

Identification of Activated Cdc42-Associated Kinase Inhibitors as Potential Anticancer Agents Using Pharmacoinformatic Approaches

 ,
,  ,
,  , ,
, ,
Abstract
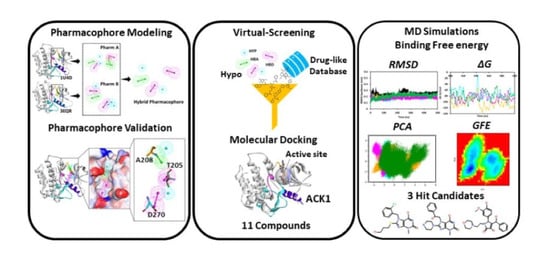
:
1. Introduction
2. Materials and Methods
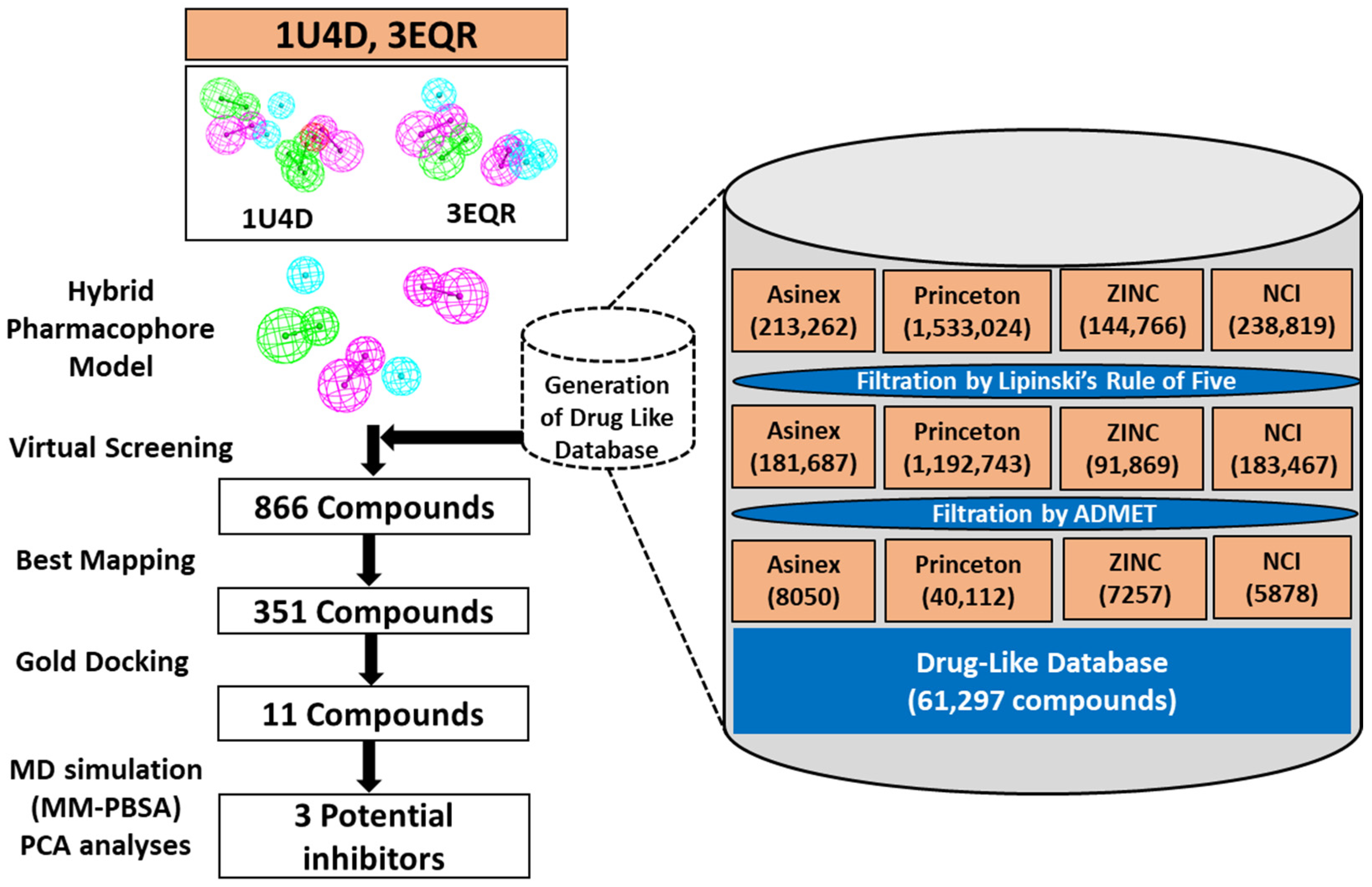
2.1. Structure-Based Pharmacophore Generation
2.2. Validation of Pharmacophore
2.3. Virtual Screening
2.4. Molecular Docking
2.5. Molecular Dynamics Simultaions
2.6. Binding Free Energy Analysis
2.7. Principal Component and Free Energy Landscape Analyses
3. Results
3.1. Active Site Analysis of ACK1
3.2. Structure-Based Pharmacophore Modeling
3.3. Validation of Pharmacophore
3.4. Virtual Screening
3.5. Molecular Docking
3.6. Molecular Dynamics Simulations
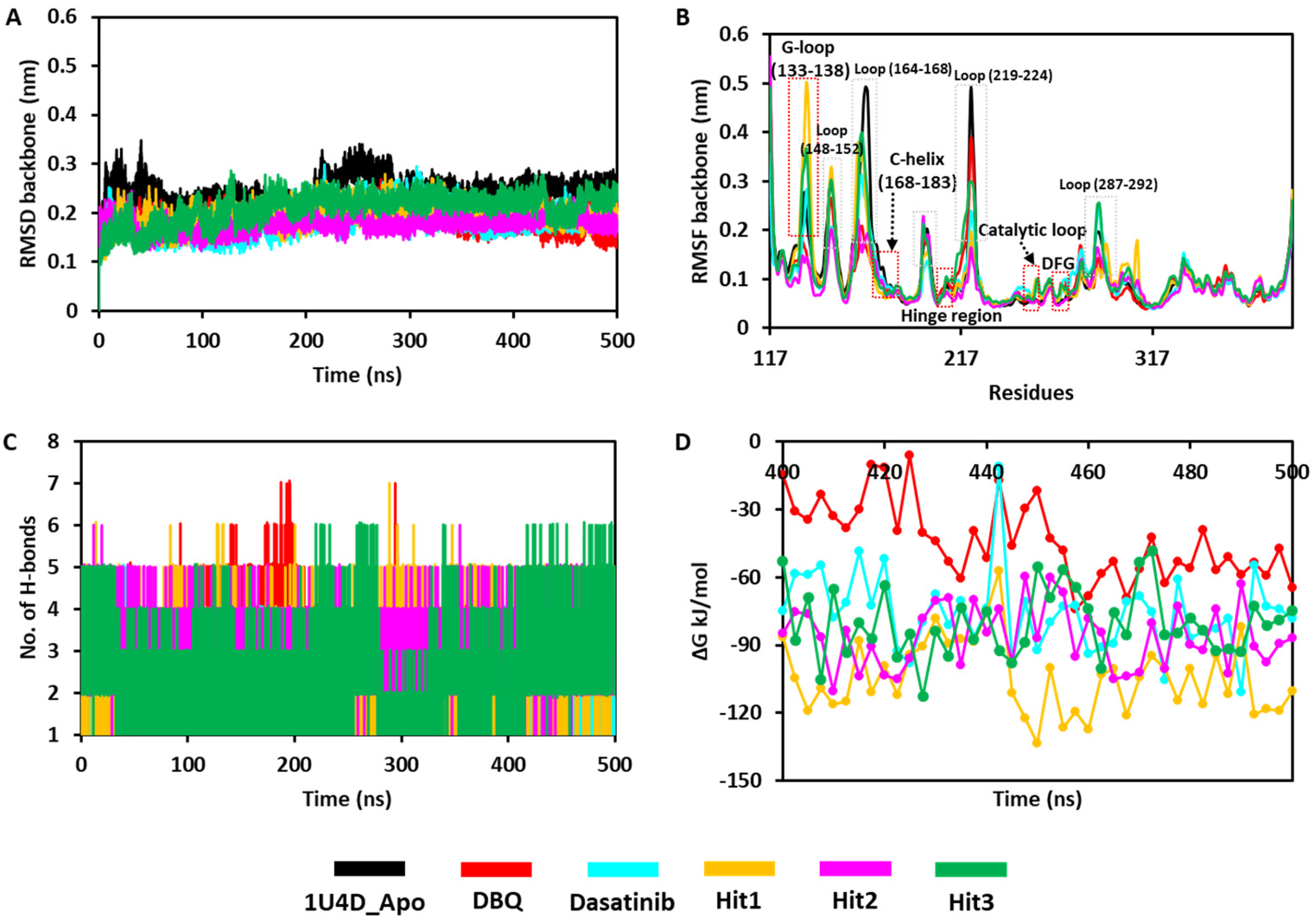
3.6.1. Stability of the Simulation Complexes
3.6.2. Binding Free Energy
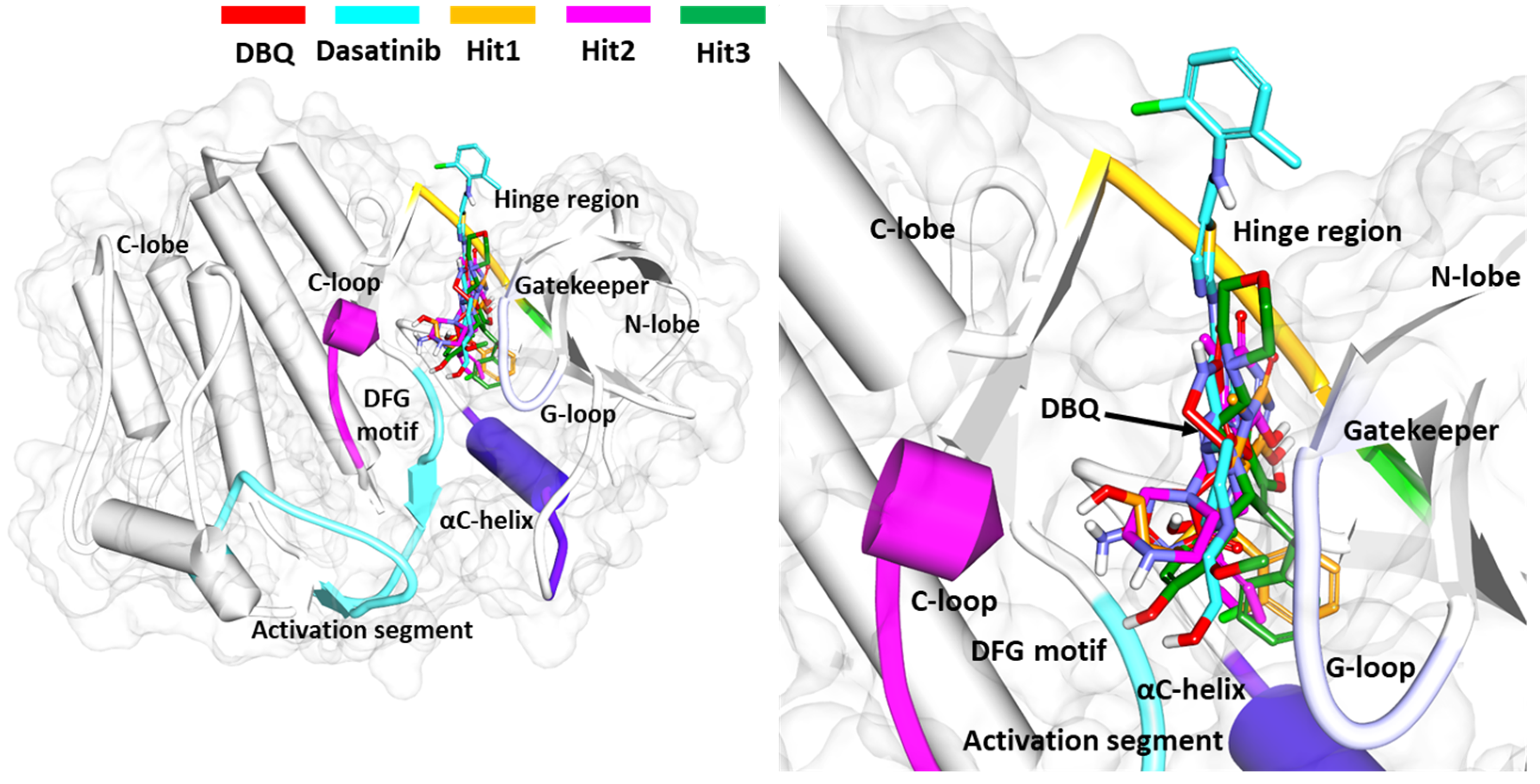
3.6.3. Binding Mode Analysis
3.6.4. Hydrogen Bond Analysis
3.6.5. Principal Component Analysis
3.6.6. Gibbs Free Energy Landscape
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Mahajan, N.P.; Whang, Y.E.; Mohler, J.L.; Earp, H.S. Activated Tyrosine Kinase Ack1 Promotes Prostate Tumorigenesis: Role of Ack1 in Polyubiquitination of Tumor Suppressor Wwox. Cancer Res. 2005, 65, 10514–10523. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lemmon, M.A.; Schlessinger, J. Cell Signaling by Receptor Tyrosine Kinases. Cell 2010, 141, 1117–1134. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, M.; Baek, M.; Kim, D.J. Protein Tyrosine Signaling and Its Potential Therapeutic Implications in Carcinogenesis. Curr. Pharm. Des. 2017, 23, 4226. [Google Scholar] [CrossRef] [PubMed]
- Lahiry, P.; Torkamani, A.; Schork, N.J.; Hegele, R.A. Kinase Mutations in Human Disease: Interpreting Genotype–Phenotype Relationships. Nat. Rev. Genet. 2010, 11, 60–74. [Google Scholar] [CrossRef]
- Siveen, K.S.; Prabhu, K.S.; Achkar, I.W.; Kuttikrishnan, S.; Shyam, S.; Khan, A.Q.; Merhi, M.; Dermime, S.; Uddin, S. Role of Non Receptor Tyrosine Kinases in Hematological Malignances and Its Targeting by Natural Products. Mol. Cancer 2018, 17, 31. [Google Scholar] [CrossRef] [Green Version]
- Roque, L.; Rodrigues, R.; Pinto, A.; Moura-Nunes, V.; Soares, J. Chromosome Imbalances in Thyroid Follicular Neoplasms: A Comparison between Follicular Adenomas and Carcinomas. Genes Chromosomes Cancer 2003, 36, 292–302. [Google Scholar] [CrossRef]
- Van der Horst, E.H.; Degenhardt, Y.Y.; Strelow, A.; Slavin, A.; Chinn, L.; Orf, J.; Rong, M.; Li, S.; See, L.H.; Nguyen, K.Q.C.; et al. Metastatic Properties and Genomic Amplification of the Tyrosine Kinase Gene ACK1. Proc. Natl. Acad. Sci. USA 2005, 102, 15901–15906. [Google Scholar] [CrossRef] [Green Version]
- Galisteo, M.L.; Yang, Y.; Ureña, J.; Schlessinger, J. Activation of the Nonreceptor Protein Tyrosine Kinase Ack by Multiple Extracellular Stimuli. Proc. Natl. Acad. Sci. USA 2006, 103, 9796–9801. [Google Scholar] [CrossRef] [Green Version]
- Mahajan, K.; Mahajan, N.P. ACK1/TNK2 Tyrosine Kinase: Molecular Signaling and Evolving Role in Cancers. Oncogene 2014, 34, 4162–4167. [Google Scholar] [CrossRef] [Green Version]
- Mahajan, K.; Coppola, D.; Chen, Y.A.; Zhu, W.; Lawrence, H.R.; Lawrence, N.J.; Mahajan, N.P. Ack1 Tyrosine Kinase Activation Correlates with Pancreatic Cancer Progression. Am. J. Pathol. 2012, 180, 1386–1393. [Google Scholar] [CrossRef]
- Mahajan, K.; Mahajan, N.P. ACK1 Tyrosine Kinase: Targeted Inhibition to Block Cancer Cell Proliferation. Cancer Lett. 2013, 338, 185. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, B.; Xu, T.; Liu, J.; Zang, S.; Gao, L.; Huang, A. Overexpression of Activated Cdc42-Associated Kinase1 (Ack1) Predicts Tumor Recurrence and Poor Survival in Human Hepatocellular Carcinoma. Pathol. Res. Pract. 2014, 210, 787–792. [Google Scholar] [CrossRef] [PubMed]
- Mahajan, K.; Coppola, D.; Challa, S.; Fang, B.; Chen, Y.A.; Zhu, W.; Lopez, A.S.; Koomen, J.; Engelman, R.W.; Rivera, C.; et al. Ack1 Mediated AKT/PKB Tyrosine 176 Phosphorylation Regulates Its Activation. PLoS ONE 2010, 5, e9646. [Google Scholar] [CrossRef] [PubMed]
- Mahajan, N.P.; Liu, Y.; Majumder, S.; Warren, M.R.; Parker, C.E.; Mohler, J.L.; Earp, H.S.; Whang, Y.E. Activated Cdc42-Associated Kinase Ack1 Promotes Prostate Cancer Progression via Androgen Receptor Tyrosine Phosphorylation. Proc. Natl. Acad. Sci. USA 2007, 104, 8438–8443. [Google Scholar] [CrossRef] [Green Version]
- Wang, A.; Pei, J.; Shuai, W.; Lin, C.; Feng, L.; Wang, Y.; Lin, F.; Ouyang, L.; Wang, G. Small Molecules Targeting Activated Cdc42-Associated Kinase 1 (ACK1/TNK2) for the Treatment of Cancers. J. Med. Chem. 2021, 64, 16328–16348. [Google Scholar] [CrossRef] [PubMed]
- Phatak, S.S.; Zhang, S. A Novel Multi-Modal Drug Repurposing Approach for Identification of Potent ACK1 Inhibitors. Biocomputing 2013, 2013, 29–40. [Google Scholar] [CrossRef] [Green Version]
- Park, C.H.; Choe, H.; Jang, I.Y.; Kwon, S.Y.; Latif, M.; Lee, H.K.; Lee, H.J.; Yang, E.H.; Yun, J.I.; Chae, C.H.; et al. Novel Bis-Ortho-Alkoxy-Para-Piperazinesubstituted-2,4-Dianilinopyrimidines (KRCA-0008) as Potent and Selective ALK Inhibitors for Anticancer Treatment. Bioorganic Med. Chem. Lett. 2013, 23, 6192–6196. [Google Scholar] [CrossRef]
- Mahajan, K.; Challa, S.; Coppola, D.; Lawrence, H.; Luo, Y.; Gevariya, H.; Zhu, W.; Chen, Y.A.; Lawrence, N.J.; Mahajan, N.P.; et al. Effect of Ack1 Tyrosine Kinase Inhibitor on Ligand-Independent Androgen Receptor Activity. Prostate 2010, 70, 1274–1285. [Google Scholar] [CrossRef] [Green Version]
- Macalino, S.J.Y.; Gosu, V.; Hong, S.; Choi, S. Role of Computer-Aided Drug Design in Modern Drug Discovery. Arch. Pharmacal Res. 2015, 38, 1686–1701. [Google Scholar] [CrossRef]
- Prada-Gracia, D.; Huerta-Yépez, S.; Moreno-Vargas, L.M. Application of Computational Methods for Anticancer Drug Discovery, Design, and Optimization. Boletín Médico Hosp. Infant. México 2016, 73, 411–423. [Google Scholar] [CrossRef]
- Lougheed, J.C.; Chen, R.H.; Mak, P.; Stout, T.J. Crystal Structures of the Phosphorylated and Unphosphorylated Kinase Domains of the Cdc42-Associated Tyrosine Kinase ACK1. J. Biol. Chem. 2004, 279, 44039–44045. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kopecky, D.J.; Hao, X.; Chen, Y.; Fu, J.; Jiao, X.Y.; Jaen, J.C.; Cardozo, M.G.; Liu, J.; Wang, Z.; Walker, N.P.C.; et al. Identification and Optimization of N3,N6-Diaryl-1H-Pyrazolo[3,4-d]Pyrimidine-3,6-Diamines as a Novel Class of ACK1 Inhibitors. Bioorganic Med. Chem. Lett. 2008, 18, 6352–6356. [Google Scholar] [CrossRef]
- Arooj, M.; Kim, S.; Sakkiah, S.; Cao, G.P.; Lee, Y.; Lee, K.W. Molecular Modeling Study for Inhibition Mechanism of Human Chymase and Its Application in Inhibitor Design. PLoS ONE 2013, 8, e62740. [Google Scholar] [CrossRef] [Green Version]
- Sakkiah, S.; Senese, S.; Yang, Q.; Lee, K.W.; Torres, J.Z. Dynamic and Multi-Pharmacophore Modeling for Designing Polo-Box Domain Inhibitors. PLoS ONE 2014, 9, e101405. [Google Scholar] [CrossRef]
- Guner, O. History and Evolution of the Pharmacophore Concept in Computer-Aided Drug Design. Curr. Top. Med. Chem. 2005, 2, 1321–1332. [Google Scholar] [CrossRef] [PubMed]
- Fei, J.; Zhou, L.; Liu, T.; Tang, X.Y. Pharmacophore Modeling, Virtual Screening, and Molecular Docking Studies for Discovery of Novel Akt2 Inhibitors. Int. J. Med. Sci. 2013, 10, 265–275. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Babu, S.; Nagarajan, S.K.; Sathish, S.; Negi, V.S.; Sohn, H.; Madhavan, T. Identification of Potent and Selective JAK1 Lead Compounds Through Ligand-Based Drug Design Approaches. Front. Pharmacol. 2022, 13, 837369. [Google Scholar] [CrossRef] [PubMed]
- Kumar, V.; Kumar, R.; Parate, S.; Yoon, S.; Lee, G.; Kim, D.; Lee, K.W. Identification of ACK1 Inhibitors as Anticancer Agents by Using Computer-Aided Drug Designing. J. Mol. Struct. 2021, 1235, 130200. [Google Scholar] [CrossRef]
- Arooj, M.; Sakkiah, S.; Kim, S.; Arulalapperumal, V.; Lee, K.W. A Combination of Receptor-Based Pharmacophore Modeling & QM Techniques for Identification of Human Chymase Inhibitors. PLoS ONE 2013, 8, e63030. [Google Scholar] [CrossRef] [Green Version]
- Zeb, A.; Son, M.; Yoon, S.; Kim, J.H.; Park, S.J.; Lee, K.W. Computational Simulations Identified Two Candidate Inhibitors of Cdk5/P25 to Abrogate Tau-Associated Neurological Disorders. Comput. Struct. Biotechnol. J. 2019, 17, 579–590. [Google Scholar] [CrossRef]
- Jones, G.; Willett, P.; Glen, R.C.; Leach, A.R.; Taylor, R. Development and Validation of a Genetic Algorithm for Flexible Docking. J. Mol. Biol. 1997, 267, 727–748. [Google Scholar] [CrossRef] [Green Version]
- Kumar, V.; Parate, S.; Thakur, G.; Lee, G.; Ro, H.S.; Kim, Y.; Kim, H.J.; Kim, M.O.; Lee, K.W. Identification of Cdk7 Inhibitors from Natural Sources Using Pharmacoinformatics and Molecular Dynamics Simulations. Biomedicines 2021, 9, 1197. [Google Scholar] [CrossRef]
- Parate, S.; Kumar, V.; Chan Hong, J.; Lee, K.W. Investigating Natural Compounds against Oncogenic RET Tyrosine Kinase Using Pharmacoinformatic Approaches for Cancer Therapeutics. RSC Adv. 2021, 12, 1194–1207. [Google Scholar] [CrossRef] [PubMed]
- Kumar, V.; Parate, S.; Yoon, S.; Lee, G.; Lee, K.W. Computational Simulations Identified Marine-Derived Natural Bioactive Compounds as Replication Inhibitors of SARS-CoV-2. Front. Microbiol. 2021, 12, 583. [Google Scholar] [CrossRef] [PubMed]
- Van der Spoel, D.; Lindahl, E.; Hess, B.; Groenhof, G.; Mark, A.E.; Berendsen, H.J.C. GROMACS: Fast, Flexible, and Free. J. Comput. Chem. 2005, 26, 1701–1718. [Google Scholar] [CrossRef] [PubMed]
- Abraham, M.J.; Murtola, T.; Schulz, R.; Páll, S.; Smith, J.C.; Hess, B.; Lindah, E. GROMACS: High Performance Molecular Simulations through Multi-Level Parallelism from Laptops to Supercomputers. SoftwareX 2015, 1, 19–25. [Google Scholar] [CrossRef] [Green Version]
- Foloppe, N.; Mackerell, A.D. All-Atom Empirical Force Field for Nucleic Acids: I. Parameter Optimization Based on Small Molecule and Condensed Phase Macromolecular Target Data. J. Comput. Chem. 2000, 21, 86–104. [Google Scholar] [CrossRef]
- Zoete, V.; Cuendet, M.A.; Grosdidier, A.; Michielin, O. SwissParam: A Fast Force Field Generation Tool for Small Organic Molecules. J. Comput. Chem. 2011, 32, 2359–2368. [Google Scholar] [CrossRef]
- Huang, Y.M.M.; Chen, W.; Potter, M.J.; Chang, C.E.A. Insights from Free-Energy Calculations: Protein Conformational Equilibrium, Driving Forces, and Ligand-Binding Modes. Biophys. J. 2012, 103, 342–351. [Google Scholar] [CrossRef] [Green Version]
- King, E.; Aitchison, E.; Li, H.; Luo, R. Recent Developments in Free Energy Calculations for Drug Discovery. Front. Mol. Biosci. 2021, 8, 775. [Google Scholar] [CrossRef]
- Homeyer, N.; Gohlke, H. Free Energy Calculations by the Molecular Mechanics Poisson−Boltzmann Surface Area Method. Mol. Inform. 2012, 31, 114–122. [Google Scholar] [CrossRef]
- Kumari, R.; Kumar, R.; Lynn, A. G_mmpbsa—A GROMACS Tool for High-Throughput MM-PBSA Calculations. J. Chem. Inf. Model. 2014, 54, 1951–1962. [Google Scholar] [CrossRef] [PubMed]
- Kumbhar, N.; Nimal, S.; Barale, S.; Kamble, S.; Bavi, R.; Sonawane, K.; Gacche, R. Identification of Novel Leads as Potent Inhibitors of HDAC3 Using Ligand-Based Pharmacophore Modeling and MD Simulation. Sci. Rep. 2022, 12, 1712. [Google Scholar] [CrossRef] [PubMed]
- Warnault, P.; Yasri, A.; Coisy-Quivy, M.; Cheve, G.; Bories, C.; Fauvel, B.; Benhida, R. Recent Advances in Drug Design of Epidermal Growth Factor Receptor Inhibitors. Curr. Med. Chem. 2013, 20, 2043–2067. [Google Scholar] [CrossRef] [PubMed]
- Cho, H.; Shin, I.; Ju, E.; Choi, S.; Hur, W.; Kim, H.; Hong, E.; Kim, N.D.; Choi, H.G.; Gray, N.S.; et al. First SAR Study for Overriding NRAS Mutant Driven Acute Myeloid Leukemia. J. Med. Chem. 2018, 61, 8353–8373. [Google Scholar] [CrossRef]
- Jiao, X.; Kopecky, D.J.; Liu, J.; Liu, J.; Jaen, J.C.; Cardozo, M.G.; Sharma, R.; Walker, N.; Wesche, H.; Li, S.; et al. Synthesis and Optimization of Substituted Furo[2,3-d]-Pyrimidin-4-Amines and 7H-Pyrrolo[2,3-d]Pyrimidin-4-Amines as ACK1 Inhibitors. Bioorganic Med. Chem. Lett. 2012, 22, 6212–6217. [Google Scholar] [CrossRef]
- Jin, M.; Wang, J.; Kleinberg, A.; Kadalbajoo, M.; Siu, K.W.; Cooke, A.; Bittner, M.A.; Yao, Y.; Thelemann, A.; Ji, Q.; et al. Discovery of Potent, Selective and Orally Bioavailable Imidazo[1,5-a]Pyrazine Derived ACK1 Inhibitors. Bioorganic Med. Chem. Lett. 2013, 23, 979–984. [Google Scholar] [CrossRef]
- BIOVIA Product Portfolio—BIOVIA—Dassault Systèmes®. Available online: https://www.3ds.com/products-services/biovia/products/ (accessed on 20 October 2022).
- DiMauro, E.F.; Newcomb, J.; Nunes, J.J.; Bemis, J.E.; Boucher, C.; Buchanan, J.L.; Buckner, W.H.; Cheng, A.; Faust, T.; Hsieh, F.; et al. Discovery of 4-Amino-5,6-Biaryl-Furo[2,3-d]Pyrimidines as Inhibitors of Lck: Development of an Expedient and Divergent Synthetic Route and Preliminary SAR. Bioorganic Med. Chem. Lett. 2007, 17, 2305–2309. [Google Scholar] [CrossRef]
- Lawrence, H.R.; Mahajan, K.; Luo, Y.; Zhang, D.; Tindall, N.; Huseyin, M.; Gevariya, H.; Kazi, S.; Ozcan, S.; Mahajan, N.P.; et al. Development of Novel ACK1/TNK2 Inhibitors Using a Fragment-Based Approach. J. Med. Chem. 2015, 58, 2746–2763. [Google Scholar] [CrossRef] [Green Version]
- US8481733B2—Substituted Imidazopyr- and Imidazotri-Azines—Google Patents. Available online: https://patents.google.com/patent/US8481733B2/en (accessed on 20 June 2022).
- Kumar, R.; Kumar, V.; Lee, K.W. A Computational Drug Repurposing Approach in Identifying the Cephalosporin Antibiotic and Anti-Hepatitis C Drug Derivatives for COVID-19 Treatment. Comput. Biol. Med. 2021, 130, 104186. [Google Scholar] [CrossRef]
- Amadei, A.; Linssen, A.B.M.; Berendsen, H.J.C. Essential Dynamics of Proteins. Proteins Struct. Funct. Bioinform. 1993, 17, 412–425. [Google Scholar] [CrossRef] [PubMed]
- David, C.C.; Jacobs, D.J. Principal Component Analysis: A Method for Determining the Essential Dynamics of Proteins. Methods Mol. Biol. 2014, 1084, 193–226. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cohen, P.; Cross, D.; Jänne, P.A. Kinase Drug Discovery 20 Years after Imatinib: Progress and Future Directions. Nat. Rev. Drug Discov. 2021, 20, 551–569. [Google Scholar] [CrossRef]
- Attwood, M.M.; Fabbro, D.; Sokolov, A.V.; Knapp, S.; Schiöth, H.B. Trends in Kinase Drug Discovery: Targets, Indications and Inhibitor Design. Nat. Rev. Drug Discov. 2021, 20, 839–861. [Google Scholar] [CrossRef] [PubMed]
- Rajendran, V.; Gopalakrishnan, C.; Sethumadhavan, R. Pathological Role of a Point Mutation (T315I) in BCR-ABL1 Protein—A Computational Insight. J. Cell. Biochem. 2018, 119, 918–925. [Google Scholar] [CrossRef]
- Rajendran, V. Structural Analysis of Oncogenic Mutation of Isocitrate Dehydrogenase 1. Mol. Biosyst. 2016, 12, 2276–2287. [Google Scholar] [CrossRef]
- Bhardwaj, V.K.; Purohit, R. Targeting the Protein-Protein Interface Pocket of Aurora-A-TPX2 Complex: Rational Drug Design and Validation. J. Biomol. Struct. Dyn. 2020, 39, 3882–3891. [Google Scholar] [CrossRef]
- Snow, R.J.; Cardozo, M.G.; Morwick, T.M.; Busacca, C.A.; Dong, Y.; Eckner, R.J.; Jacober, S.; Jakes, S.; Kapadia, S.; Lukas, S.; et al. Discovery of 2-Phenylamino-Imidazo[4,5-h]Isoquinolin-9-Ones: A New Class of Inhibitors of Lck Kinase. J. Med. Chem. 2002, 45, 3394–3405. [Google Scholar] [CrossRef]
- Zhao, H.; Caflisch, A. Molecular Dynamics in Drug Design. Eur. J. Med. Chem. 2015, 91, 4–14. [Google Scholar] [CrossRef]
- De Vivo, M.; Masetti, M.; Bottegoni, G.; Cavalli, A. Role of Molecular Dynamics and Related Methods in Drug Discovery. J. Med. Chem. 2016, 59, 4035–4061. [Google Scholar] [CrossRef] [PubMed]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| PDB ID | Ligands Id | Resolution (Å) | Active Sire Residues | References | ||||||
|---|---|---|---|---|---|---|---|---|---|---|
| A208 | T205 | D270 | E206 | E177 | D134 | K158 | ||||
| 1U4D | DBQ | 2.10 | √ | √ | √ | √ | √ | [21] | ||
| 3EQR | T74 | 2.00 | √ | √ | [22] | |||||
| 3EQP | T95 | 2.00 | √ | √ | ||||||
| 4EWH | T77 | 2.50 | √ | [46] | ||||||
| 4ID7 | 1G0 | 3.00 | √ | √ | [47] | |||||
| 5ZXB | 9KO | 2.20 | √ | √ | √ | [45] | ||||
| S. No. | Parameters | Calculated Values |
|---|---|---|
| 1 | Total no. of molecules in the database (D) | 140 |
| 2 | Total number of active molecules in the database (A) | 20 |
| 3 | Total number of active molecules in the retrieved hits (Ht) | 22 |
| 4 | Number of retrieved hits by pharmacophore (Ha) | 17 |
| 5 | % Yield of actives [(Ha/Ht) × 100] | 77.27% |
| 6 | % Ratio of actives [(Ha/A × 100) | 85% |
| 7 | False-negative [A-Ha] | 3 |
| 8 | False-positive [Ht-Ha] | 5 |
| 9 | Goodness of fit | 0.76 |
| 10 | Enrichment factor (EF) | 5.40 |
| Lipinski’s Rule of Five | ADMET Descriptors | ||
|---|---|---|---|
| Parameters | Threshold value | Parameters | Threshold value |
| Number of hydrogen bond donors | ≤5 | Absorption level | 0 (Good) |
| Number of hydrogen bond acceptors | ≤10 | Solubility level | 3 (Good) |
| Molecular weight (Da) | ≤500 | Blood-brain barrier level | 3 (Low) |
| AlogP value | ≤5 | CYP2D6 prediction | False |
| Hepatotoxic prediction | False | ||
| Name | Hydrogen Bond Interactions | Van Der Waals Interactions | π-π/π-Alkyl Interactions | |||
|---|---|---|---|---|---|---|
| Amino Acid | Amino Acid Atom | Ligand Atom | Distance (<3.5 Å) | |||
| Hit1 | T205 | OG1 | H28 | 2.70 | D134, L207, S212, N257, G269 | L132, V140, A156, K158, M181, I190, M203, L259, F271 |
| E206 | O | H28 | 1.79 | |||
| A208 | HN | O12 | 2.56 | |||
| R256 | O | H35 | 1.95 | |||
| D270 | HN | S13 | 2.25 | |||
| Hit2 | E206 | O | H32 | 1.83 | G133, G135, K158, T205, L207, G211, N257, G269, F271 | L132, V140, A156, M181, I190, L259 |
| A208 | HN | O12 | 1.74 | |||
| D270 | HN | O18 | 1.93 | |||
| Hit3 | T205 | HG1 | O17 | 2.01 | G133, V140, A156, I190, E177, M203, P209, G211, L259, G269, F271 | L132, K158, L207, M181 |
| E206 | O | H38 | 1.73 | |||
| A208 | HN | O12 | 2.30 | |||
| D270 | OD2 | H42 | 1.65 | |||
| A208 | HN | N8 | 2.42 | |||
| Dasatinib | D270 | OD1 | H47 | 2.82 | G133, 135, S136, E206, L207, P209, G211, S212 | L132, V140, A156, I190, L259 |
| T205 | OG1 | H28 | 2.70 | |||
| DBQ | A208 | HN | N7 | 2.48 | L132, V140, I190, E206, G211, L207, N257, G269 | A156, L259 |
| D270 | HN | N16 | 2.83 | |||
| Hits | 2D Structure | IUPAC Name |
|---|---|---|
| Hit1 |  | 7-[(2-chlorophenyl)methyl]-6-hydroxy-8-(3-hydroxypropylsulfanyl)-3-methyl-purin-2-one |
| Hit2 |  | 6-hydroxy-7-[(2R)-2-hydroxy-2-phenyl-ethyl]-3-methyl-8-piperazin-1-yl-purin-2-one |
| Hit3 |  | (2S)-4-hydroxy-2-(4-hydroxy-3-methoxy-phenyl)-3-[(S)-hydroxy(phenyl)methyl]-1-(2-morpholinoethyl)-2H-pyrrol-5-one |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kumar, V.; Kumar, R.; Parate, S.; Danishuddin; Lee, G.; Kwon, M.; Jeong, S.-H.; Ro, H.-S.; Lee, K.W.; Kim, S.-W. Identification of Activated Cdc42-Associated Kinase Inhibitors as Potential Anticancer Agents Using Pharmacoinformatic Approaches. Biomolecules 2023, 13, 217. https://doi.org/10.3390/biom13020217
Kumar V, Kumar R, Parate S, Danishuddin, Lee G, Kwon M, Jeong S-H, Ro H-S, Lee KW, Kim S-W. Identification of Activated Cdc42-Associated Kinase Inhibitors as Potential Anticancer Agents Using Pharmacoinformatic Approaches. Biomolecules. 2023; 13(2):217. https://doi.org/10.3390/biom13020217
Chicago/Turabian StyleKumar, Vikas, Raj Kumar, Shraddha Parate, Danishuddin, Gihwan Lee, Moonhyuk Kwon, Seong-Hee Jeong, Hyeon-Su Ro, Keun Woo Lee, and Seon-Won Kim. 2023. "Identification of Activated Cdc42-Associated Kinase Inhibitors as Potential Anticancer Agents Using Pharmacoinformatic Approaches" Biomolecules 13, no. 2: 217. https://doi.org/10.3390/biom13020217