1. Introduction
Computational drug-likeness prediction aims to assess the chance for a compound to become a marketed drug and is one of the essential metrics for screening drug candidates. Early drug-likeness predictions were made by extracting rules based on physicochemical properties. Lipinski et al. proposed ‘the Rule of 5’ (Ro5) to exclude compounds with poor absorption or permeation, which are unbeneficial for drug-likeness [
1]. The Ro5 states that a compound is likely to have poor absorption or permeation when it has more than 5 hydrogen-bond donors, more than 10 hydrogen-bond acceptors, a molecular weight greater than 500 Da, and a calculated octanol–water partition coefficient greater than 5. Ghose et al. proposed an additional physicochemical property, molar refractivity, to complete the Ro5 [
2]. Similarly, Veber et al. included the number of rotatable bonds and the polar surface area to account for molecular flexibility [
3]. Although rule-based methods are convenient to implement, they are not applicable to all drug molecules, because they are based on simple criteria. According to a previous study, 16% of 771 oral drugs from the ChEMBL database of bioactive molecules with drug-like properties violate at least one component of the Ro5, and 6% violate more than two [
4]. For a more quantitative estimation of drug-likeness, Bickerton et al. introduced the Quantitative Estimation of Drug-Likeness (QED) score [
4] based on desirability functions fitted to the distributions of eight physicochemical properties, including properties used by Veber filters [
3], the number of aromatic rings, and the number of structural alerts. QED not only outperforms the Ro5 and other rules but also can modulate thresholds according to specific requirements, providing more flexibility for screening drug candidates. Besides physicochemical properties, drug-likeness is also related to chemical absorption, distribution, metabolism, excretion, and toxicity (ADMET) properties. Recently, Guan et al. developed the ADMET-score to evaluate the drug-likeness of compounds based on 18 weighted ADMET properties [
5]. The ADMET-score was able to distinguish withdrawn drugs from approved drugs with statistically significant accuracy.
Accurate prediction of drug-likeness is more complex than a simple counting of molecular properties. To improve the accuracy of drug-likeness prediction, researchers have adopted machine learning algorithms based on various molecular descriptors that enable more concrete representations of molecular information than basic chemical properties. Examples of these molecular descriptors include extended atom types [
6], Ghose–Crippen fragment descriptors [
7], molecular operating environment physicochemical descriptors [
7], topological pharmacophore descriptors [
7], ECFP4 fingerprints [
8], LCFP6 fingerprints [
9], and their combinations [
7]. These descriptors, combined with various machine learning methods including support-vector machines [
7,
8,
10], artificial neural networks [
7], decision trees [
6], naïve Bayesian classifiers [
11], and recursive partitioning [
11], have increased the accuracy of methods to discriminate drug-like and non-drug-like compounds to about 90%. In recent years, deep learning as a powerful machine learning method has been applied for drug-likeness prediction. Hu et al. proposed a deep autoencoder model to classify drug-like and non-drug-like compounds based on the synthetic minority oversampling technique (SMOTE), which achieved 96% accuracy [
12]. Seyed et al. proposed a deep belief model based on a combination of fingerprints including MACCS, PubChem fingerprints, and ECFP4 [
13], which yielded similar accuracy. Beker et al. combined different deep learning models using a Bayesian neural network and then chose the predictions of drug-like molecules with the lowest variance. According to their findings, the combination of graph convolution neural network (GCNN) and autoencoder yielded the highest accuracy of 93% [
14]. Lee et al. generated a drug-likeness score function using unsupervised learning, which can predict drug-likeness with a continuous value rather than a rigid cut-off [
15]. Their model also obtained over 90% accuracy in classifying drugs and non-drug molecules.
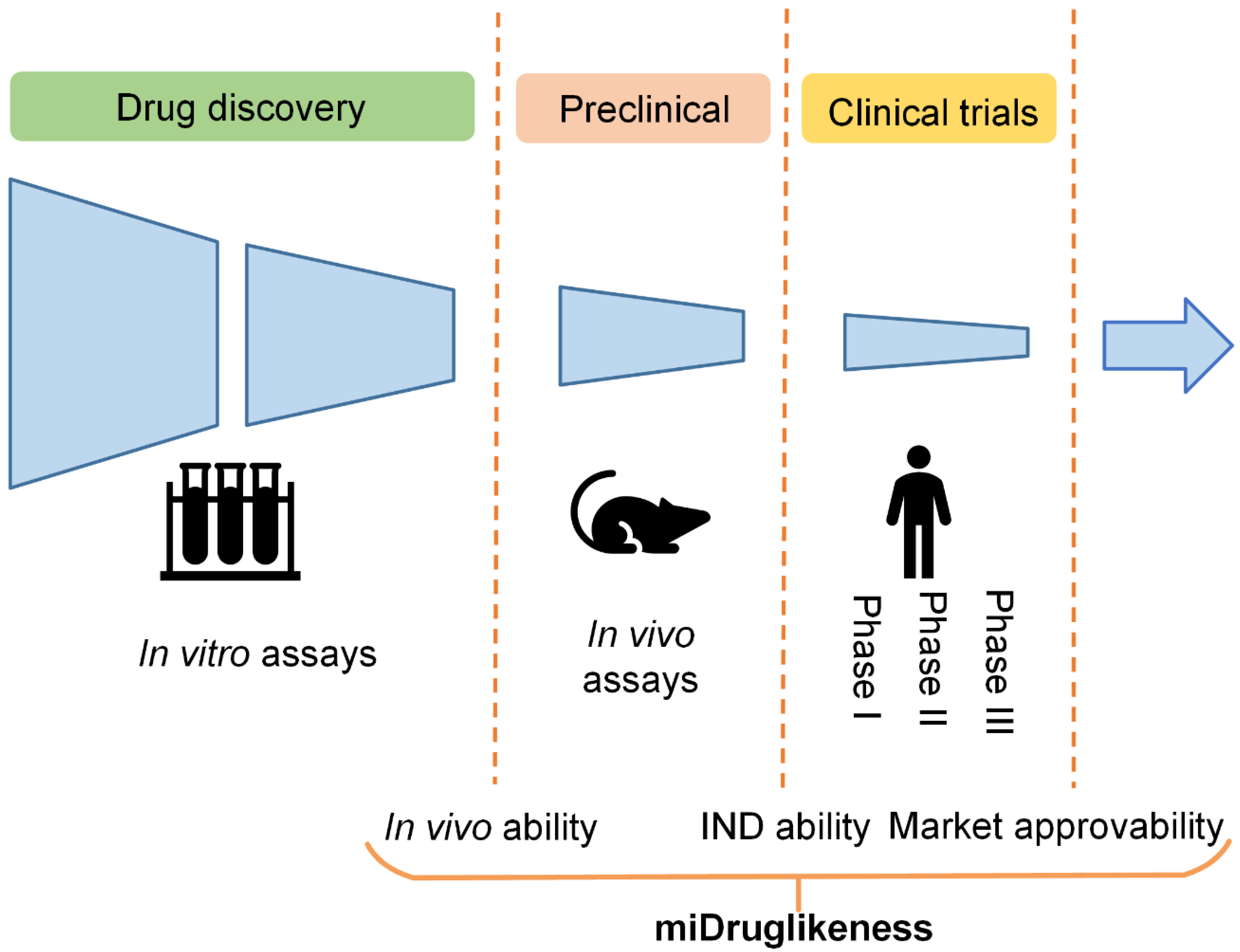
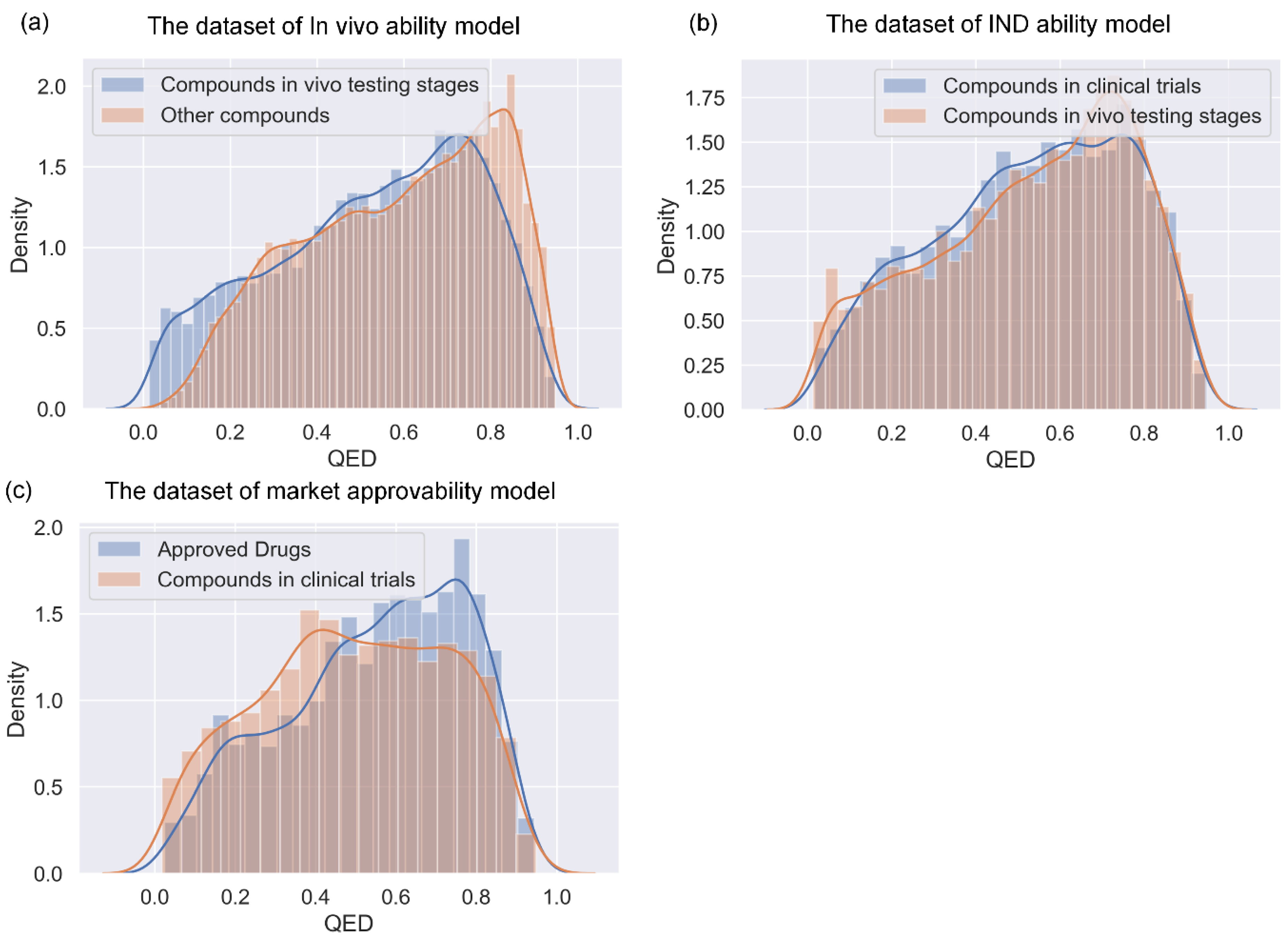
The aforementioned drug-likeness prediction models are typically trained using approved drugs (or a combination of approved drugs and candidate drugs in clinical trials) as positive samples and purchasable compounds as negative samples. However, there are situations where we are interested in determining the likelihood that a compound that is initially identified as being drug-like will be successful in the in vivo testing stage (referred to here as ‘in vivo ability’). Similarly, we may wish to forecast whether a compound in the in vivo testing stage will become an investigational new drug (referred to here as ‘IND ability’), or whether an investigational new drug will ultimately be approved to become a marketed drug (referred to here as ‘market approvability’). Failure in drug development can occur in various stages. For example, the clinical failure rate from phase I clinical trials to the successful launching of a drug on the market is over 90% [
16]. The subdivisional drug-likeness prediction including market approvability prediction can therefore be useful for boosting success rates in clinical trials and other stages.
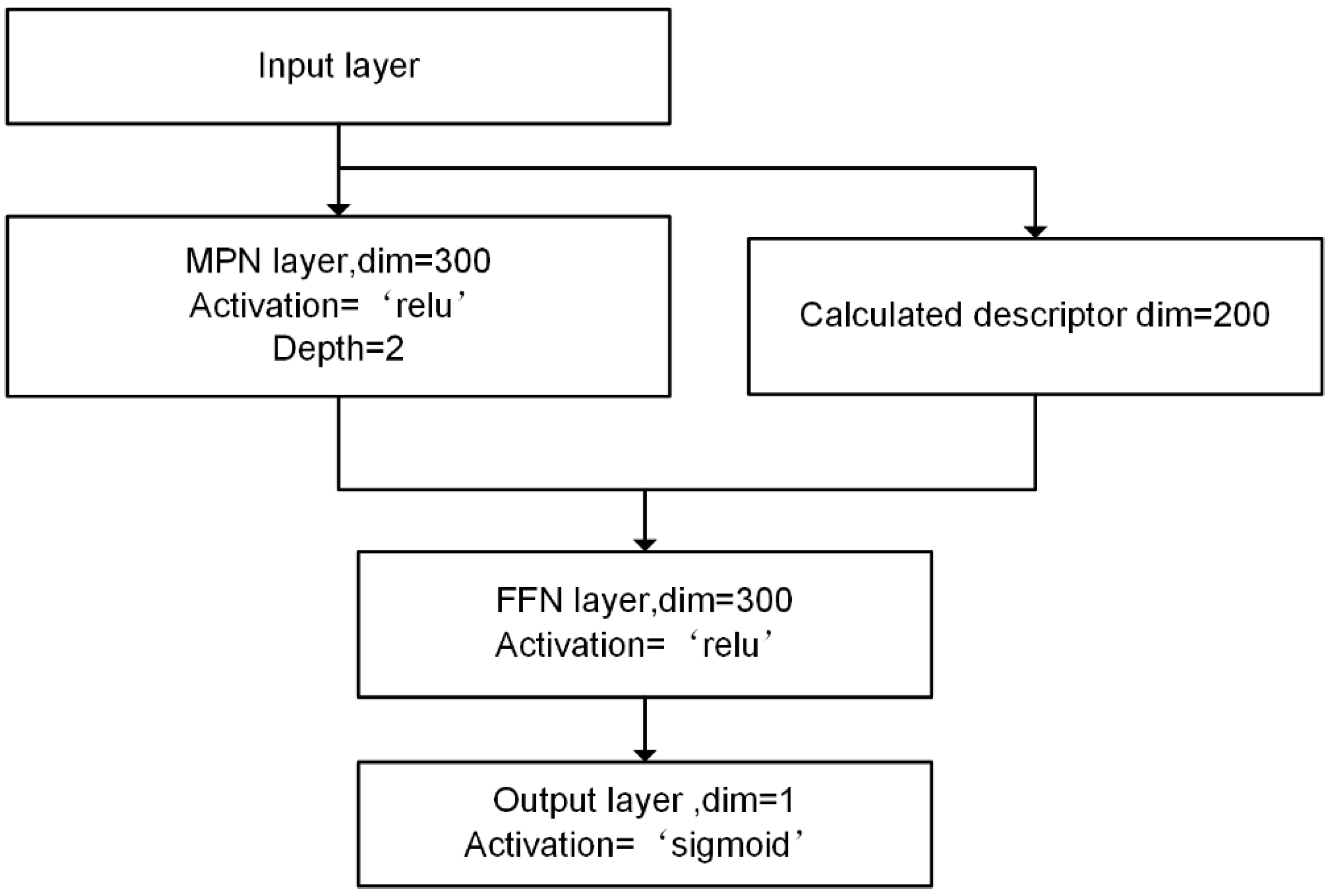
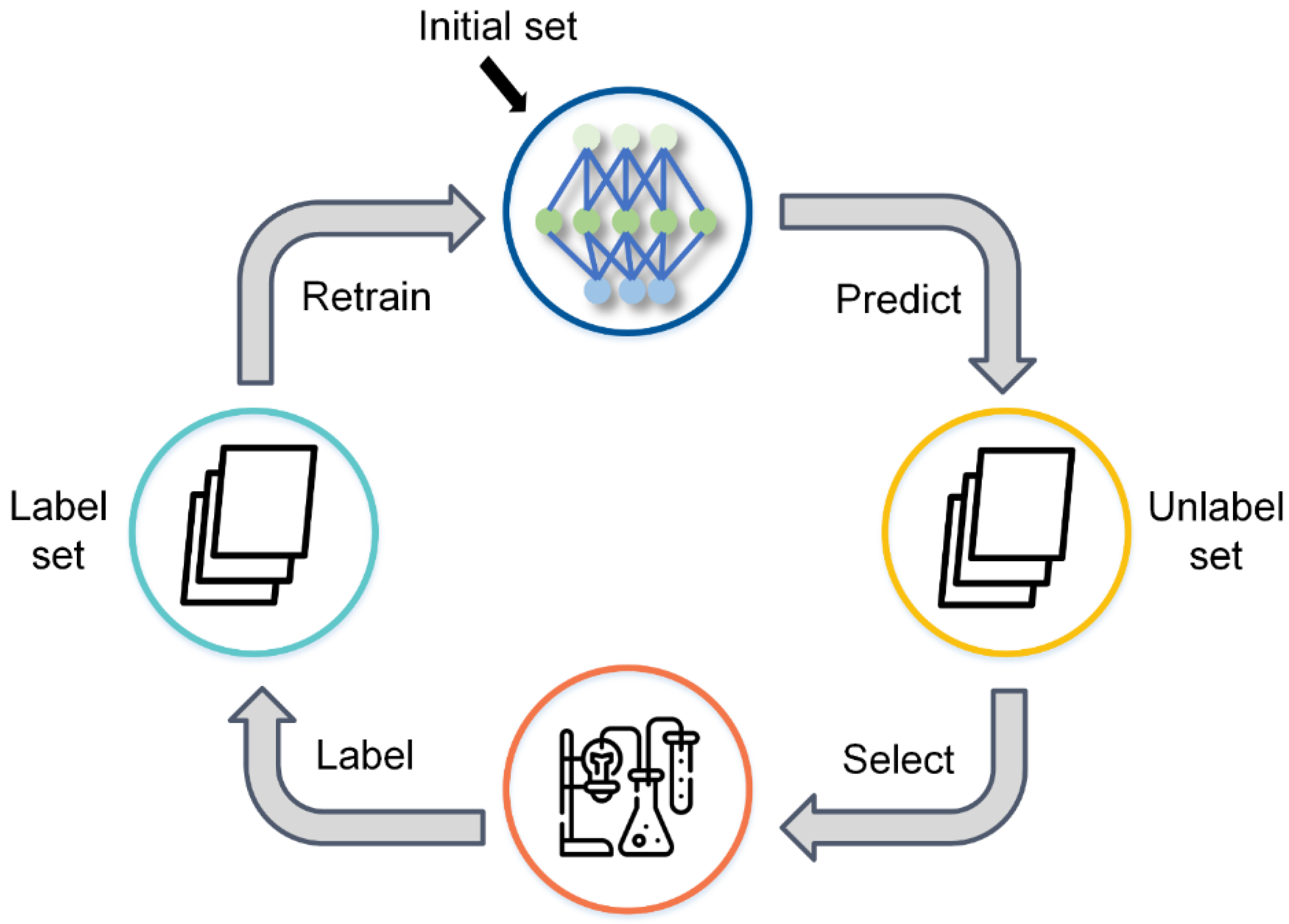
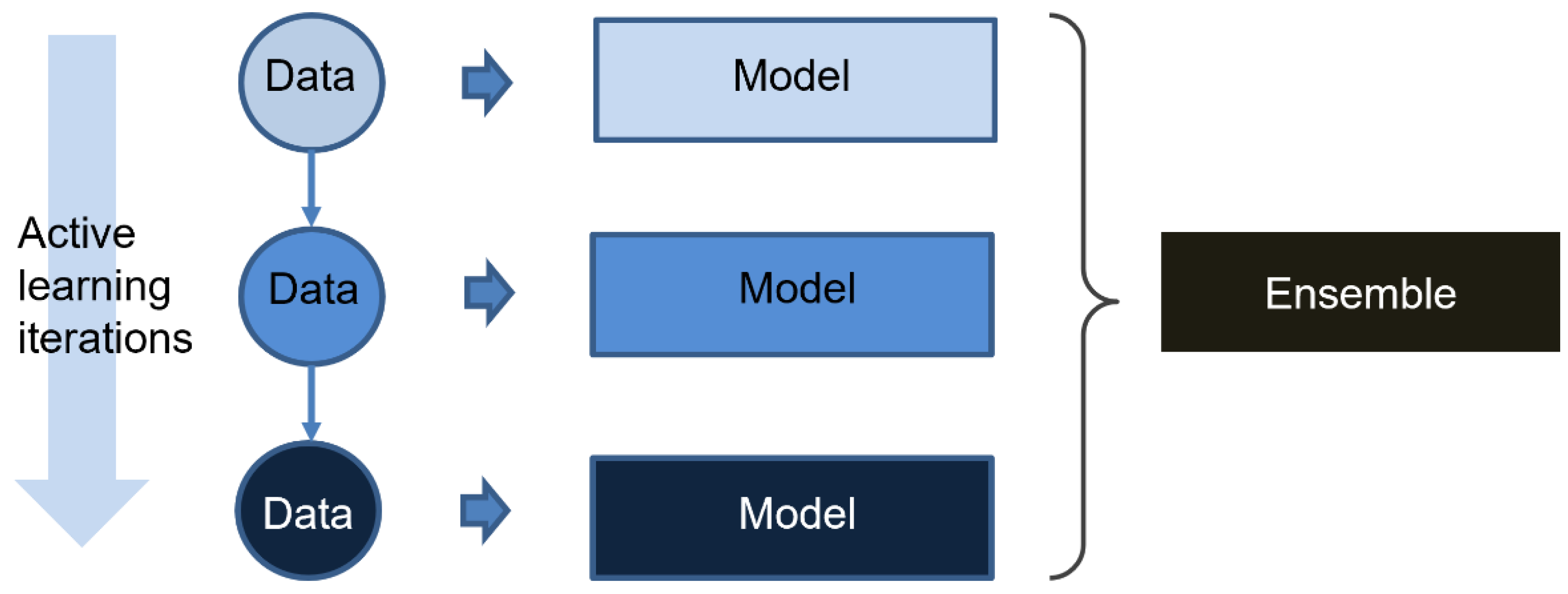
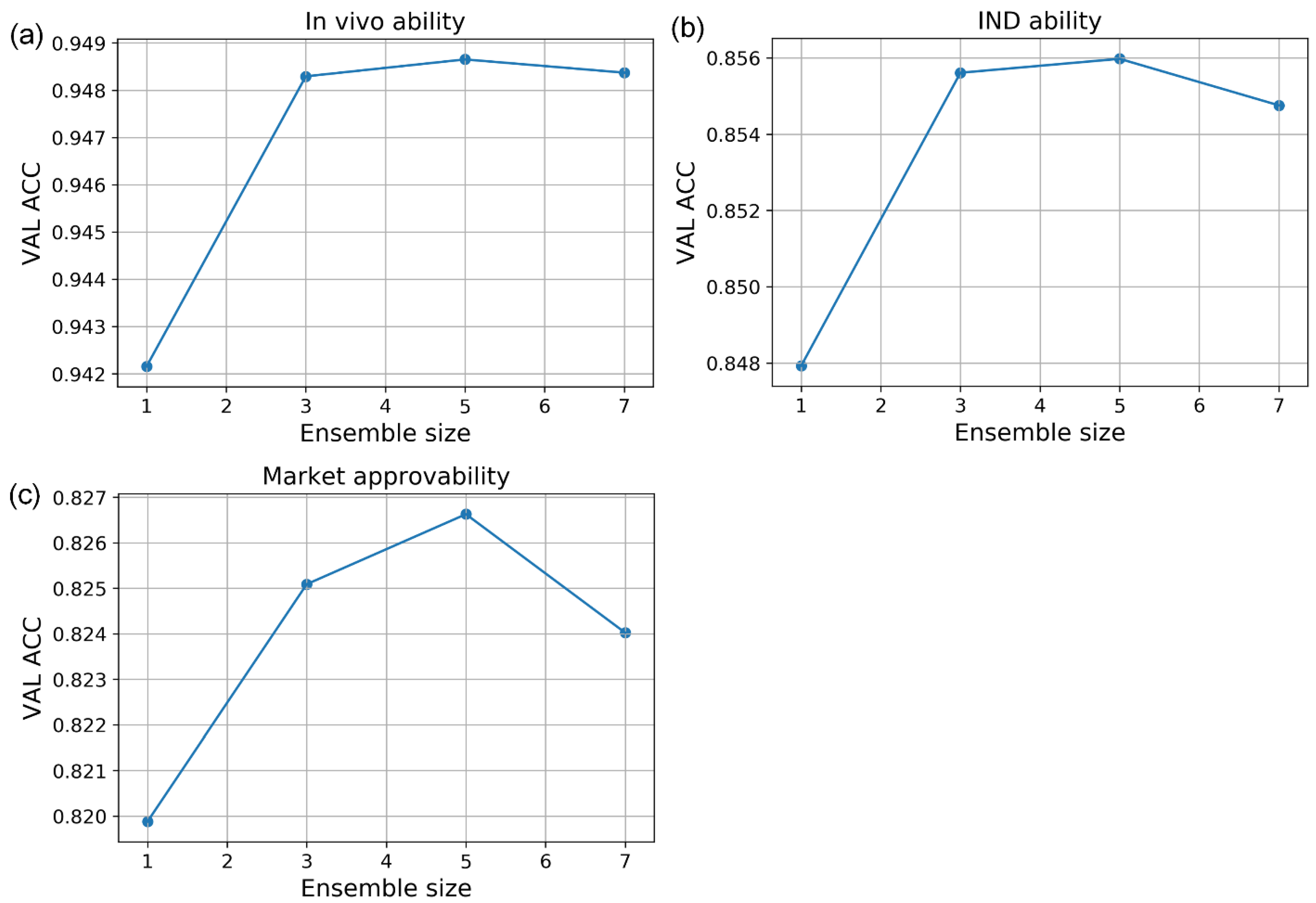
In this study, we developed a miDruglikeness system including three subdivisional drug-likeness prediction models to deal with the abovementioned problems (
Figure 1). The in vivo ability, IND ability, and market approvability models were respectively built using graph neural networks (GNNs). To enhance the predictive ability of these models, we combined active learning with ensemble learning. The miDruglikeness system showed satisfying performance in internal test datasets and self-collected external test datasets. The performance of miDruglikeness in the virtual screening test was significantly better than QED and state-of-the-art protein-ligand scoring-based methods. Besides, we also employed Shapley Additive exPlanation (SHAP) [
17] to interpret these models and built a webserver of miDruglikeness for public usage.
4. Discussion
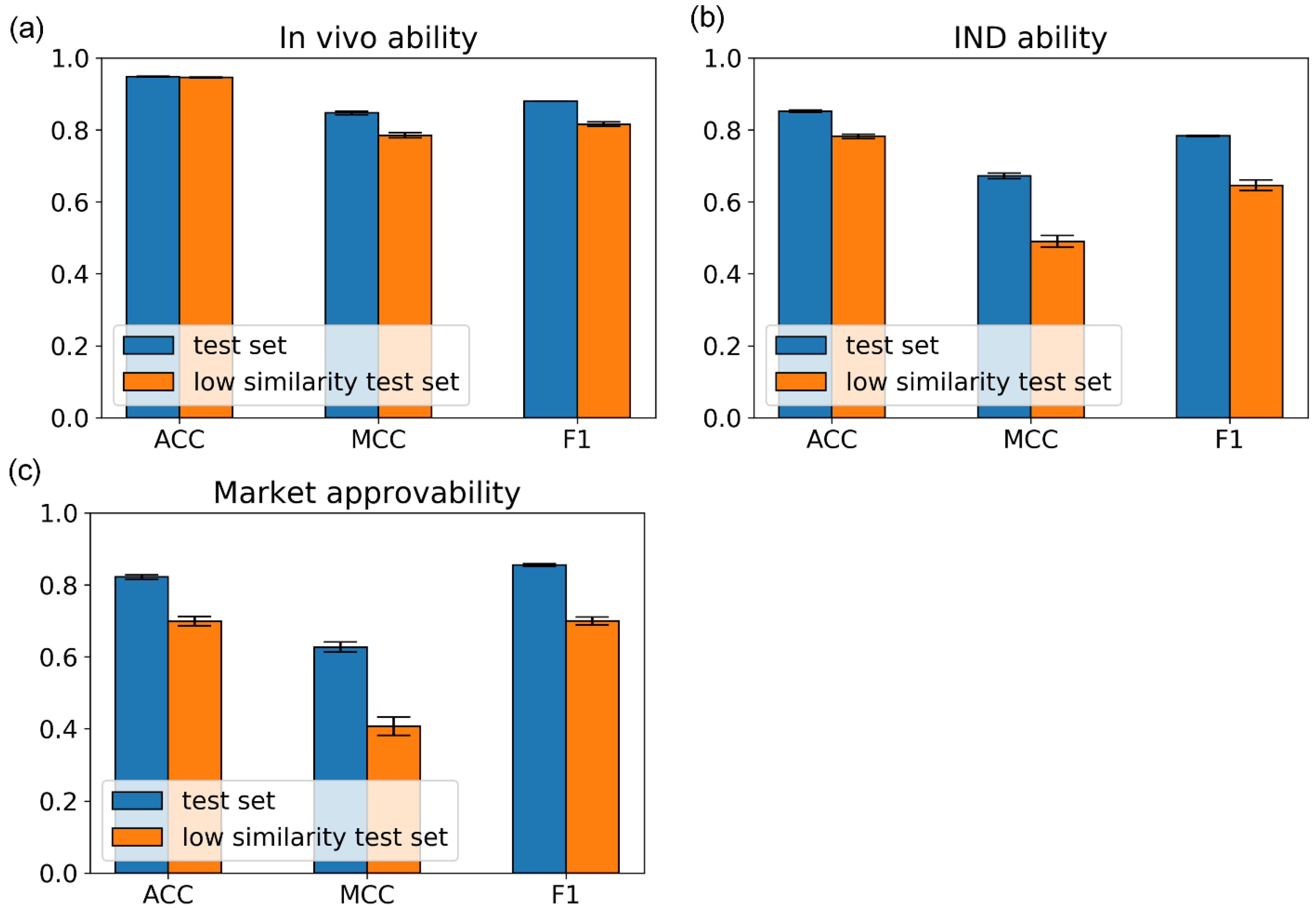
In this work, we first proposed and implemented three subdivisional models to individually predict the possibility of a compound entering the in vivo testing, clinical trials, and market approval stages. By making drug-likeness predictions in this subtle manner, we can predict the probability of a compound entering the next stage of drug discovery and development. Our testing results demonstrated the robustness and the generalization ability of miDruglikeness models. Although docking approaches based on target information are popular for virtual screening, their performance is inadequate. Our in vivo ability model showed a very good performance for virtual screening. Since in vivo ability model is target independent, it would serve as an orthogonal complementary to target-based virtual screening methods and is anticipated to considerably increase the success rate of virtual screening. Many researchers utilize QED as a filter in virtual screening. However, we discovered that our in vivo ability model worked far better than QED. Thus, we highly recommend that users can employ in vivo ability as a strong filter in their virtual screening campaigns.
The miDruglikeness models are mainly based on the structural information of compounds. Nevertheless, drug approval is a complicated process that is also affected by biological and commercial factors. Thus, the IND ability prediction task and market approvability prediction are more challenging than in vivo ability prediction task. To further improve the predictive performance of the IND ability model and the market approvability model, additional information such as biological data from in vivo assays and clinical trials should also be considered.
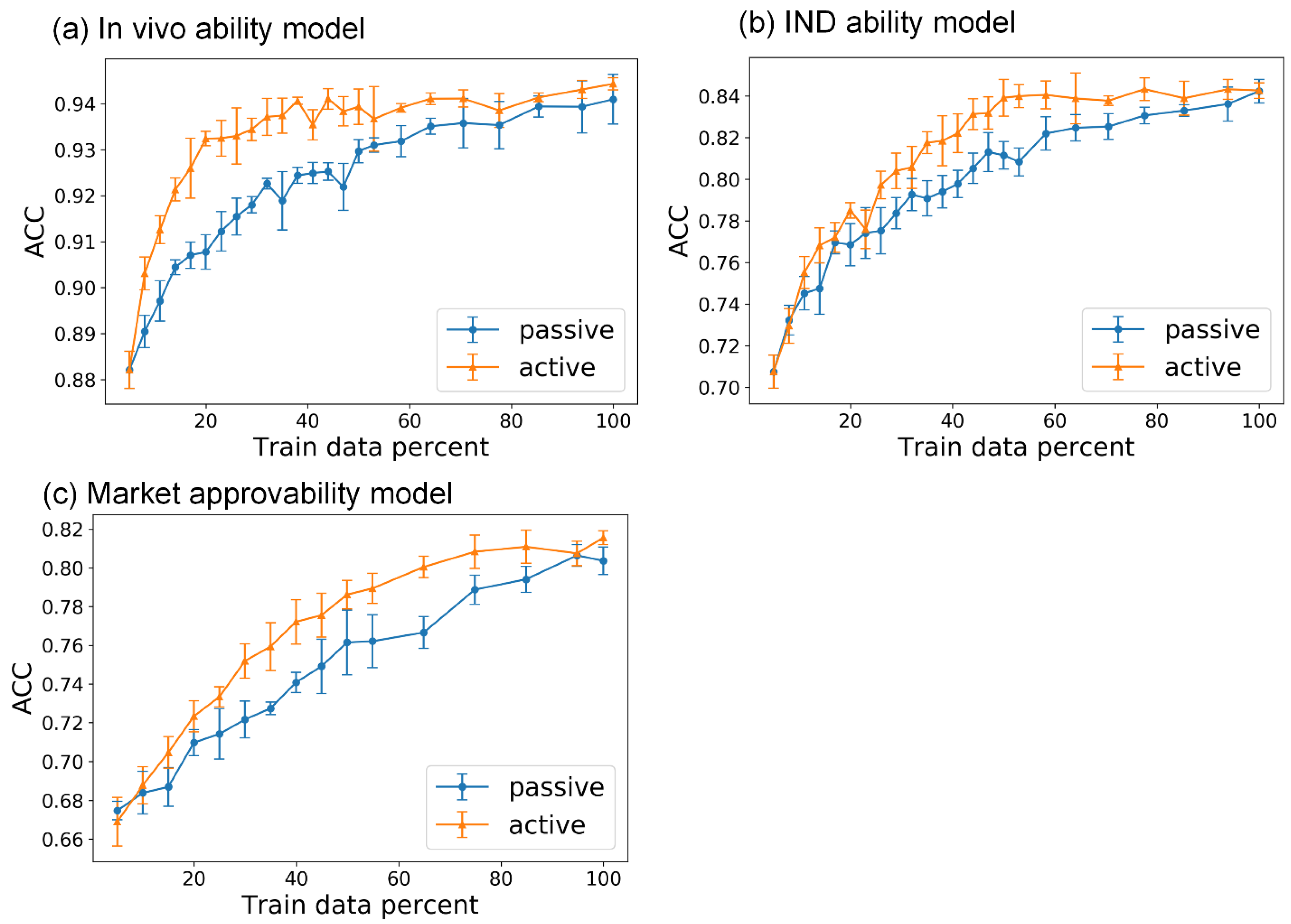
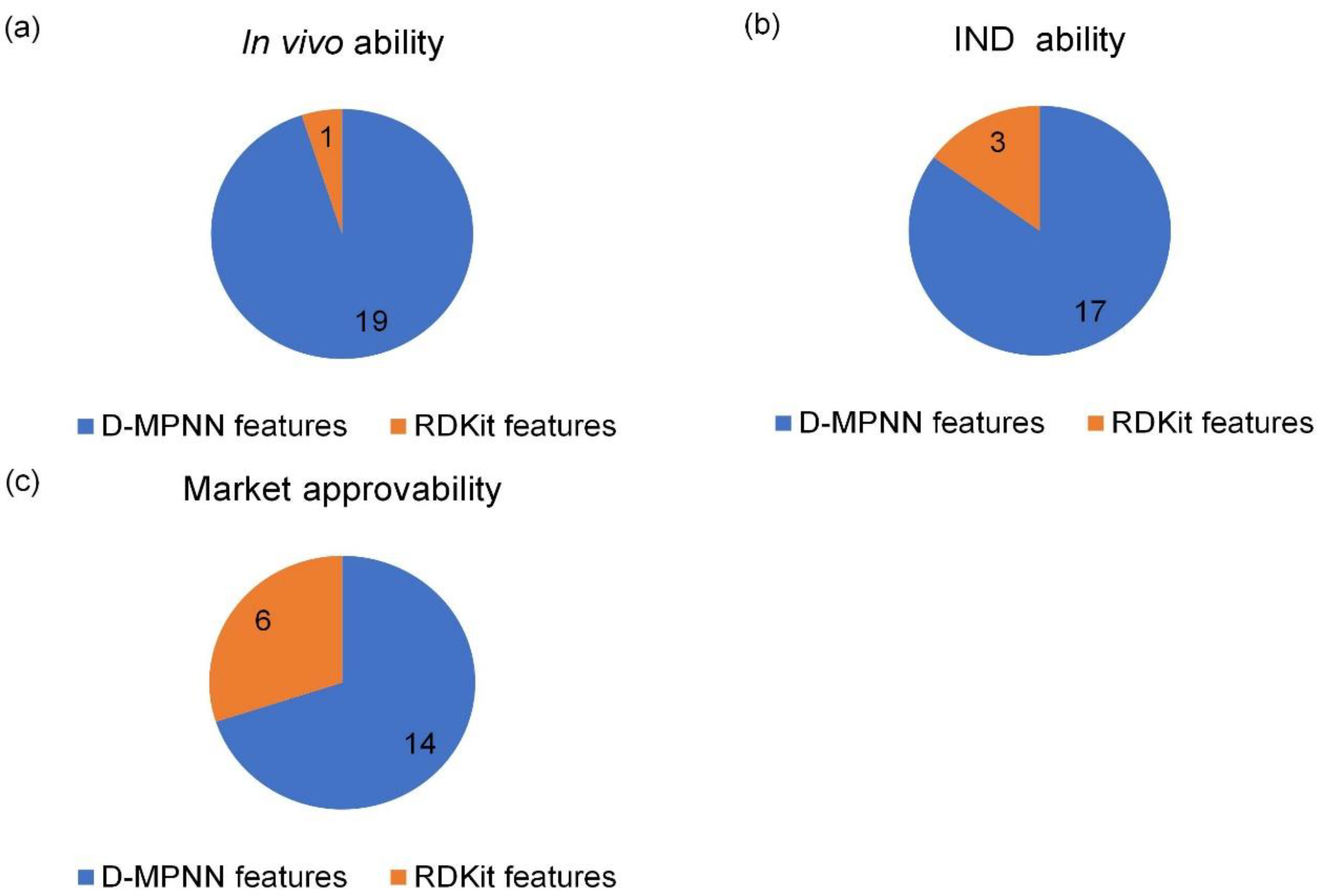
We developed the active ensemble learning method for miDruglikeness models in this study. Additionally, active ensemble learning is a general strategy for machine learning models with uncertainty and can be used for a variety of different tasks, such as the prediction of bioactivities. Active ensemble learning can combinedly be used with other methods such as XGBoost and DNN. From
Figure S7, we also discovered that active learning could maintain data balance at a certain range in the data selection. It could be applied for scenarios when sampling imbalanced data.


 ,
, 









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}