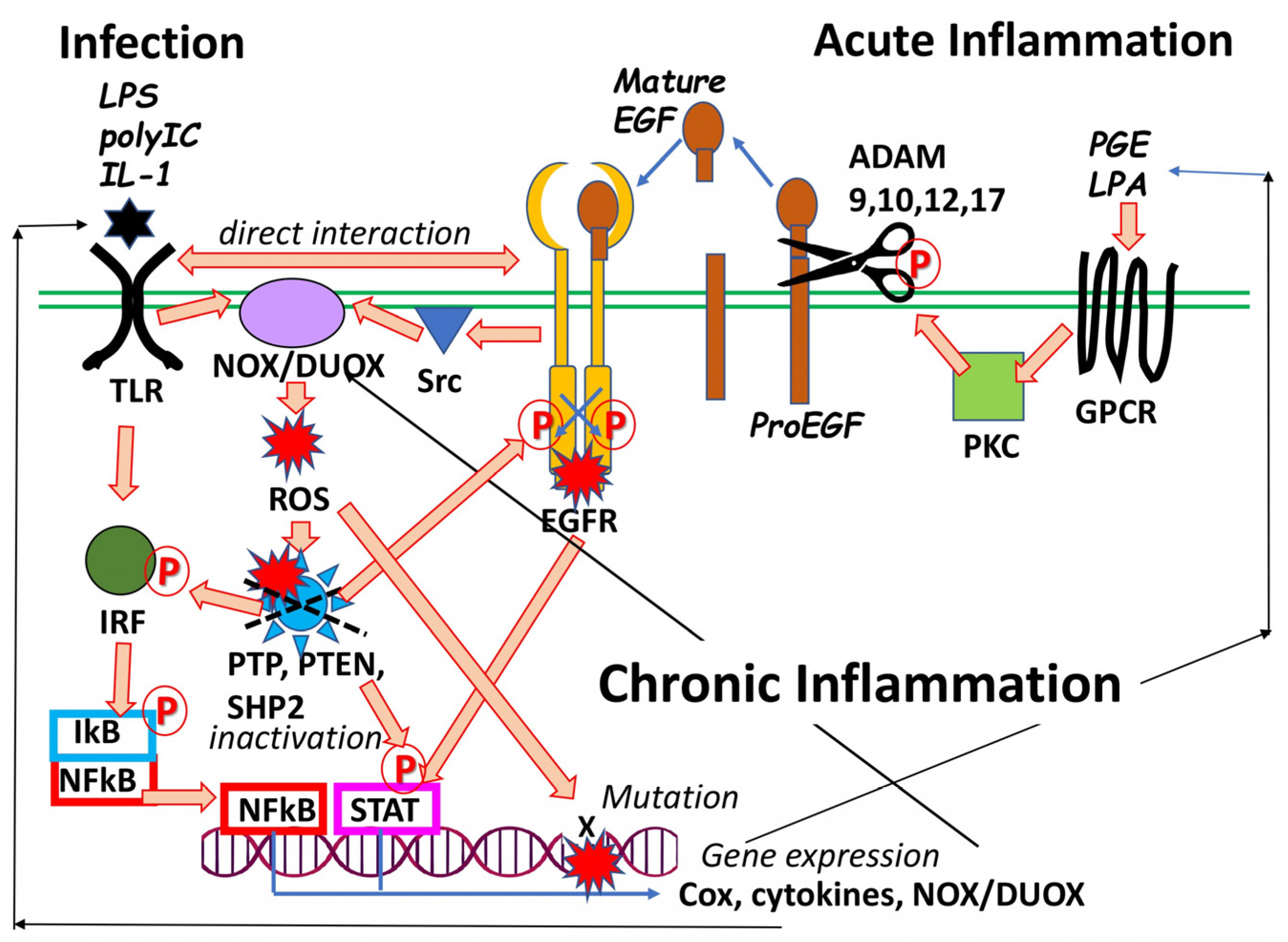
1.2. Phenotypic Association of The EGF Ligand Family with Schizophrenia
2. Method for Literature Selection
3. Neurotrophic Functions of EGF and Neuregulins in Dopaminergic Neurons
3.1. Distributions of ErbB1 (EGFR) and ErbB4 in the Midbrain
3.2. Influences of EGF/Neuregulin Signaling on Dopamine Metabolism
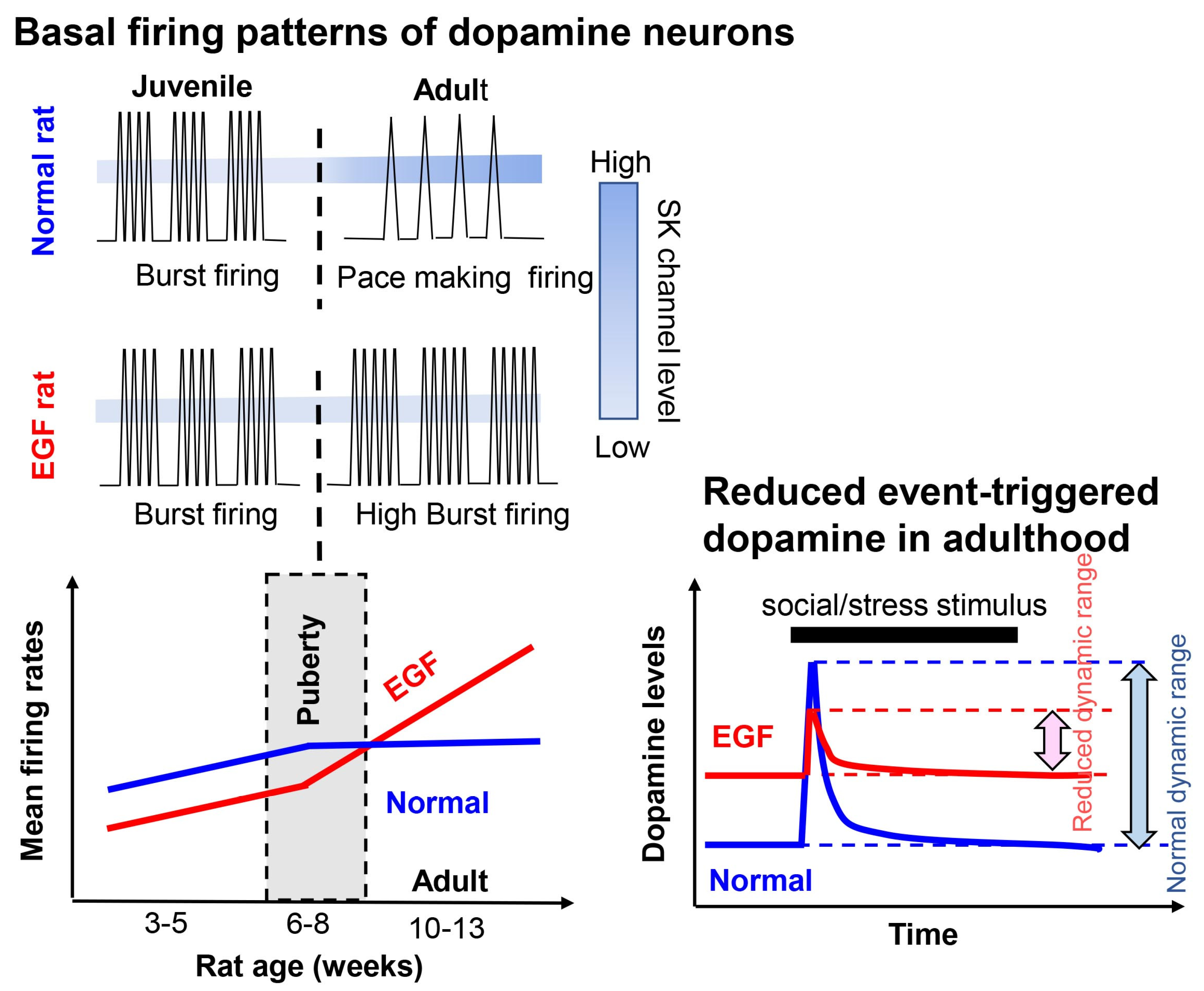
3.3. Post-Pubertal Elevation of Dopaminergic Activity in the EGF/Neuregulin Models
4. Chronic Hyper-Dopaminergic Influences on Animal Models and Patients with Schizophrenia
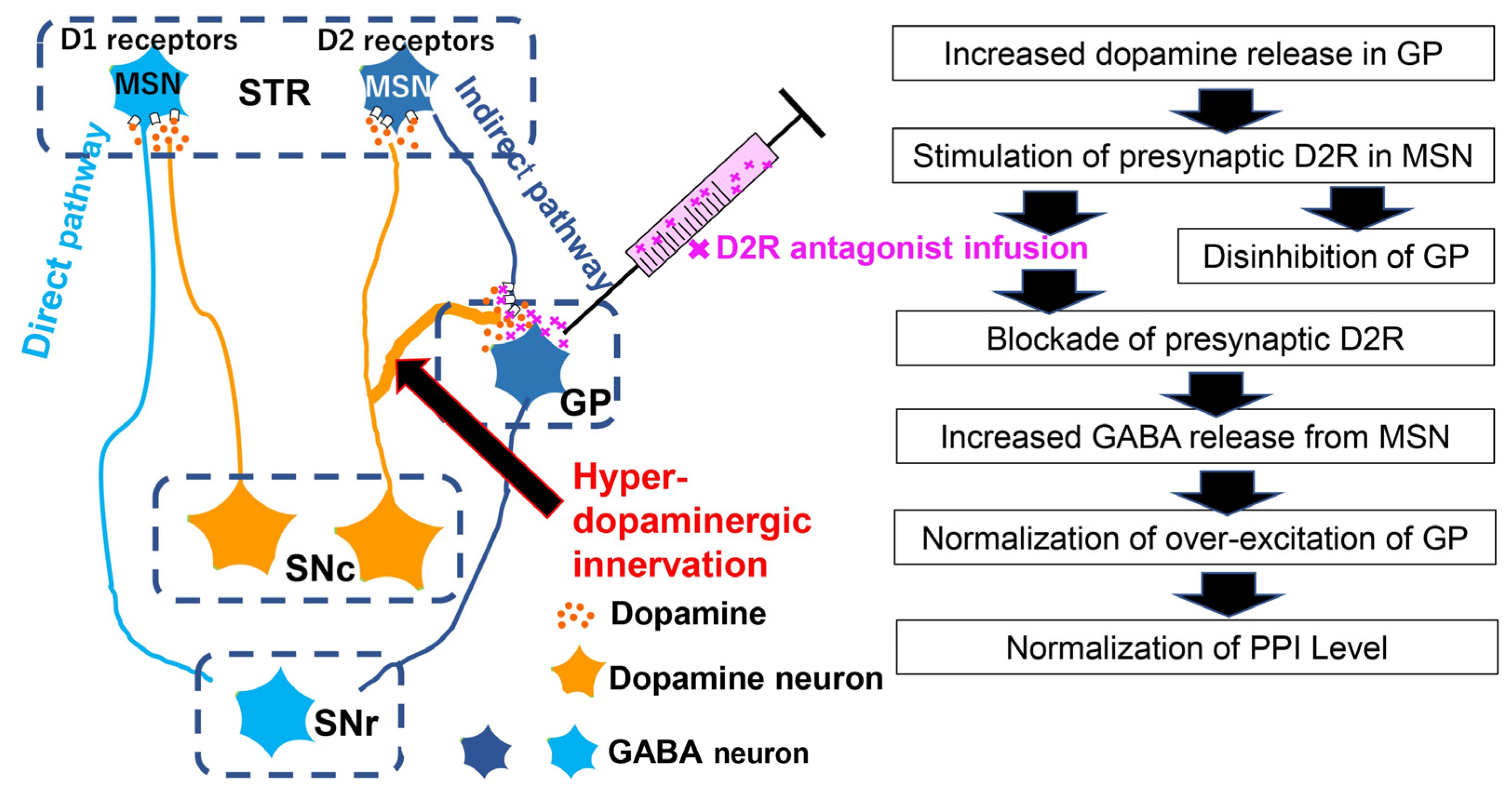
4.1. Interaction of A10 Dopaminergic Activity with Acoustic Prepulse Inhibition
4.2. Chronic Hyper-Dopaminergic States Impair Social Interaction
5. Drug Development Targeting EGF/ErbB Signals
6. Provisional Conclusion
7. Future Directions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
References
- Cohen, S. Origins of growth factors: NGF and EGF. J. Biol. Chem. 2008, 283, 33793–33797. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yarden, Y.; Pines, G. The ERBB network: At last, cancer therapy meets systems biology. Nat. Rev. Cancer 2012, 12, 553–563. [Google Scholar] [CrossRef] [PubMed]
- Kumar, A.; Petri, E.T.; Halmos, B.; Boggon, T.J. Structure and clinical relevance of the epidermal growth factor receptor in human cancer. J. Clin. Oncol. 2008, 26, 1742–1751. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gregory, H. In vivo aspects of urogastrone-epidermal growth factor. J. Cell Sci. Suppl. 1985, 3, 11–17. [Google Scholar] [CrossRef] [Green Version]
- Chao, M.V. Neurotrophins and their receptors: A convergence point for many signalling pathways. Nat. Rev. Neurosci. 2003, 4, 299–309. [Google Scholar] [CrossRef]
- Higashiyama, S.; Horikawa, M.; Yamada, K.; Ichino, N.; Nakano, N.; Nakagawa, T.; Miyagawa, J.; Matsushita, N.; Nagatsu, T.; Taniguchi, N.; et al. A novel brain-derived member of the epidermal growth factor family that interacts with ErbB3 and ErbB4. J. Biochem. 1997, 122, 675–680. [Google Scholar] [CrossRef]
- Harris, R.C.; Chung, E.; Coffey, J. The EGF Receptor Family: Biologic Mechanisms and Role in Cancer; Academic Press: London, UK, 2012; pp. 3–14. [Google Scholar]
- Vullhorst, D.; Ahmad, T.; Karavanova, I.; Keating, C.; Buonanno, A. Structural similarities between neuregulin 1-3 isoforms determine their subcellular distribution and signaling mode in central neurons. J. Neurosci. 2017, 37, 5232–5249. [Google Scholar] [CrossRef] [Green Version]
- Weickert, C.S.; Blum, M. Striatal TGF-alpha: Postnatal developmental expression and evidence for a role in the proliferation of subependymal cells. Brain Res. Dev. 1995, 86, 203–216. [Google Scholar] [CrossRef]
- Nakagawa, T.; Sasahara, M.; Hayase, Y.; Haneda, M.; Yasuda, H.; Kikkawa, R.; Higashiyama, S.; Hazama, F. Neuronal and glial expression of heparin-binding EGF-like growth factor in central nervous system of prenatal and early-postnatal rat. Brain Res. Dev. Brain Res. 1998, 108, 263–272. [Google Scholar] [CrossRef]
- Eilam, R.; Pinkas-Kramarski, R.; Ratzkin, B.J.; Segal, M.; Yarden, Y. Activity-dependent regulation of Neu differentiation factor/neuregulin expression in rat brain. Proc. Natl. Acad. Sci. USA 1998, 95, 1888–1893. [Google Scholar] [CrossRef]
- Kao, W.T.; Wang, Y.; Kleinman, J.E.; Lipska, B.K.; Hyde, T.M.; Weinberger, D.R.; Law, A.J. Common genetic variation in Neuregulin 3 (NRG3) influences risk for schizophrenia and impacts NRG3 expression in human brain. Proc. Natl. Acad. Sci. USA 2010, 107, 15619–15624. [Google Scholar] [CrossRef] [Green Version]
- Mei, L.; Xiong, W.-C. Neuregulin 1 in neural development, synaptic plasticity and schizophrenia. Nat. Rev. Neurosci. 2008, 9, 437–452. [Google Scholar] [CrossRef]
- Scalabrino, G. Epidermal growth factor in the CNS: A beguiling journey from integrated cell biology to multiple sclerosis. An extensive translationaloverview. Cell. Mol. Neurobiol. 2022, 42, 891–916. [Google Scholar] [CrossRef]
- Kwon, O.B.; Paredes, D.; Gonzalez, C.M.; Neddens, J.; Hernandez, L.; Vullhorst, D.; Buonanno, A. Neuregulin-1 regulates LTP at CA1 hippocampal synapses through activation of dopamine D4 receptors. Proc. Natl. Acad. Sci. USA 2008, 105, 15587–15592. [Google Scholar] [CrossRef] [Green Version]
- Wang, R.; Iwakura, Y.; Araki, K.; Keino-Masu, K.; Masu, M.; Wang, X.Y.; Takei, N.; Higashiyama, S.; Nawa, H. ErbB2 dephosphorylation and anti-proliferative effects of neuregulin-1 in ErbB2-overexpressing cells; re-evaluation of their low-affinity interaction. Sci. Rep. 2013, 3, 1402. [Google Scholar] [CrossRef] [Green Version]
- Eppstein, D.A.; Marsh, Y.V.; Schreiber, A.B.; Newman, S.R.; Todaro, G.J.; Nestor, J.J. Epidermal growth factor receptor occupancy inhibits vaccinia virus infection. Nature 1985, 318, 663–665. [Google Scholar] [CrossRef]
- Kim, M.; Yang, H.; Kim, S.K.; Reche, P.A.; Tirabassi, R.S.; Hussey, R.E.; Chishti, Y.; Rheinwald, J.G.; Morehead, T.J.; Zech, T.; et al. Biochemical and functional analysis of smallpox growth factor (SPGF) and anti-SPGF monoclonal antibodies. J. Biol. Chem. 2004, 279, 25838–25848. [Google Scholar] [CrossRef]
- Conrad, M.; Goldstein, D.; Andresson, T.; Schlegel, R. The E5 protein of HPV-6, but not HPV-16, associates efficiently with cellular growth factor receptors. Virology 1994, 200, 796–800. [Google Scholar] [CrossRef]
- Greten, F.R.; Grivennikov, S.I. Inflammation and cancer: Triggers, mechanisms, and consequences. Immunity 2019, 51, 27–41. [Google Scholar] [CrossRef]
- Berasain, C.; Castillo, J.; Perugorria, M.J.; Latasa, M.U.; Prieto, J.; Avila, M.A. Inflammation and liver cancer: New molecular links. Ann. N. Y. Acad. Sci. 2009, 1155, 206–221. [Google Scholar] [CrossRef]
- Yamashita, M.; Chattopadhyay, S.; Fensterl, V.; Saikia, P.; Wetzel, J.L.; Sen, G.C. Epidermal growth factor receptor is essential for Toll-like receptor 3 signaling. Sci. Signal. 2012, 5, ra50. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fukata, M.; Chen, A.; Vamadevan, A.S.; Cohen, J.; Breglio, K.; Krishnareddy, S.; Hsu, D.; Xu, R.; Harpaz, N.; Dannenberg, A.J.; et al. Toll-like receptor-4 promotes the development of colitis-associated colorectal tumors. Gastroenterology 2007, 133, 1869–1881. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Damiano, V.; Caputo, R.; Bianco, R.; D’Armiento, F.P.; Leonardi, A.; De Placido, S.; Bianco, A.R.; Agrawal, S.; Ciardiello, F.; Tortora, G. Novel toll-like receptor 9 agonist induces epidermal growth factor receptor (EGFR) inhibition and synergistic antitumor activity with EGFR inhibitors. Clin. Cancer Res. 2006, 12, 577–583. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Singh, A.B.; Harris, R.C. Autocrine, paracrine and juxtacrine signaling by EGFR ligands. Cell. Signal. 2005, 17, 1183–1193. [Google Scholar] [CrossRef] [PubMed]
- Shostak, K.; Chariot, A. EGFR and NF-κB: Partners in cancer. Trends Mol. Med. 2015, 21, 385–393. [Google Scholar] [CrossRef]
- Venkataraman, T.; Frieman, M.B. The role of epidermal growth factor receptor (EGFR) signaling in SARS coronavirus-induced pulmonary fibrosis. Antiviral Res. 2017, 143, 142–150. [Google Scholar] [CrossRef]
- Kalinowski, A.; Galen, B.T.; Ueki, I.F.; Sun, Y.; Mulenos, A.; Osafo-Addo, A.; Clark, B.; Joerns, J.; Liu, W.; Nadel, J.A.; et al. Respiratory syncytial virus activates epidermal growth factor receptor to suppress interferon regulatory factor 1-dependent interferon-lambda and antiviral defense in airway epithelium. Mucosal Immunol. 2018, 11, 958–967. [Google Scholar] [CrossRef] [Green Version]
- Watanabe, Y.; Someya, T.; Nawa, H. Cytokine hypothesis of schizophrenia pathogenesis: Evidence from human studies and animal models. Psychiatry Clin. Neurosci. 2010, 64, 217–230. [Google Scholar] [CrossRef]
- Khandaker, G.M.; Cousins, L.; Deakin, J.; Lennox, B.R.; Yolken, R.; Jones, P.B. Inflammation and immunity in schizophrenia: Implications for pathophysiology and treatment. Lancet Psychiatry 2015, 2, 258–270. [Google Scholar] [CrossRef] [Green Version]
- Ermakov, E.A.; Melamud, M.M.; Buneva, V.N.; Ivanova, S.A. Immune system abnormalities in schizophrenia: An integrative view and translational perspectives. Front. Psychiatry 2022, 13, 880568. [Google Scholar] [CrossRef]
- Nakamura, J.P.; Schroeder, A.; Gibbons, A.; Sundram, S.; Hill, R.A. Timing of maternal immune activation and sex influence schizophrenia-relevant cognitive constructs and neuregulin and GABAergic pathways. Brain. Behav. Immun. 2022, 100, 70–82. [Google Scholar] [CrossRef]
- Idrizi, R.; Malcolm, P.; Weickert, C.S.; Zavitsanou, K.; Sundram, S. Striatal but not frontal cortical up-regulation of the epidermal growth factor receptor in rats exposed to immune activation in utero and cannabinoid treatment in adolescence. Psychiatry Res. 2016, 240, 260–264. [Google Scholar] [CrossRef]
- Martins-Filho, P.R.; Tanajura, D.M.; Santos, H.P., Jr.; Santos, V.S. COVID-19 during pregnancy: Potential risk for neurodevelopmental disorders in neonates? Eur. J. Obstet. Gynecol. Reprod. Biol. 2020, 250, 255–256. [Google Scholar] [CrossRef]
- Figueiredo, C.P.; Fontes-Dantas, F.L.; da Poian, A.T.; Clarke, J.R. SARS-CoV-2-associated cytokine storm during pregnancy as a possible risk factor for neuropsychiatric disorder development in post-pandemic infants. Neuropharmacology 2021, 201, 108841. [Google Scholar] [CrossRef]
- Rhoades, R.; Solomon, S.; Johnson, C.; Teng, S. Impact of SARS-CoV-2 on host factors involved in mental disorders. Front. Microbiol. 2022, 13, 845559. [Google Scholar] [CrossRef]
- Yamaguchi, R.; Yamamoto, T.; Sakamoto, A.; Narahara, S.; Sugiuchi, H.; Yamaguchi, Y. Neutrophil elastase enhances IL-12p40 production by lipopolysaccharide-stimulated macrophages via transactivation of the PAR-2/EGFR/TLR4 signaling pathway. Blood Cells Mol. Dis. 2016, 59, 1–7. [Google Scholar] [CrossRef]
- Manea, A.; Tanase, L.I.; Raicu, M.; Simionescu, M. Jak/STAT signaling pathway regulates nox1 and nox4-based NADPH oxidase in human aortic smooth muscle cells. Arterioscler. Thromb. Vasc. Biol. 2010, 30, 105–112. [Google Scholar] [CrossRef] [Green Version]
- Ketharanathan, T.; Pereira, A.; Reets, U.; Walker, D.; Sundram, S. Brain Changes in NF-ΚB1 and epidermal growth factor system markers at peri-pubescence in the spiny mouse following maternal immune activation. Psychiatry Res. 2021, 295, 113564. [Google Scholar] [CrossRef]
- Zunke, F.; Rose-John, S. The shedding protease ADAM17: Physiology and pathophysiology. Biochim. Biophys. Acta Mol. Cell Res. 2017, 1864, 2059–2070. [Google Scholar] [CrossRef]
- Bernstein, H.-G.; Keilhoff, G.; Dobrowolny, H.; Lendeckel, U.; Steiner, J. From putative brain tumor marker to high cognitive abilities: Emerging roles of a disintegrin and metalloprotease (ADAM) 12 in the brain. J. Chem. Neuroanat. 2020, 109, 101846. [Google Scholar] [CrossRef]
- Roskoski, R. ErbB/HER protein-tyrosine kinases: Structures and small molecule inhibitors. Pharmacol. Res. 2014, 87, 42–59. [Google Scholar] [CrossRef] [PubMed]
- Iwakura, Y.; Nawa, H. ErbB1-4-dependent EGF/neuregulin signals and their cross talk in the central nervous system: Pathological implications in schizophrenia and Parkinson’s disease. Front. Cell. Neurosci. 2013, 7, 4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, P.; Cleveland IV, T.E.; Bouyain, S.; Byrne, P.O.; Longo, P.A.; Leahy, D.J. A single ligand is sufficient to activate EGFR dimers. Proc. Natl. Acad. Sci. USA 2012, 109, 10861–10866. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abe, Y.; Namba, H.; Zheng, Y.; Nawa, H. In situ hybridization reveals developmental regulation of ErbB1-4 mRNA expression in mouse midbrain: Implication of ErbB receptors for dopaminergic neurons. Neuroscience 2009, 161, 95–110. [Google Scholar] [CrossRef] [PubMed]
- Seroogy, K.B.; Gall, C.M.; Lee, D.C.; Kornblum, H.I. Proliferative zones of postnatal rat brain express epidermal growth factor receptor mRNA. Brain Res. 1995, 670, 157–164. [Google Scholar] [CrossRef]
- Nagano, T.; Namba, H.; Abe, Y.; Aoki, H.; Takei, N.; Nawa, H. In vivo administration of epidermal growth factor and its homologue attenuates developmental maturation of functional excitatory synapses in cortical GABAergic neurons. Eur. J. Neurosci. 2007, 25, 380–390. [Google Scholar] [CrossRef]
- Iwakura, Y.; Zheng, Y.; Sibilia, M.; Abe, Y.; Piao, Y.S.; Yokomaku, D.; Wang, R.; Ishizuka, Y.; Takei, N.; Nawa, H. Qualitative and quantitative re-evaluation of epidermal growth factor-ErbB1 action on developing midbrain dopaminergic neurons in vivo and in vitro: Target-derived neurotrophic signaling (Part 1). J. Neurochem. 2011, 118, 45–56. [Google Scholar] [CrossRef]
- Vullhorst, D.; Neddens, J.; Karavanova, I.; Tricoire, L.; Petralia, R.S.; McBain, C.J.; Buonanno, A. Selective expression of ErbB4 in interneurons, but not pyramidal cells, of the rodent hippocampus. J. Neurosci. 2009, 29, 12255–12264. [Google Scholar] [CrossRef] [Green Version]
- Makinodan, M.; Rosen, K.M.; Ito, S.; Corfas, G. A critical period for social experience-dependent oligodendrocyte maturation and myelination. Science 2012, 337, 1357–1360. [Google Scholar] [CrossRef] [Green Version]
- Futamura, T.; Toyooka, K.; Iritani, S.; Niizato, K.; Nakamura, R.; Tsuchiya, K.; Someya, T.; Kakita, A.; Takahashi, H.; Nawa, H. Abnormal expression of epidermal growth factor and its receptor in the forebrain and serum of schizophrenic patients. Mol. Psychiatry 2002, 7, 673–682. [Google Scholar] [CrossRef]
- Zhang, X.; Xiao, W.; Chen, K.; Zhao, Y.; Ye, F.; Tang, X.; Du, X. Decreased serum EGF in first-episode and chronic schizophrenia patients: Negative correlation with psychopathology. Sci. Rep. 2020, 10, 6506. [Google Scholar] [CrossRef] [Green Version]
- Ikeda, Y.; Yahata, N.; Ito, I.; Nagano, M.; Toyota, T.; Yoshikawa, T.; Okubo, Y.; Suzuki, H. Low serum levels of brain-derived neurotrophic factor and epidermal growth factor in patients with chronic schizophrenia. Schizophr. Res. 2008, 101, 58–66. [Google Scholar] [CrossRef]
- Hashimoto, K.; Shimizu, E.; Komatsu, N.; Watanabe, H.; Shinoda, N.; Nakazato, M.; Kumakiri, C.; Okada, S.; Takei, N.; Iyo, M. No changes in serum epidermal growth factor levels in patients with schizophrenia. Psychiatry Res. 2005, 135, 257–260. [Google Scholar] [CrossRef]
- Stefansson, H.; Sigurdsson, E.; Steinthorsdottir, V.; Bjornsdottir, S.; Sigmundsson, T.; Ghosh, S.; Brynjolfsson, J.; Gunnarsdottir, S.; Ivarsson, O.; Chou, T.T.; et al. Neuregulin 1 and susceptibility to schizophrenia. Am. J. Hum. Genet. 2002, 71, 877–892. [Google Scholar] [CrossRef] [Green Version]
- Munafò, M.R.; Thiselton, D.L.; Clark, T.G.; Flint, J. Association of the NRG1 gene and schizophrenia: A meta-analysis. Mol. Psychiatry 2006, 11, 539–546. [Google Scholar] [CrossRef] [Green Version]
- Anttila, S.; Illi, A.; Kampman, O.; Mattila, K.M.; Lehtimäki, T.; Leinonen, E. Association of EGF polymorphism with schizophrenia in Finnish men. Neuroreport 2004, 15, 1215–1218. [Google Scholar] [CrossRef]
- Hänninen, K.; Katila, H.; Anttila, S.; Rontu, R.; Maaskola, J.; Hurme, M.; Lehtimäki, T. Epidermal growth factor a61g polymorphism is associated with the age of onset of schizophrenia in male patients. J. Psychiatr. Res. 2007, 41, 8–14. [Google Scholar] [CrossRef]
- Watanabe, Y.; Fukui, N.; Muratake, T.; Kaneko, N.; Someya, T. No association of EGF polymorphism with schizophrenia in a Japanese population. Neuroreport 2005, 16, 403–405. [Google Scholar] [CrossRef]
- Groenestege, W.M.T.; Thébault, S.; Van Der Wijst, J.; Van Den Berg, D.; Janssen, R.; Tejpar, S.; Van Den Heuvel, L.P.; Van Cutsem, E.; Hoenderop, J.G.; Knoers, N.V.; et al. Impaired basolateral sorting of pro-EGF causes isolated recessive renal hypomagnesemia. J. Clin. Investig. 2007, 117, 2260–2267. [Google Scholar] [CrossRef]
- Hahn, C.G.; Wang, H.Y.; Cho, D.S.; Talbot, K.; Gur, R.E.; Berrettini, W.H.; Bakshi, K.; Kamins, J.; Borgmann-Winter, K.E.; Siegel, S.J.; et al. Altered neuregulin 1-erbB4 signaling contributes to NMDA receptor hypofunction in schizophrenia. Nat. Med. 2006, 12, 824–828. [Google Scholar] [CrossRef]
- Law, A.J.; Lipska, B.K.; Weickert, C.S.; Hyde, T.M.; Straub, R.E.; Hashimoto, R.; Harrison, P.J.; Kleinman, J.E.; Weinberger, D.R. Neuregulin 1 transcripts are differentially expressed in schizophrenia and regulated by 5′ SNPs associated with the disease. Proc. Natl. Acad. Sci. USA 2006, 103, 6747–6752. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hashimoto, R.; Straub, R.E.; Weickert, C.S.; Hyde, T.M.; Kleinman, J.E.; Weinberger, D.R. Expression analysis of neuregulin-1 in the dorsolateral prefrontal cortex in schizophrenia. Mol. Psychiatry 2004, 9, 299–307. [Google Scholar] [CrossRef] [PubMed]
- Shibuya, M.; Komi, E.; Wang, R.; Kato, T.; Watanabe, Y.; Sakai, M.; Ozaki, M.; Someya, T.; Nawa, H. Measurement and comparison of serum neuregulin 1 immunoreactivity in control subjects and patients with schizophrenia: An influence of its genetic polymorphism. J. Neural Transm. 2010, 117, 887–895. [Google Scholar] [CrossRef] [PubMed]
- Nawa, H.; Sotoyama, H.; Iwakura, Y.; Takei, N.; Namba, H. Neuropathologic implication of peripheral neuregulin-1 and EGF signals in dopaminergic dysfunction and behavioral deficits relevant to schizophrenia: Their target cells and time window. Biomed. Res. Int. 2014, 2014, 697935. [Google Scholar] [CrossRef] [Green Version]
- Patlola, S.R.; Donohoe, G.; McKernan, D.P. The relationship between inflammatory biomarkers and cognitive dysfunction in patients with schizophrenia: A systematic review and meta-analysis. Prog. Neuropsychopharmacol. Biol. Psychiatry 2023, 121, 110668. [Google Scholar] [CrossRef]
- Goldsmith, D.R.; Bekhbat, M.; Mehta, N.D.; Felger, J.C. Inflammation-related functional and structural dysconnectivity as a pathway to psychopathology. Biol. Psychiatry 2023, 93, 405–418. [Google Scholar] [CrossRef]
- Ishikawa, Y.; Furuyashiki, T. The impact of stress on immune systems and its relevance to mental illness. Neurosci. Res. 2022, 175, 16–24. [Google Scholar] [CrossRef]
- Meyer, U. Prenatal poly(i:C) exposure and other developmental immune activation models in rodent systems. Biol. Psychiatry 2014, 75, 307–315. [Google Scholar] [CrossRef]
- Nawa, H.; Takei, N. Recent progress in animal modeling of immune inflammatory processes in schizophrenia: Implication of specific cytokines. Neurosci. Res. 2006, 56, 2–13. [Google Scholar] [CrossRef]
- Tohmi, M.; Tsuda, N.; Watanabe, Y.; Kakita, A.; Nawa, H. Perinatal inflammatory cytokine challenge results in distinct neurobehavioral alterations in rats: Implication in psychiatric of developmental origin. Neurosci. Res. 2004, 50, 67–75. [Google Scholar] [CrossRef]
- Shi, L.; Fatemi, S.H.; Sidwell, R.W.; Patterson, P.H. Maternal influenza infection causes marked behavioral and pharmacological changes in the offspring. J. Neurosci. 2003, 23, 297–302. [Google Scholar] [CrossRef]
- Fortier, M.È.; Joober, R.; Luheshi, G.N.; Boksa, P. Maternal exposure to bacterial endotoxin during pregnancy enhances amphetamine-induced locomotion and startle responses in adult rat offspring. J. Psychiatr. Res. 2004, 38, 335–345. [Google Scholar] [CrossRef]
- Aoki, H. Neurocognitive impairments of offspring induced by maternal cytokine challenges. Niigata Med. J. 2008, 122, 262–270. [Google Scholar]
- Hsiao, E.Y.; Patterson, P.H. Activation of the maternal immune system induces endocrine changes in the placenta via IL-6. Brain Behav. Immun. 2011, 25, 604–615. [Google Scholar] [CrossRef] [Green Version]
- Tsuda, N.; Mizuno, M.; Yamanaka, T.; Komurasaki, T.; Yoshimoto, M.; Nawa, H. Common behavioral influences of the ErbB1 ligands transforming growth factor alpha and epiregulin administered to mouse neonates. Brain Dev. 2008, 30, 533–543. [Google Scholar] [CrossRef]
- Futamura, T.; Kakita, A.; Tohmi, M.; Sotoyama, H.; Takahashi, H.; Nawa, H. Neonatal perturbation of neurotrophic signaling results in abnormal sensorimotor gating and social interaction in adults: Implication for epidermal growth factor in cognitive development. Mol. Psychiatry 2003, 8, 19–29. [Google Scholar] [CrossRef] [Green Version]
- Tohmi, M.; Tsuda, N.; Zheng, Y.; Mizuno, M.; Sotoyama, H.; Shibuya, M.; Kawamura, M.; Kakita, A.; Takahashi, H.; Nawa, H. The cellular and behavioral consequences of interleukin-1 alpha penetration through the blood-brain barrier of neonatal rats: A critical period for efficacy. Neuroscience 2007, 150, 234–250. [Google Scholar] [CrossRef]
- Kato, T.; Abe, Y.; Sotoyama, H.; Kakita, A.; Kominami, R.; Hirokawa, S.; Ozaki, M.; Takahashi, H.; Nawa, H. Transient exposure of neonatal mice to neuregulin-1 results in hyperdopaminergic states in adulthood: Implication in neurodevelopmental hypothesis for schizophrenia. Mol. Psychiatry 2011, 16, 307–320. [Google Scholar] [CrossRef] [Green Version]
- Kato, T.; Abe, Y.; Hirokawa, S.; Iwakura, Y.; Mizuno, M.; Namba, H.; Nawa, H. Neurobehavioral differences between mice receiving distinct neuregulin variants as neonates; impact on sensitivity to MK-801. Curr. Mol. Med. 2015, 15, 222–236. [Google Scholar] [CrossRef] [Green Version]
- Tohmi, M.; Tsuda, N.; Mizuno, M.; Takei, N.; Frankland, P.W.; Nawa, H. Distinct influences of neonatal epidermal growth factor challenge on adult neurobehavioral traits in four mouse strains. Behav. Genet. 2005, 35, 615–629. [Google Scholar] [CrossRef]
- Mizuno, M.; Malta, R.S.; Nagano, T.; Nawa, H. Conditioned place preference and locomotor sensitization after repeated administration of cocaine or methamphetamine in rats treated with epidermal growth factor during the neonatal period. Ann. N. Y. Acad. Sci. 2004, 1025, 612–618. [Google Scholar] [CrossRef] [PubMed]
- Sotoyama, H.; Zheng, Y.; Iwakura, Y.; Mizuno, M.; Aizawa, M.; Shcherbakova, K.; Wang, R.; Namba, H.; Nawa, H. Pallidal hyperdopaminergic innervation underlying D2 receptor-dependent behavioral deficits in the schizophrenia animal model established by EGF. PLoS ONE 2011, 6, e25831. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sakai, M.; Kashiwahara, M.; Kakita, A.; Nawa, H. An attempt of non-human primate modeling of schizophrenia with neonatal challenges of epidermal growth factor. J. Addict. Res. Ther. 2014, 5, 170. [Google Scholar]
- Jodo, E.; Inaba, H.; Narihara, I.; Sotoyama, H.; Kitayama, E.; Yabe, H.; Namba, H.; Eifuku, S.; Nawa, H. Neonatal exposure to an inflammatory cytokine, epidermal growth factor, results in the deficits of mismatch negativity in rats. Sci. Rep. 2019, 9, 7503. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Inaba, H.; Kai, R.; Namba, H.; Sotoyama, H.; Jodo, E.; Nin, F.; Hibino, H.; Yabe, H.; Eifuku, S.; Horii, A.; et al. Perinatal epidermal growth factor signal perturbation results in the series of abnormal auditory oscillations and responses relevant to schizophrenia. Schizophr. Bull. Open 2021, 2, sgaa070. [Google Scholar] [CrossRef]
- Narihara, I.; Kitajo, K.; Namba, H.; Sotoyama, H.; Inaba, H.; Watanabe, D.; Nawa, H. Rat call-evoked electrocorticographic responses and intercortical phase synchrony impaired in a cytokine-induced animal model for schizophrenia. Neurosci. Res. 2022, 175, 62–72. [Google Scholar] [CrossRef]
- Iwakura, Y.; Piao, Y.S.; Mizuno, M.; Takei, N.; Kakita, A.; Takahashi, H.; Nawa, H. Influences of dopaminergic lesion on epidermal growth factor-ErbB signals in Parkinson’s disease and its model: Neurotrophic implication in nigrostriatal neurons. J. Neurochem. 2005, 93, 974–983. [Google Scholar] [CrossRef]
- Cohen, A.D.; Zigmond, M.J.; Smith, A.D. Effects of intrastriatal GDNF on the response of dopamine neurons to 6-hydroxydopamine: Time course of protection and neurorestoration. Brain Res. 2011, 1370, 80–88. [Google Scholar] [CrossRef] [Green Version]
- Hadjiconstantinou, M.; Fitkin, J.G.; Dalia, A.; Neff, N.H. Epidermal growth factor enhances striatal dopaminergic parameters in the 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine-treated mouse. J. Neurochem. 1991, 57, 479–482. [Google Scholar] [CrossRef]
- Schneider, J.S.; DiStefano, L. Enhanced restoration of striatal dopamine concentrations by combined GM1 ganglioside and neurotrophic factor treatments. Brain Res. 1995, 674, 260–264. [Google Scholar] [CrossRef]
- Gerhardt, G.A.; Cass, W.A.; Huettl, P.; Brock, S.; Zhang, Z.; Gash, D.M. GDNF improves dopamine function in the substantia nigra but not the putamen of unilateral MPTP-lesioned rhesus monkeys. Brain Res. 1999, 817, 163–171. [Google Scholar] [CrossRef]
- Yu, Z.Q.; Zha, J.H.; Liu, H.M.; Ding, Y.X.; Wang, Y.Q.; Wang, H.J.; Gao, D.S. Effect of intranigral injection of GDNF and EGF on the survival and possible differentiation fate of progenitors and immature neurons in 6-OHDA-lesioned rats. Neurochem. Res. 2009, 34, 2089–2101. [Google Scholar] [CrossRef]
- Hidalgo-Figueroa, M.; Bonilla, S.; Gutierrez, F.; Pascual, A.; Lopez-Barneo, J. GDNF is predominantly expressed in the PV+ neostriatal interneuronal ensemble in normal mouse and after injury of the nigrostriatal pathway. J. Neurosci. 2012, 32, 864–872. [Google Scholar] [CrossRef] [Green Version]
- Kumar, A.; Kopra, J.; Varendi, K.; Porokuokka, L.L.; Panhelainen, A.; Kuure, S.; Marshall, P.; Karalija, N.; Härma, M.A.; Vilenius; et al. GDNF overexpression from the native locus reveals its role in the nigrostriatal dopaminergic system function. PLoS Genet. 2015, 11, e1005710. [Google Scholar] [CrossRef] [Green Version]
- Carlsson, T.; Schindler, F.R.; Höllerhage, M.; Depboylu, C.; Arias-Carrión, O.; Schnurrbusch, S.; Rösler, T.W.; Wozny, W.; Schwall, G.P.; Groebe, K.; et al. Systemic administration of neuregulin-1β1 protects dopaminergic neurons in a mouse model of Parkinson’s disease. J. Neurochem. 2011, 117, 1066–1074. [Google Scholar] [CrossRef]
- Depboylu, C.; Rösler, T.W.; de Andrade, A.; Oertel, W.H.; Höglinger, G.U. Systemically administered neuregulin-1β1 rescues nigral dopaminergic neurons via the ErbB4 receptor tyrosine kinase in MPTP mouse models of Parkinson’s disease. J. Neurochem. 2015, 133, 590–597. [Google Scholar] [CrossRef]
- Thuret, S.; Alavian, K.N.; Gassmann, M.; Lloyd, C.K.; Smits, S.M.; Smidt, M.P.; Klein, R.; Dyck, R.H.; Simon, H.H. The neuregulin receptor, ErbB4, is not required for normal development and adult maintenance of the substantia nigra pars compacta. J. Neurochem. 2004, 91, 1302–1311. [Google Scholar] [CrossRef]
- Namba, H.; Nagano, T.; Jodo, E.; Eifuku, S.; Horie, M.; Takebayashi, H.; Iwakura, Y.; Sotoyama, H.; Takei, N.; Nawa, H. Epidermal growth factor signals attenuate phenotypic and functional development of neocortical GABA neurons. J. Neurochem. 2017, 142, 886–900. [Google Scholar] [CrossRef] [Green Version]
- Abe, Y.; Namba, H.; Kato, T.; Iwakura, Y.; Nawa, H. Neuregulin-1 signals from the periphery regulate AMPA receptor sensitivity and expression in GABAergic interneurons in developing neocortex. J. Neurosci. 2011, 31, 5699–5709. [Google Scholar] [CrossRef] [Green Version]
- Ting, A.K.; Chen, Y.; Wen, L.; Yin, D.M.; Shen, C.; Tao, Y.; Liu, X.; Xiong, W.C.; Mei, L. Neuregulin 1 promotes excitatory synapse development and function in GABAergic interneurons. J. Neurosci. 2011, 31, 15–25. [Google Scholar] [CrossRef] [Green Version]
- Yang, J.M.; Zhang, J.; Chen, X.J.; Geng, H.Y.; Ye, M.; Spitzer, N.C.; Luo, J.H.; Duan, S.M.; Li, X.M. Development of GABA circuitry of fast-spiking basket interneurons in the medial prefrontal cortex of erbb4-mutant mice. J. Neurosci. 2013, 33, 19724–19733. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Skirzewski, M.; Karavanova, I.; Shamir, A.; Erben, L.; Garcia-Olivares, J.; Shin, J.H.; Vullhorst, D.; Alvarez, V.A.; Amara, S.G.; Buonanno, A. ErbB4 signaling in dopaminergic axonal projections increases extracellular dopamine levels and regulates spatial/working memory behaviors. Mol. Psychiatry 2018, 23, 2227–2237. [Google Scholar] [CrossRef] [PubMed]
- Yurek, D.M.; Zhang, L.; Fletcher-Turner, A.; Seroogy, K.B. Supranigral injection of neuregulin1-beta induces striatal dopamine overflow. Brain Res. 2004, 1028, 116–119. [Google Scholar] [CrossRef] [PubMed]
- Roy, K.; Murtie, J.C.; El-Khodor, B.F.; Edgar, N.; Sardi, S.P.; Hooks, B.M.; Benoit-Marand, M.; Chen, C.; Moore, H.; O’Donnell, P.; et al. Loss of erbB signaling in oligodendrocytes alters myelin and dopaminergic function, a potential mechanism for neuropsychiatric disorders. Proc. Natl. Acad. Sci. USA 2007, 104, 8131–8136. [Google Scholar] [CrossRef] [Green Version]
- Ledonne, A.; Nobili, A.; Latagliata, E.C.; Cavallucci, V.; Guatteo, E.; Puglisi-Allegra, S.; D’Amelio, M.; Mercuri, N.B. Neuregulin 1 signalling modulates mGluR1 function inmesencephalic dopaminergic neurons. Mol. Psychiatry 2015, 20, 959–973. [Google Scholar] [CrossRef] [Green Version]
- Ledonne, A.; Mercuri, N.B. mGluR1-dependent long term depression in rodent midbrain dopamine neurons is regulated by neuregulin 1/ErbB signaling. Front. Mol. Neurosci. 2018, 11, 346. [Google Scholar] [CrossRef] [Green Version]
- Erben, L.; Welday, J.P.; Cronin, M.E.; Murphy, R.; Skirzewski, M.; Vullhorst, D.; Carroll, S.L.; Buonanno, A. Developmental, neurochemical, and behavioral analyses of ErbB4 Cyt-1 knockout mice. J. Neurochem. 2022, 161, 435–452. [Google Scholar] [CrossRef]
- Skirzewski, M.; Cronin, M.E.; Murphy, R.; Fobbs, W.; Kravitz, A.V.; Buonanno, A. ErbB4 null mice display altered mesocorticolimbic and nigrostriatal dopamine levels as well as deficits in cognitive and motivational behaviors. eNeuro 2020, 7. [Google Scholar] [CrossRef]
- Oyagi, A.; Oida, Y.; Kakefuda, K.; Shimazawa, M.; Shioda, N.; Moriguchi, S.; Kitaichi, K.; Nanba, D.; Yamaguchi, K.; Furuta, Y.; et al. Generation and characterization of conditional heparin-binding EGF-like growth factor knockout mice. PLoS ONE 2009, 4, e7461. [Google Scholar] [CrossRef] [Green Version]
- Golani, I.; Tadmor, H.; Buonanno, A.; Kremer, I.; Shamir, A. Disruption of the ErbB signaling in adolescence increases striatal dopamine levels and affects learning and hedonic-like behavior in the adult mouse. Eur. Neuropsychopharmacol. 2014, 24, 1808–1818. [Google Scholar] [CrossRef]
- Huang, C.M.; Kao, L.S. Nerve growth factor, epidermal growth factor, and insulin differentially potentiate ATP-induced [Ca2+]i rise and dopamine secretion in PC12 cells. J. Neurochem. 1996, 66, 124–130. [Google Scholar] [CrossRef]
- Farkas, L.M.; Krieglstein, K. Heparin-binding epidermal growth factor-like growth factor (HB-EGF) regulates survival of midbrain dopaminergic neurons. J. Neural Transm. 2002, 109, 267–277. [Google Scholar] [CrossRef]
- Mizuno, M.; Sotoyama, H.; Narita, E.; Kawamura, H.; Namba, H.; Zheng, Y.; Eda, T.; Nawa, H. A cyclooxygenase-2 inhibitor ameliorates behavioral impairments induced by striatal administration of epidermal growth factor. J. Neurosci. 2007, 27, 10116–10127. [Google Scholar] [CrossRef] [Green Version]
- Fallon, J.H.; Loughlin, S.E. Substantia nigra. In The Rat Nervous System; Pasxinos, G., Ed.; Academic Press: San Diego, CA, USA, 1995; p. 229. [Google Scholar]
- Namba, H.; Tomiyama, K.; Nawa, H. Abnormal development of nigral dopamine activities in a cytokine-induced schizophrenia model; implication for its postpubertal onset. Soc. Neurosci. Abstr. 2017, 47, 258.13. [Google Scholar]
- Namba, H.; Nawa, H. Post-pubertal difference in nigral dopaminergic cells firing in the schizophrenia model prepared by perinatal challenges of a cytokine, EGF. Neuroscience 2020, 441, 22–32. [Google Scholar] [CrossRef]
- McCutcheon, J.E.; Marinelli, M. Age matters. Eur. J. Neurosci. 2009, 29, 997–1014. [Google Scholar] [CrossRef]
- McCutcheon, J.E.; Conrad, K.L.; Carr, S.B.; Ford, K.A.; McGehee, D.S.; Marinelli, M. Dopamine neurons in the ventral tegmental area fire faster in adolescent rats than in adults. J. Neurophysiol. 2012, 108, 1620–1630. [Google Scholar] [CrossRef] [Green Version]
- Namba, H.; Okubo, T.; Nawa, H. Perinatal exposure to neuregulin-1 results in disinhibition of adult midbrain dopaminergic neurons: Implication in schizophrenia modeling. Sci. Rep. 2016, 6, 22606. [Google Scholar] [CrossRef] [Green Version]
- Giannopoulou, I.; Pagida, M.A.; Briana, D.D.; Panayotacopoulou, M.T. Perinatal hypoxia as a risk factor for psychopathology later in life: The role of dopamine and neurotrophins. Hormones 2018, 17, 25–32. [Google Scholar] [CrossRef] [Green Version]
- Lodge, D.J.; Grace, A.A. Divergent activation of ventromedial and ventrolateral dopamine systems in animal models of amphetamine sensitization and schizophrenia. Int. J. Neuropsychopharmacol. 2012, 15, 69–76. [Google Scholar] [CrossRef] [Green Version]
- Winter, C.; Djodari-Irani, A.; Sohr, R.; Morgenstern, R.; Feldon, J.; Juckel, G.; Meyer, U. Prenatal immune activation leads to multiple changes in basal neurotransmitter levels in the adult brain: Implications for brain disorders of neurodevelopmental origin such as schizophrenia. Int. J. Neuropsychopharmacol. 2009, 12, 513–524. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Swerdlow, N.R.; Mansbach, R.S.; Geyer, M.A.; Pulvirenti, L.; Koob, G.F.; Braff, D.L. Amphetamine disruption of prepulse inhibition of acoustic startle is reversed by depletion of mesolimbic dopamine. Psychopharmacology 1990, 100, 413–416. [Google Scholar] [CrossRef] [PubMed]
- Martinez, Z.A.; Ellison, G.D.; Geyer, M.A.; Swerdlow, N.R. Effects of sustained cocaine exposure on sensorimotor gating of startle in rats. Psychopharmacology 1999, 142, 253–260. [Google Scholar] [CrossRef] [PubMed]
- Swerdlow, N.R.; Keith, V.A.; Braff, D.L.; Geyer, M.A. Effects of spiperone, raclopride, SCH 23390 and clozapine on apomorphine inhibition of sensorimotor gating of the startle response in the rat. J. Pharmacol. Exp. Ther. 1991, 256, 530–536. [Google Scholar] [PubMed]
- Swerdlow, N.R.; Geyer, M.A. Clozapine and haloperidol in an animal model of sensorimotor gating deficits in schizophrenia. Pharmacol. Biochem. Behav. 1993, 44, 741–744. [Google Scholar] [CrossRef]
- Wan, F.J.; Taaid, N.; Swerdlow, N.R. Do D1/D2 interactions regulate prepulse inhibition in rats? Neuropsychopharmacology 1996, 14, 265–274. [Google Scholar]
- Geyer, M.A.; Krebs-Thomson, K.; Braff, D.L.; Swerdlow, N.R. Pharmacological studies of prepulse inhibition models of sensorimotor gating deficits in schizophrenia: A decade in review. Psychopharmacology 2001, 156, 117–154. [Google Scholar] [CrossRef]
- Ralph, R.J.; Paulus, M.P.; Fumagalli, F.; Caron, M.G.; Geyer, M.A. Prepulse inhibition deficits and perseverative motor patterns in dopamine transporter knock-out mice: Differential effects of D1 and D2 receptor antagonists. J. Neurosci. 2001, 21, 305–313. [Google Scholar] [CrossRef] [Green Version]
- Ralph, R.J.; Varty, G.B.; Kelly, M.A.; Wang, Y.M.; Caron, M.G.; Rubinstein, M.; Grandy, D.K.; Low, M.J.; Geyer, M.A. The dopamine D2, but not D3 or D4, receptor subtype is essential for the disruption of prepulse inhibition produced by amphetamine in mice. J. Neurosci. 1999, 19, 4627–4633. [Google Scholar] [CrossRef] [Green Version]
- Wan, F.J.; Geyer, M.A.; Swerdlow, N.R. Accumbens D2 modulation of sensorimotor gating in rats: Assessing anatomical localization. Pharmacol. Biochem. Behav. 1994, 49, 155–163. [Google Scholar] [CrossRef]
- Swerdlow, N.R.; Braff, D.L.; Masten, V.L.; Geyer, M.A. Schizophrenic-like sensorimotor gating abnormalities in rats following dopamine infusion into the nucleus accumbens. Psychopharmacology 1990, 101, 414–420. [Google Scholar] [CrossRef]
- Wan, F.J.; Swerdlow, N.R. Intra-accumbens infusion of quinpirole impairs sensorimotor gating of acoustic startle in rats. Psychopharmacology 1993, 113, 103–109. [Google Scholar] [CrossRef]
- Swerdlow, N.R.; Braff, D.L.; Geyer, M.A. GABAergic projection from nucleus accumbens to ventral pallidum mediates dopamine-induced sensorimotor gating deficits of acoustic startle in rats. Brain Res. 1990, 532, 146–150. [Google Scholar] [CrossRef]
- Swerdlow, N.R.; Caine, S.B.; Geyer, M.A. Regionally selective effects of intracerebral dopamine infusion on sensorimotor gating of the startle reflex in rats. Psychopharmacology 1992, 108, 189–195. [Google Scholar] [CrossRef]
- Sotoyama, H.; Namba, H.; Kobayashi, Y.; Hasegawa, T.; Watanabe, D.; Nakatsukasa, E.; Sakimura, K.; Furuyashiki, T.; Nawa, H. Resting-state dopaminergic cell firing in the ventral tegmental area negatively regulates affiliative social interactions in a developmental animal model of schizophrenia. Transl. Psychiatry 2021, 11, 236. [Google Scholar] [CrossRef]
- Sotoyama, H.; Inaba, H.; Iwakura, Y.; Namba, H.; Takei, N.; Sasaoka, T.; Nawa, H. The dual role of dopamine in the modulation of information processing in the prefrontal cortex underlying social behavior. FASEB J. 2022, 36, e22160. [Google Scholar] [CrossRef]
- Fauchey, V.; Jaber, M.; Caron, M.G.; Bloch, B.; Le Moine, C. Differential regulation of the dopamine D1, D2 and D3 receptor gene expression and changes in the phenotype of the striatal neurons in mice lacking the dopamine transporter. Eur. J. Neurosci. 2000, 12, 19–26. [Google Scholar] [CrossRef]
- Dumartin, B.; Jaber, M.; Gonon, F.; Caron, M.G.; Giros, B.; Bloch, B. Dopamine tone regulates D1 receptor trafficking and delivery in striatal neurons in dopamine transporter-deficient mice. Proc. Natl. Acad. Sci. USA 2000, 97, 1879–1884. [Google Scholar] [CrossRef] [Green Version]
- Gunaydin, L.A.; Grosenick, L.; Finkelstein, J.C.; Kauvar, I.V.; Fenno, L.E.; Adhikari, A.; Lammel, S.; Mirzabekov, J.J.; Airan, R.D.; Zalocusky, K.A. Natural neural projection dynamics underlying social behavior. Cell 2014, 157, 1535–1551. [Google Scholar] [CrossRef] [Green Version]
- Chaudhury, D.; Walsh, J.J.; Friedman, A.K.; Juarez, B.; Ku, S.M.; Koo, J.W.; Ferguson, D.; Tsai, H.C.; Pomeranz, L.; Christoffel, D.J.; et al. Rapid regulation of depression-related behaviours by control of midbrain dopamine neurons. Nature 2013, 493, 532–536. [Google Scholar] [CrossRef] [Green Version]
- Lipska, B.K.; Jaskiw, G.E.; Chrapusta, S.; Karoum, F.; Weinberger, D.R. Ibotenic acid lesion of the ventral hippocampus differentially affects dopamine and its metabolites in the nucleus accumbens and prefrontal cortex in the rat. Brain Res. 1992, 585, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Rueter, L.E.; Ballard, M.E.; Gallagher, K.B.; Basso, A.M.; Curzon, P.; Kohlhaas, K.L. Chronic low dose risperidone and clozapine alleviate positive but not negative symptoms in the rat neonatal ventral hippocampal lesion model of schizophrenia. Psychopharmacology 2004, 176, 312–319. [Google Scholar] [CrossRef] [PubMed]
- Coyle, J.T. Glutamate and schizophrenia: Beyond the dopamine hypothesis. Cell Mol. Neurobiol. 2006, 26, 365–384. [Google Scholar] [CrossRef] [PubMed]
- Volavka, J.; Czobor, P.; Sheitman, B.; Lindenmayer, J.P.; Citrome, L.; McEvoy, J.P.; Cooper, T.B.; Chakos, M.; Lieberman, J.A. Clozapine, olanzapine, risperidone, and haloperidol in the treatment of patients with chronic schizophrenia and schizoaffective disorder. Am. J. Psychiatry 2002, 159, 255–262. [Google Scholar] [CrossRef] [PubMed]
- Tollefson, G.D.; Sanger, T.M. Negative symptoms: A path analytic approach to a double-blind, placebo- and haloperidol-controlled clinical trial with olanzapine. Am. J. Psychiatry 1997, 154, 466–474. [Google Scholar]
- Harrow, M.; Yonan, C.A.; Sands, J.R.; Marengo, J. Depression in schizophrenia: Are neuroleptics, akinesia, or anhedonia involved? Schizophr. Bull. 1994, 20, 327–338. [Google Scholar] [CrossRef] [Green Version]
- Segura-Aguilar, J.; Paris, I.; Muñoz, P.; Ferrari, E.; Zecca, L.; Zucca, F.A. Protective and toxic roles of dopamine in parkinson’s disease. J. Neurochem. 2014, 129, 898–915. [Google Scholar] [CrossRef]
- Ferrer, I.; Alcántara, S.; Ballabriga, J.; Olivé, M.; Blanco, R.; Rivera, R.; Carmona, M.; Berruezo, M.; Pitarch, S.; Planas, A.M. Transforming growth factor-alpha (TGF-Alpha) and epidermal growth factor-receptor (EGF-R) immunoreactivity in normal and pathologic brain. Prog. Neurobiol. 1996, 49, 99–123. [Google Scholar] [CrossRef]
- Qu, W.-S.; Liu, J.-L.; Li, C.-Y.; Li, X.; Xie, M.-J.; Wang, W.; Tian, D.-S. Rapidly activated epidermal growth factor receptor mediates lipopolysaccharide-triggered migration of microglia. Neurochem. Int. 2015, 90, 85–92. [Google Scholar] [CrossRef]
- Zhang, J.; Wu, J.; Fujita, Y.; Yao, W.; Ren, Q.; Yang, C.; Li, S.; Shirayama, Y.; Hashimoto, K. Antidepressant effects of TrkB ligands on depression-like behavior and dendritic changes in mice after inflammation. Int. J. Neuropsychopharmacol. 2014, 18, pyu077. [Google Scholar] [CrossRef]
- Furuyashiki, T.; Kitaoka, S. Neural mechanisms underlying adaptive and maladaptive consequences of stress: Roles of dopaminergic and inflammatory responses. Psychiatry Clin. Neurosci. 2019, 73, 669–675. [Google Scholar] [CrossRef] [Green Version]
- Wang, H.; He, Y.; Sun, Z.; Ren, S.; Liu, M.; Wang, G.; Yang, J. Microglia in depression: An overview of microglia in the pathogenesis and treatment of depression. J. Neuroinflamm. 2022, 19, 132. [Google Scholar] [CrossRef]
- Okubo, Y.; Suhara, T.; Suzuki, K.; Kobayashi, K.; Inoue, O.; Terasaki, O.; Someya, Y.; Sassa, T.; Sudo, Y.; Matsushima, E.; et al. Decreased prefrontal dopamine D1 receptors in schizophrenia revealed by PET. Nature 1997, 385, 634–636. [Google Scholar] [CrossRef]
- Stenkrona, P.; Matheson, G.J.; Halldin, C.; Cervenka, S.; Farde, L. D1-dopamine receptor availability in first-episode neuroleptic naive psychosis patients. Int. J. Neuropsychopharmacol. 2019, 22, 415–425. [Google Scholar] [CrossRef] [Green Version]
- Kosaka, J.; Takahashi, H.; Ito, H.; Takano, A.; Fujimura, Y.; Matsumoto, R.; Nozaki, S.; Yasuno, F.; Okubo, Y.; Kishimoto, T.; et al. Decreased binding of [11C]NNC112 and [11C]SCH23390 in patients with chronic schizophrenia. Life Sci. 2010, 86, 814–818. [Google Scholar] [CrossRef]
- Hirvonen, J.; van Erp, T.G.; Huttunen, J.; Aalto, S.; Någren, K.; Huttunen, M.; Lönnqvist, J.; Kaprio, J.; Cannon, T.D.; Hietala, J. Brain dopamine d1 receptors in twins discordant for schizophrenia. Am. J. Psychiatry 2006, 163, 1747–1753. [Google Scholar] [CrossRef]
- Lindström, L.H.; Gefvert, O.; Hagberg, G.; Lundberg, T.; Bergström, M.; Hartvig, P.; Långström, B. Increased dopamine synthesis rate in medial prefrontal cortex and striatum in schizophrenia indicated by L-(beta-11C) DOPA and PET. Biol. Psychiatry 1999, 46, 681–688. [Google Scholar] [CrossRef]
- Watanabe, Y.; Tanaka, H.; Tsukabe, A.; Kunitomi, Y.; Nishizawa, M.; Hashimoto, R.; Yamamori, H.; Fujimoto, M.; Fukunaga, M.; Tomiyama, N. Neuromelanin magnetic resonance imaging reveals increased dopaminergic neuron activity in the substantia nigra of patients with schizophrenia. PLoS ONE 2014, 9, e104619. [Google Scholar] [CrossRef] [Green Version]
- Shibata, E.; Sasaki, M.; Tohyama, K.; Otsuka, K.; Endoh, J.; Terayama, Y.; Sakai, A. Use of neuromelanin-sensitive MRI to distinguish schizophrenic and depressive patients and healthy individuals based on signal alterations in the substantia nigra and locus ceruleus. Biol. Psychiatry 2008, 64, 401–406. [Google Scholar] [CrossRef]
- Mizuno, M.; Sotoyama, H.; Namba, H.; Shibuya, M.; Eda, T.; Wang, R.; Okubo, T.; Nagata, K.; Iwakura, Y.; Nawa, H. ErbB inhibitors ameliorate behavioral impairments of an animal model for schizophrenia: Implication of their dopamine-modulatory actions. Transl. Psychiatry 2013, 3, e252. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mizuno, M.; Iwakura, Y.; Shibuya, M.; Zheng, Y.; Eda, T.; Kato, T.; Takasu, Y.; Nawa, H. Antipsychotic potential of quinazoline ErbB1 inhibitors in a schizophrenia model established with neonatal hippocampal lesioning. J. Pharmacol. Sci. 2010, 114, 320–331. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tadmor, H.; Golani, I.; Doron, R.; Kremer, I.; Shamir, A. ErbB signaling antagonist ameliorates behavioral deficit induced by phencyclidine (PCP) in mice, without affecting metabolic syndrome markers. Prog. Neuropsychopharmacol. Biol. Psychiatry 2018, 82, 322–331. [Google Scholar] [CrossRef] [PubMed]
- Mizuno, M.; Kawamura, H.; Takei, N.; Nawa, H. The anthraquinone derivative Emodin ameliorates neurobehavioral deficits of a rodent model for schizophrenia. J. Neural Transm. 2008, 115, 521–530. [Google Scholar] [CrossRef]
- Kobayashi, Y.; Iwakura, Y.; Sotoyama, H.; Kitayama, E.; Takei, N.; Someya, T.; Nawa, H. Clozapine-dependent inhibition of EGF/neuregulin receptor (ErbB) kinases. Transl. Psychiatry 2019, 9, 181. [Google Scholar] [CrossRef] [Green Version]
- de Castro, G., Jr.; Awada, A. Side effects of anti-cancer molecular-targeted therapies (not monoclonal antibodies). Curr. Opin. Oncol. 2006, 18, 307–315. [Google Scholar] [CrossRef]
- Pandey, A.; Kalita, K.N. Treatment-resistant schizophrenia: How far have we traveled? Front. Psychiatry 2022, 13, 994425. [Google Scholar] [CrossRef]
- Leucht, S.; Corves, C.; Arbter, D.; Engel, R.R.; Li, C.; Davis, J.M. Second-generation versus first-generation antipsychotic drugs for schizophrenia: A meta-analysis. Lancet 2009, 373, 31–41. [Google Scholar] [CrossRef]
- Pereira, A.; Sugiharto-Winarno, A.; Zhang, B.; Malcolm, P.; Fink, G.; Sundram, S. Clozapine induction of ERK1/2 cell signalling via the EGF receptor in mouse prefrontal cortex and striatum is distinct from other antipsychotic drugs. Int. J. Neuropsychopharmacol. 2012, 15, 1149–1160. [Google Scholar] [CrossRef] [Green Version]
- Pereira, A.; Zhang, B.; Malcolm, P.; Sundram, S. Clozapine regulation of p90RSK and c-Fos signaling via the ErbB1-ERK pathway is distinct from olanzapine and haloperidol in mouse cortex and striatum. Prog. Neuropsychopharmacol. Biol. Psychiatry 2013, 40, 353–363. [Google Scholar] [CrossRef]
- Fitton, R.; Sweetman, J.; Heseltine-Carp, W.; van der Feltz-Cornelis, C. Anti-inflammatory medications for the treatment of mental disorders: A scoping review. Brain Behav. Immun. 2022, 26, 100518. [Google Scholar] [CrossRef]
- Diwanji, D.; Trenker, R.; Thaker, T.M.; Wang, F.; Agard, D.A.; Verba, K.A.; Jura, N. Structures of the HER2-HER3-NRG1β complex reveal a dynamic dimer interface. Nature 2021, 600, 339–343. [Google Scholar] [CrossRef] [PubMed]
- Haddad, Y.; Remes, M.; Adam, V.; Heger, Z. Toward structure-based drug design against the epidermal growth factor receptor (EGFR). Drug Discov. Today 2021, 26, 289–295. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
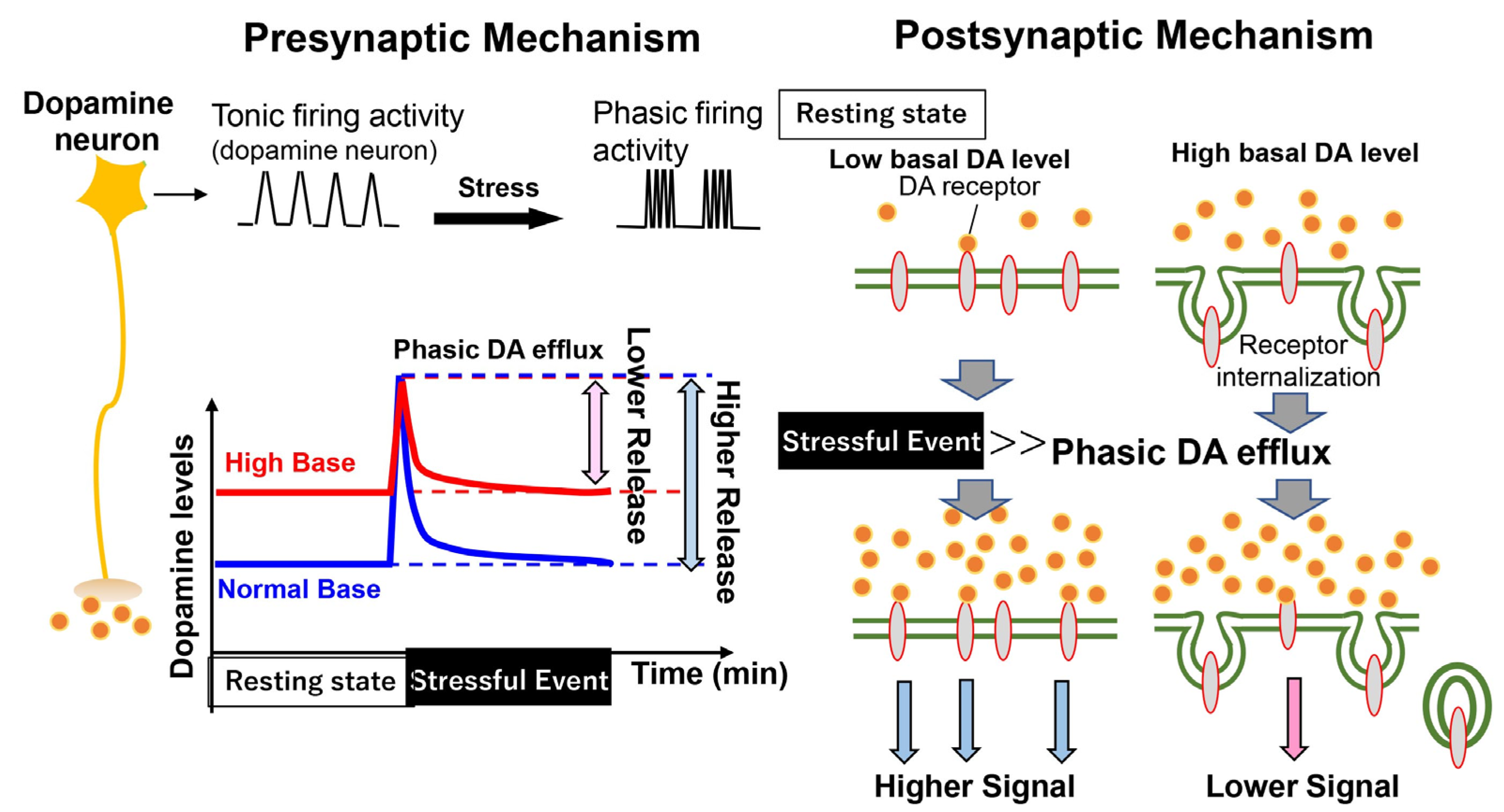{kind=link}
| Drug | Target | Assay Model | Effects | References |
|---|---|---|---|---|
| gefitinib | ErbB1 | EGF, VHL | PPI, LI | [162,163] |
| PD153035 | ErbB1/B2 | EGF, VHL | PPI | [162,163] |
| erlotinib | ErbB1 | VHL | PPI | [163] |
| JNJ28871063 | pan-ErbB | PCP | Social Interaction | [164] |
| emodin | tyrosine kinases | EGF, VHL | PPI, LI | [165] |
| clozapine | pan-ErbB | in vitro kinase | Tyr phosphorylation | [166] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sotoyama, H.; Namba, H.; Tohmi, M.; Nawa, H. Schizophrenia Animal Modeling with Epidermal Growth Factor and Its Homologs: Their Connections to the Inflammatory Pathway and the Dopamine System. Biomolecules 2023, 13, 372. https://doi.org/10.3390/biom13020372
Sotoyama H, Namba H, Tohmi M, Nawa H. Schizophrenia Animal Modeling with Epidermal Growth Factor and Its Homologs: Their Connections to the Inflammatory Pathway and the Dopamine System. Biomolecules. 2023; 13(2):372. https://doi.org/10.3390/biom13020372
Chicago/Turabian StyleSotoyama, Hidekazu, Hisaaki Namba, Manavu Tohmi, and Hiroyuki Nawa. 2023. "Schizophrenia Animal Modeling with Epidermal Growth Factor and Its Homologs: Their Connections to the Inflammatory Pathway and the Dopamine System" Biomolecules 13, no. 2: 372. https://doi.org/10.3390/biom13020372