

Identification of Novel Ribonucleotide Reductase Inhibitors for Therapeutic Application in Bile Tract Cancer: An Advanced Pharmacoinformatics Study
 , , and
, , and
Abstract
:1. Introduction
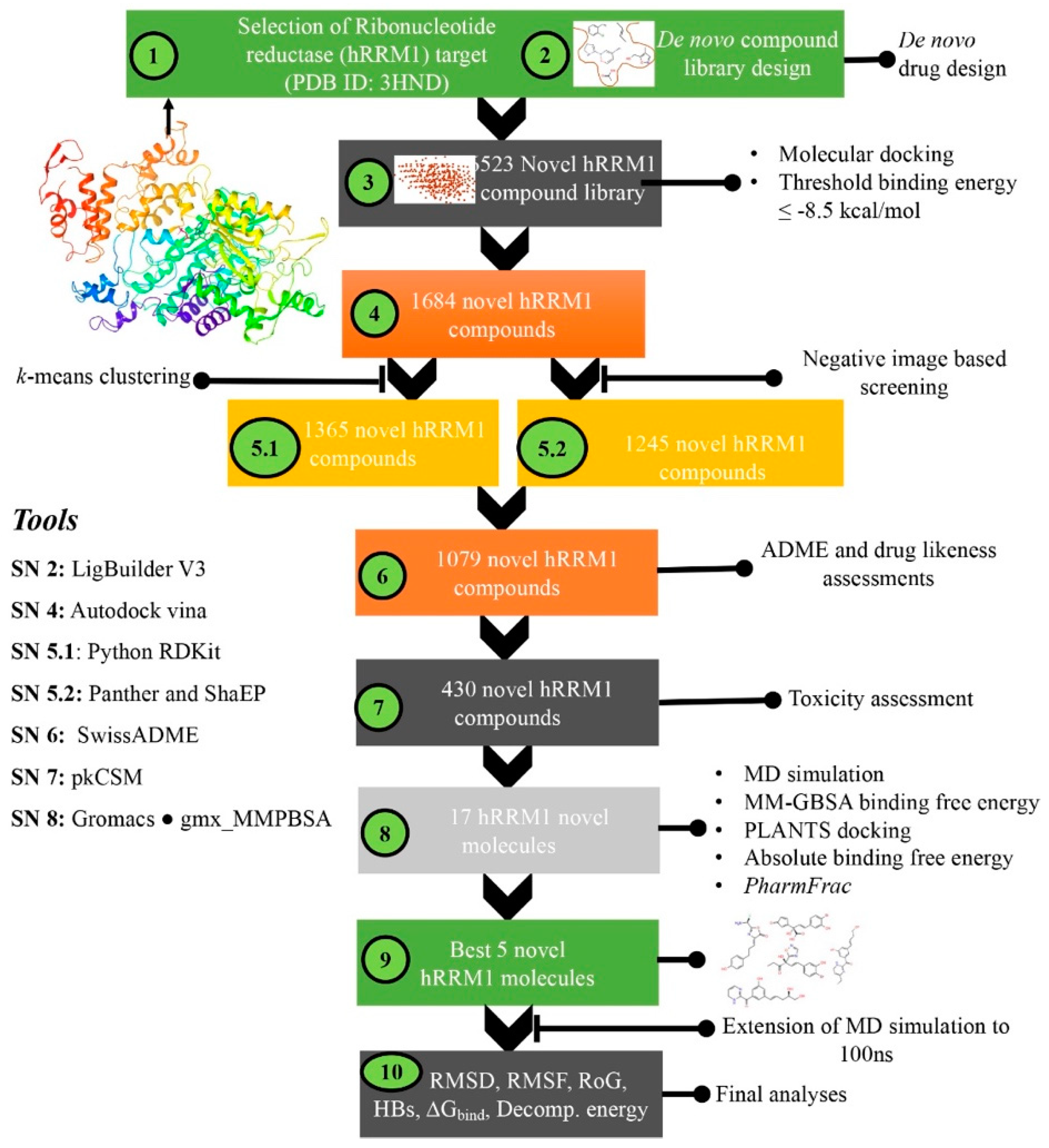
2. Materials and Methods
2.1. Protein Target Selection and Preparation
2.2. De Novo Design Using LigBuilder
2.3. Molecular Docking Using Autodock Vina
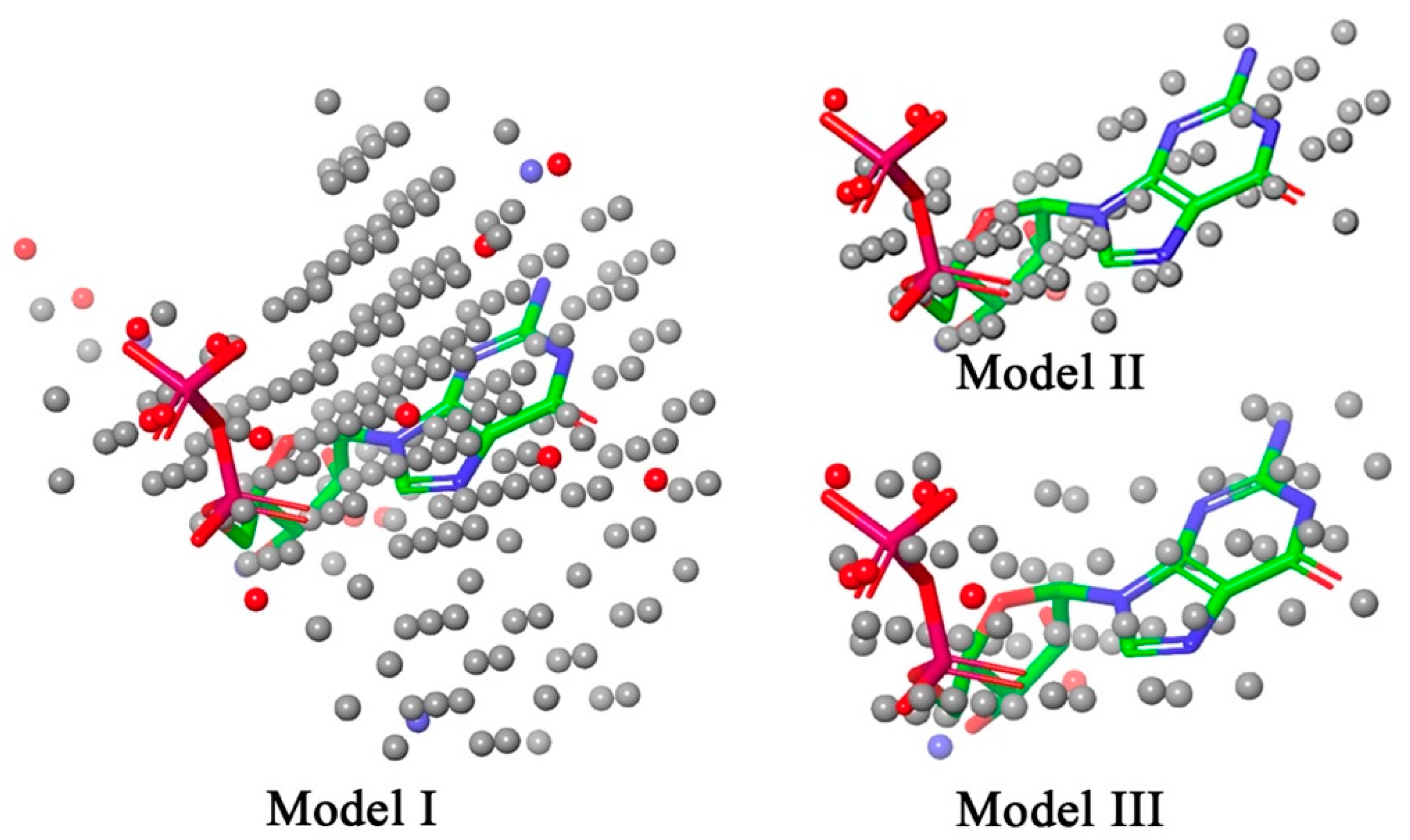
2.4. Unsupervised Pharmacophoric Features Assessment and Negative Image-Based Modeling
2.4.1. K-Means Clustering
| Algorithm1. The k-means clustering algorithm |
Repeat |
2.4.2. Negative Image-Based Modeling
2.5. Pharmacokinetics, Drug-Likeness, and Toxicity Assessment
2.6. Molecular Docking Using PLANTS and SwissDock, and Absolute Binding Free Energy Calculation Using KDEEP
2.7. Pharmacophoric Features Assessment
2.8. Molecular Dynamics Simulation
2.9. Binding Free Energy Using MM-GBSA Approach and Per-Residue Decomposition Energy Calculation
3. Results and Discussion
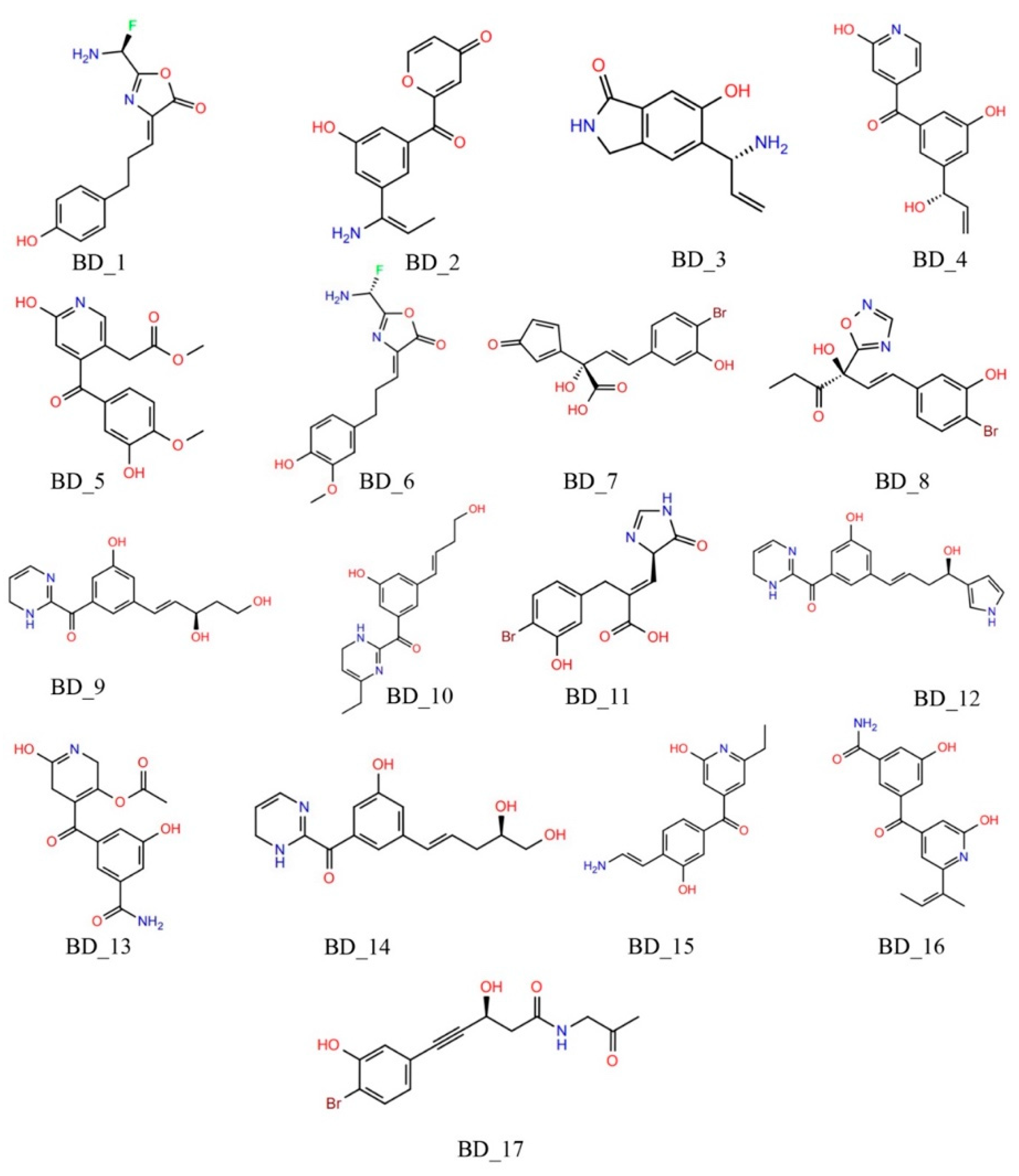
3.1. De Novo Design of hRRM1 Inhibitors and Virtual Screening
3.1.1. Molecular Docking Based Screening
3.1.2. K-Means Clustering of Pharmacophoric Features
3.1.3. Negative Image-Based Screening
3.1.4. Pharmacokinetics and Drug-Likeness Parameter Assessment
3.1.5. Toxicity Assessment
3.1.6. Molecular Docking Using PLANTS and SwissDock, and Absolute Binding Affinity Calculation through KDEEP
3.1.7. Pharmacophoric Features Assessment
3.1.8. MD Simulation and MM-GBSA Binding Free Energy Estimation
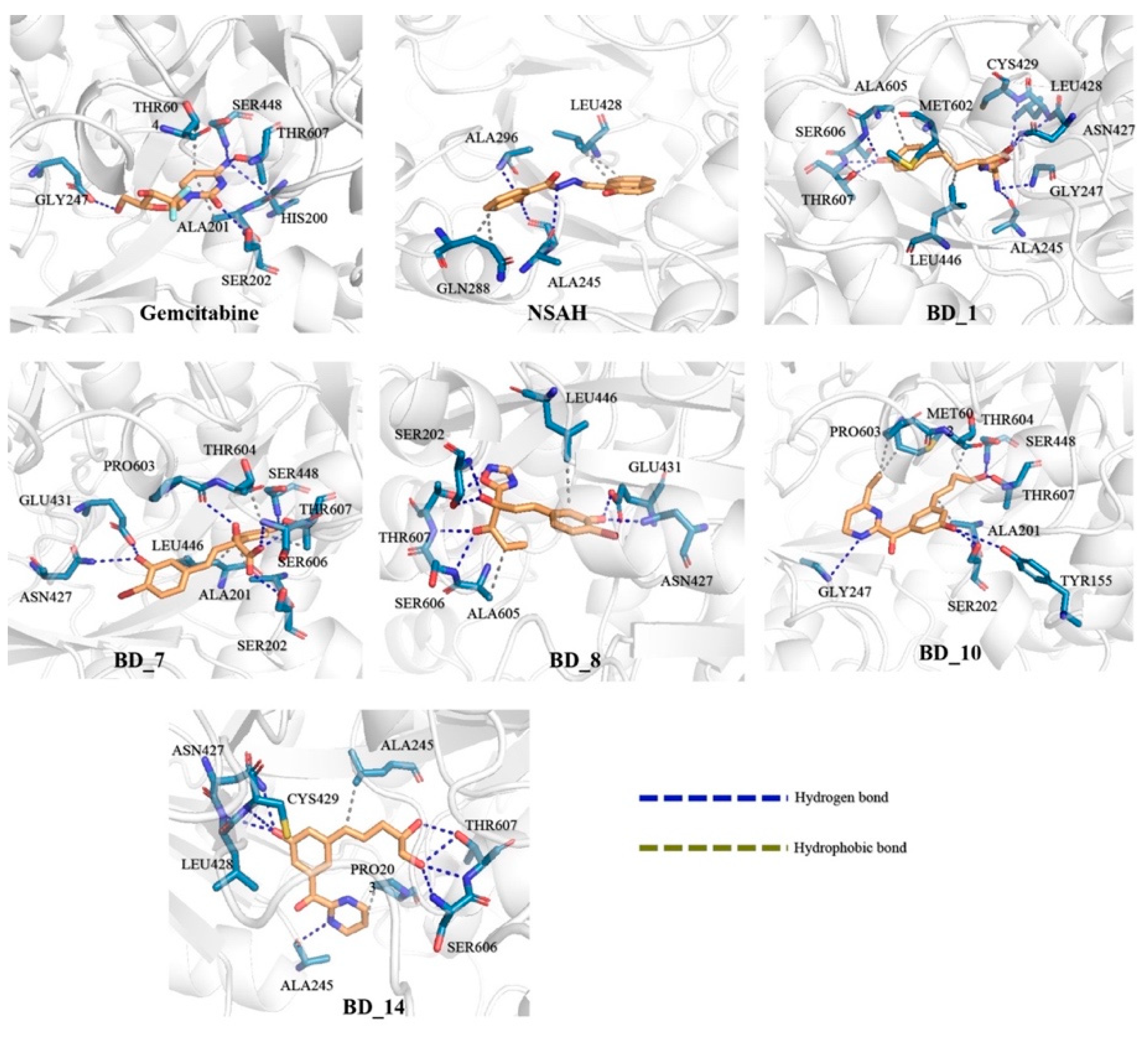
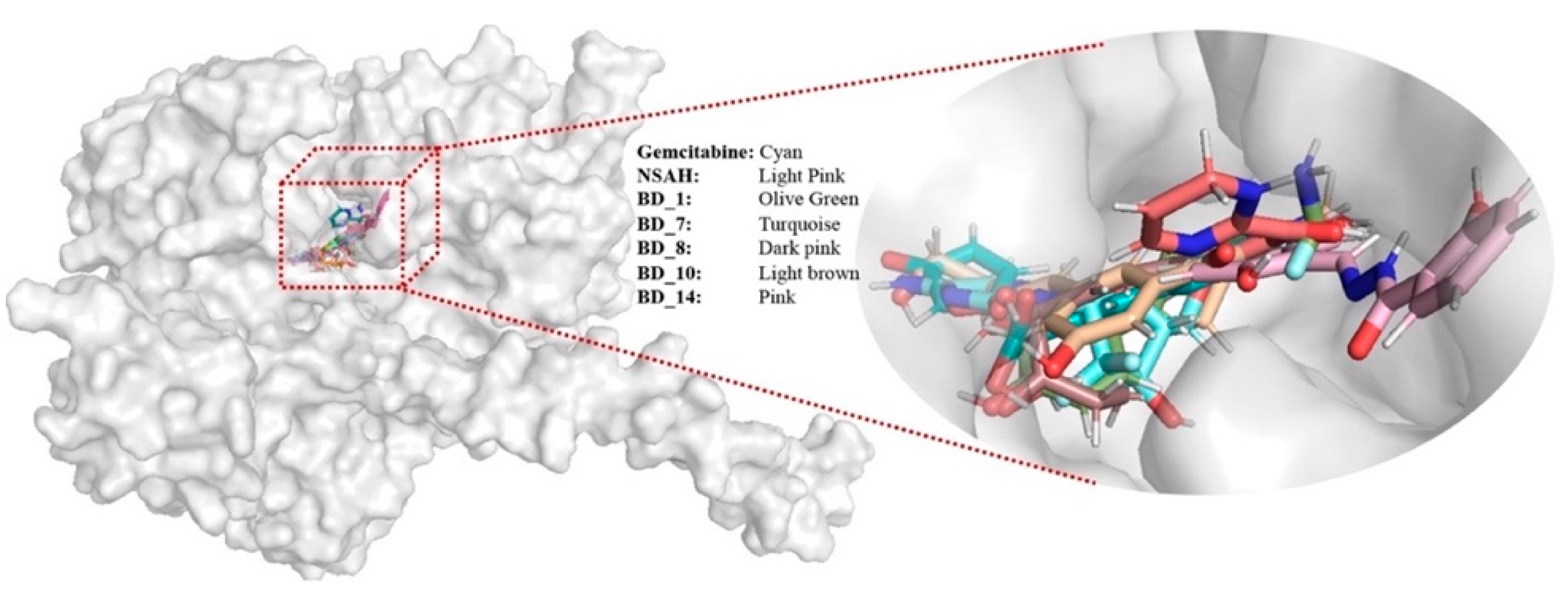
3.2. Binding Interaction Analysis
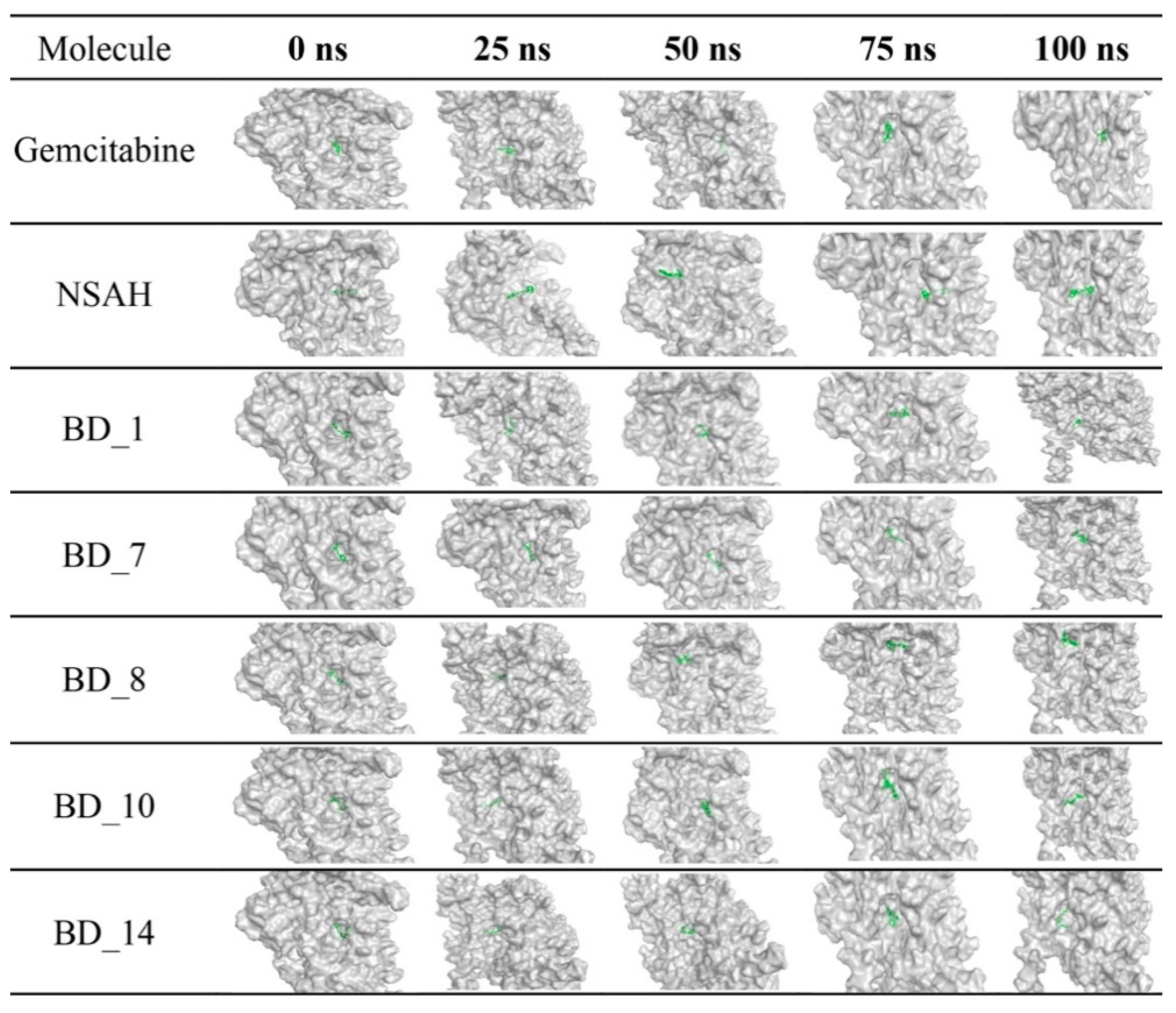
3.3. Molecular Dynamics Simulation of Top Five hRRM1 Inhibitors
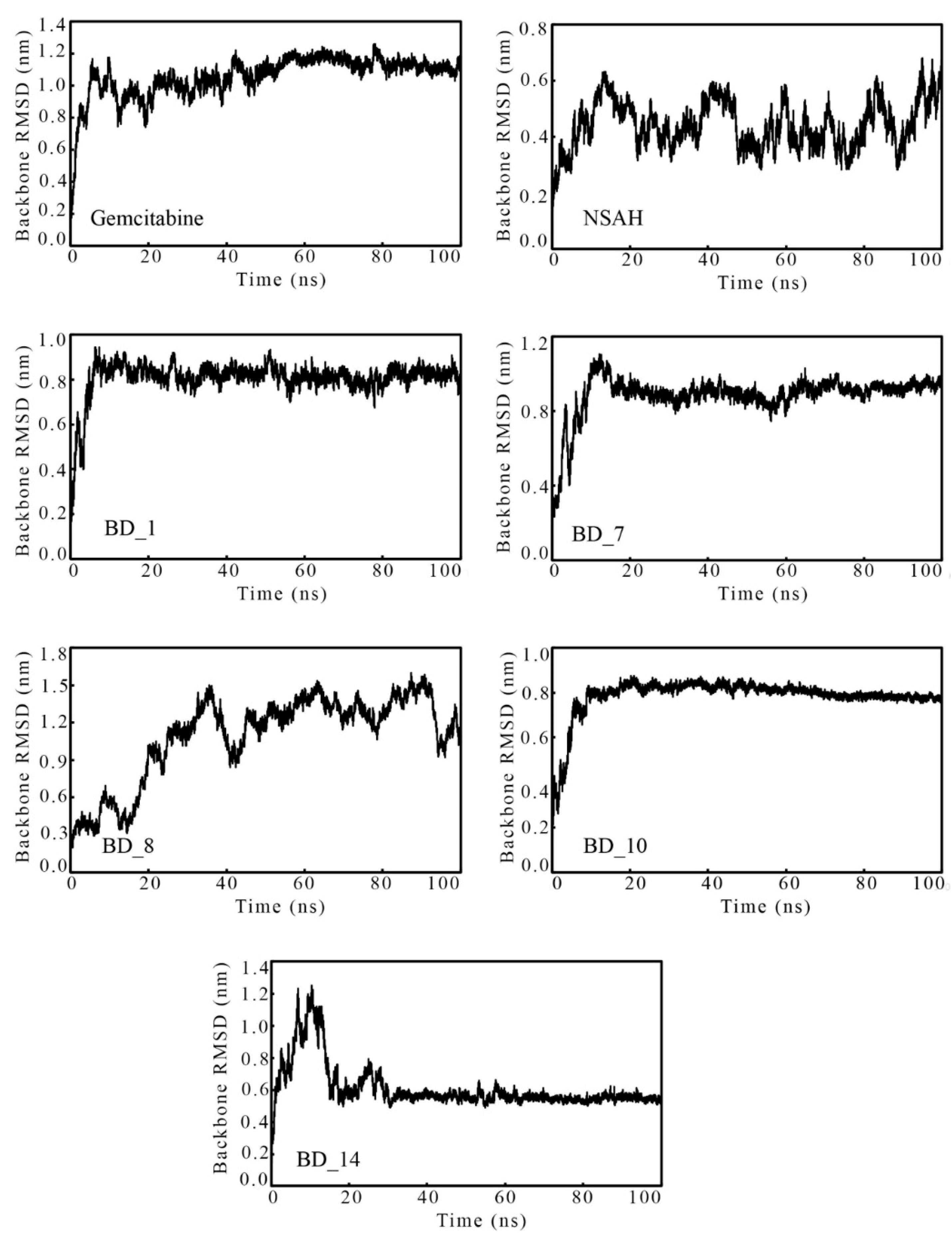
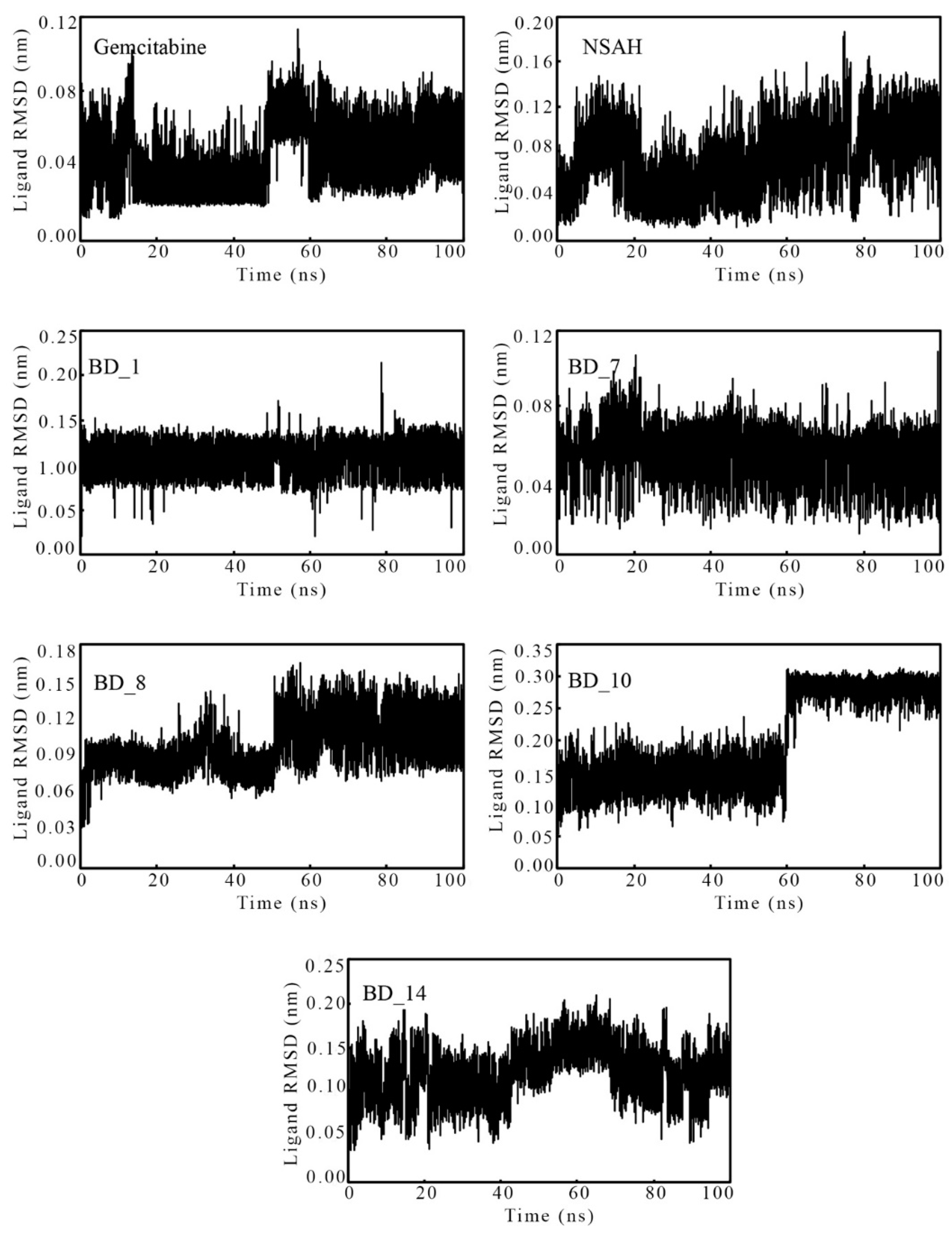
3.3.1. Root-Mean Square Deviation
3.3.2. Root-Mean Square Fluctuation
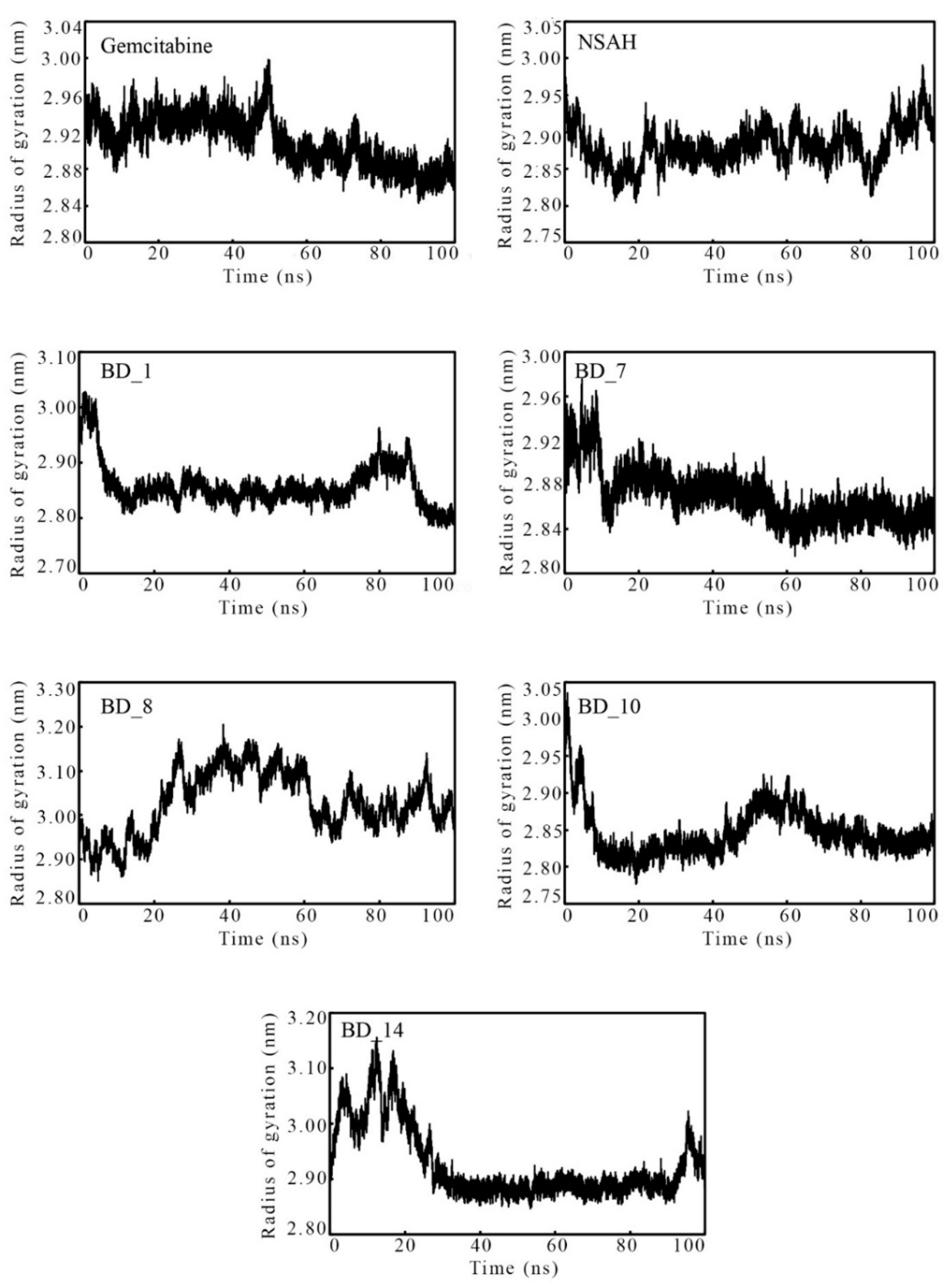
3.3.3. Radius of Gyration
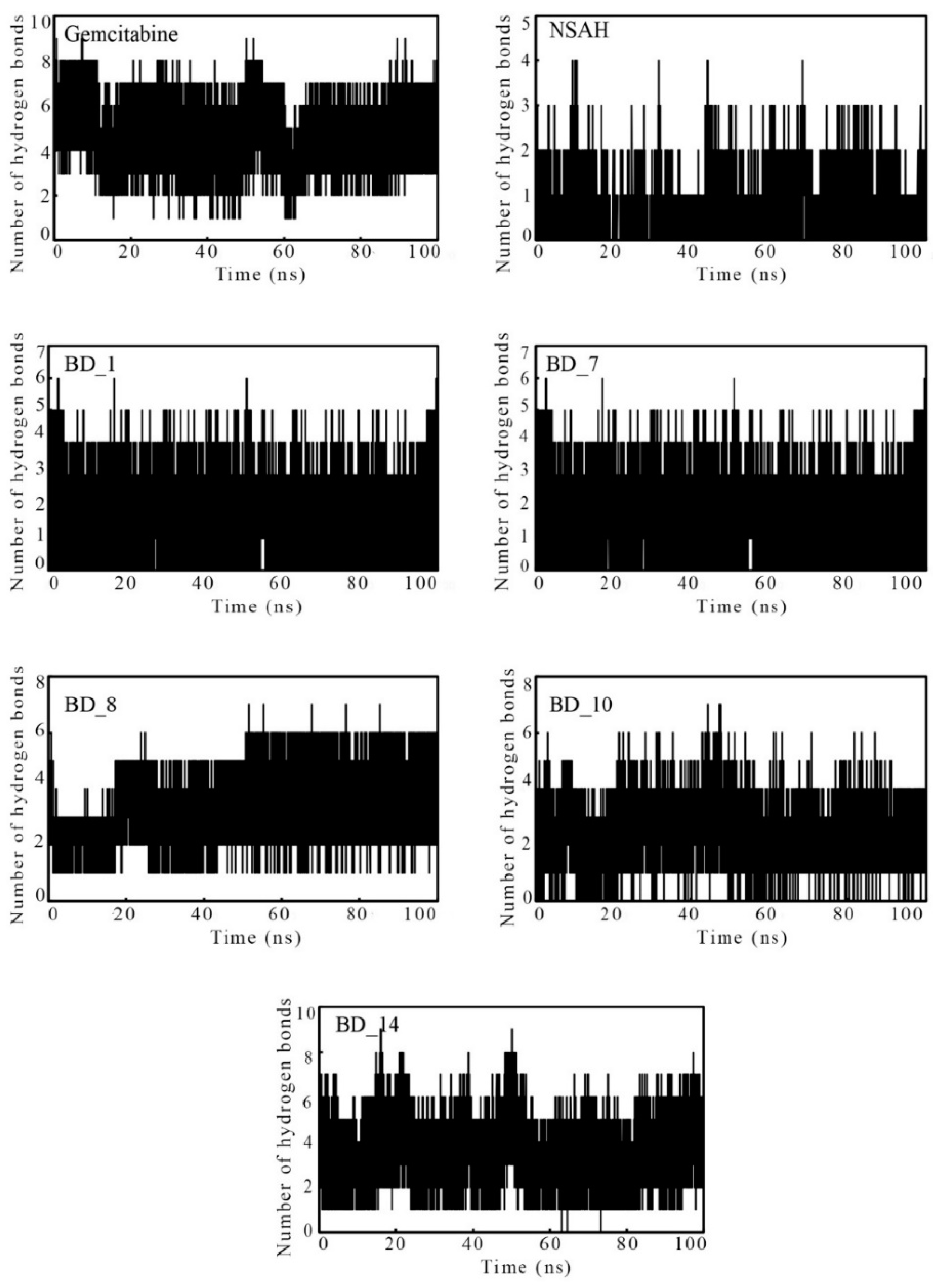
3.3.4. Intermolecular Hydrogen Bonds Analysis during MD Simulation
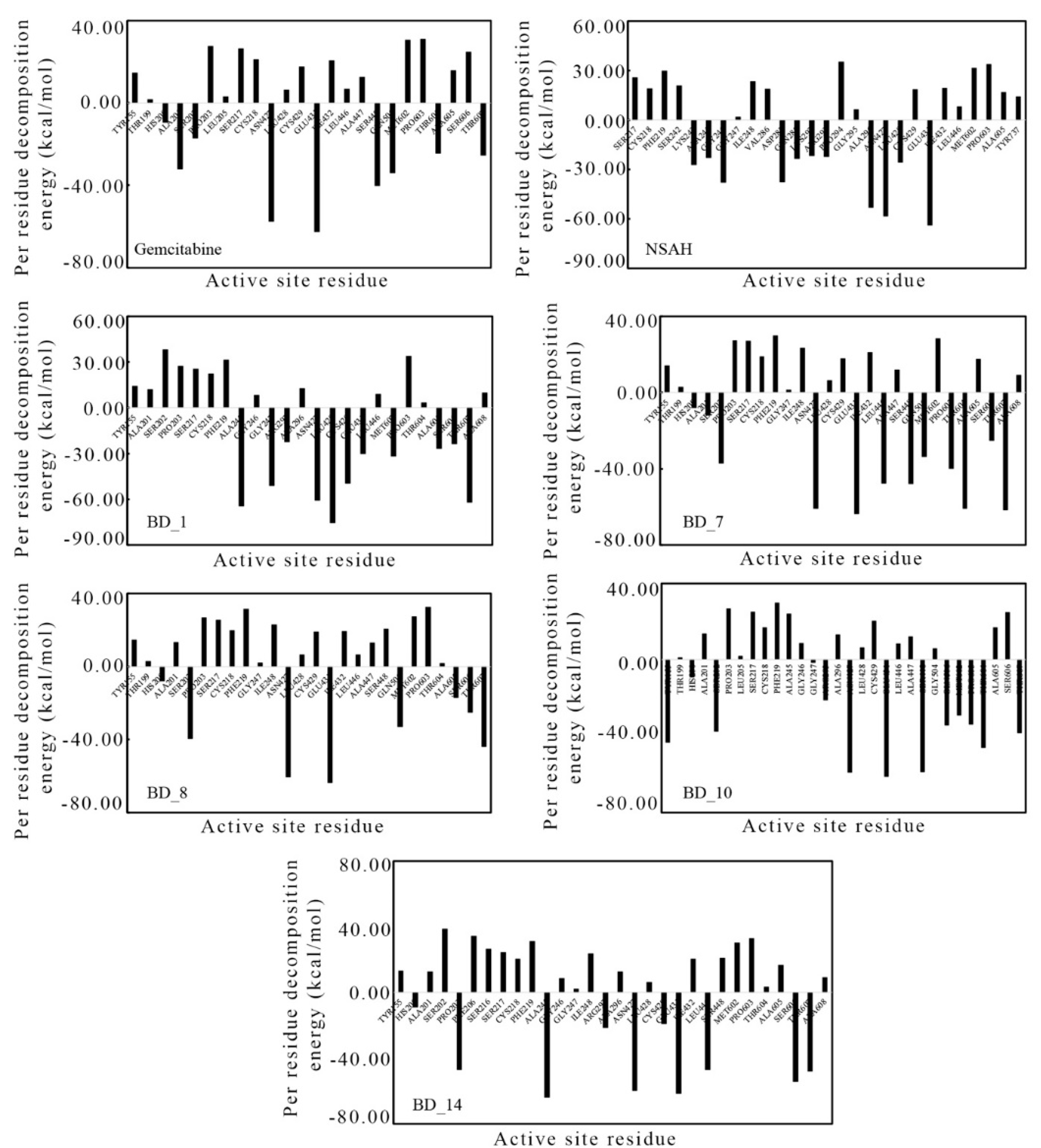
3.4. Binding Free Energy Using MM-GBSA Approach and Per-Residue Decomposition Energy
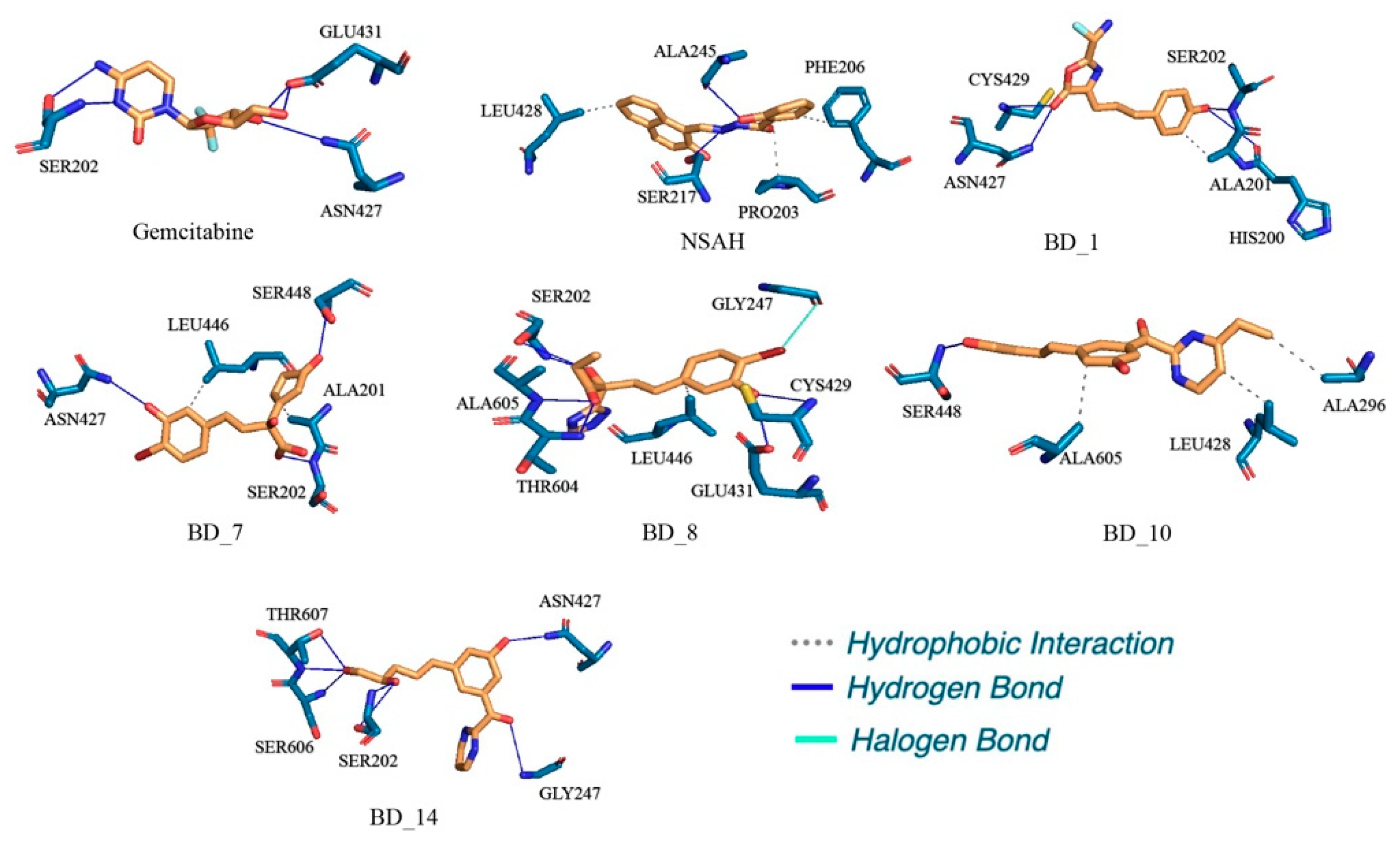
3.5. Post MD Simulation Binding Interaction Analysis
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Valle, J.W.; Borbath, I.; Khan, S.A.; Huguet, F.; Gruenberger, T.; Arnold, D. On behalf of the ESMO Guidelines Committee Biliary cancer: ESMO clinical practice guidelines for diagnosis, treatment and follow-up. Ann. Oncol. 2016, 27, v28–v37. [Google Scholar] [CrossRef] [PubMed]
- West, J.; Wood, H.; Logan, R.F.A.; Quinn, M.; Aithal, G.P. Trends in the incidence of primary liver and biliary tract cancers in England and Wales 1971–2001. Br. J. Cancer 2006, 94, 1751–1758. [Google Scholar] [CrossRef]
- Taylor-Robinson, S.D.; Toledano, M.B.; Arora, S.; Keegan, T.J.; Hargreaves, S.; Beck, A.; Khan, S.A.; Elliott, P.; Thomas, H.C. Increase in mortality rates from intrahepatic cholangiocarcinoma in England and Wales 1968–1998. Gut 2001, 48, 816–820. [Google Scholar] [CrossRef] [PubMed]
- Von Hahn, T.; Ciesek, S.; Wegener, G.; Plentz, R.R.; Weismüller, T.J.; Wedemeyer, H.; Manns, M.P.; Greten, T.F.; Malek, N.P. Epidemiological trends in incidence and mortality of hepatobiliary cancers in Germany. Scand. J. Gastroenterol. 2011, 46, 1092–1098. [Google Scholar] [CrossRef] [PubMed]
- Alvaro, D.; Crocetti, E.; Ferretti, S.; Bragazzi, M.C.; Capocaccia, R. Descriptive epidemiology of cholangiocarcinoma in Italy. Dig. Liver Dis. 2010, 42, 490–495. [Google Scholar] [CrossRef] [PubMed]
- Witjes, C.D.; Karim-Kos, H.E.; Visser, O.; de Vries, E.; Ijzermans, J.N.; de Man, R.A.; Coebergh, J.W.W.; Verhoef, C. Intrahepatic cholangiocarcinoma in a low endemic area: Rising incidence and improved survival. HPB 2012, 14, 777–781. [Google Scholar] [CrossRef]
- Lee, T.-Y.; Lin, J.-T.; Kuo, K.N.; Wu, M.-S.; Ho, H.J.; Chen, T.-T.; Wu, C.-Y. A nationwide population-based study shows increasing incidence of cholangiocarcinoma. Hepatol. Int. 2012, 7, 226–232. [Google Scholar] [CrossRef]
- Utada, M.; Ohno, Y.; Tamaki, T.; Sobue, T.; Endo, G. Long-term Trends in Incidence and Mortality of Intrahepatic and Extrahepatic Bile Duct Cancer in Japan. J. Epidemiol. 2014, 24, 193–199. [Google Scholar] [CrossRef]
- Banales, J.M.; Marin, J.J.G.; Lamarca, A.; Rodrigues, P.M.; Khan, S.A.; Roberts, L.R.; Cardinale, V.; Carpino, G.; Andersen, J.B.; Braconi, C.; et al. Cholangiocarcinoma 2020: The next horizon in mechanisms and management. Nat. Rev. Gastroenterol. Hepatol. 2020, 17, 557–588. [Google Scholar] [CrossRef]
- Banales, J.M.; Cardinale, V.; Carpino, G.; Marzioni, M.; Andersen, J.B.; Invernizzi, P.; Lind, G.E.; Folseraas, T.; Forbes, S.J.; Fouassier, L.; et al. Expert consensus document: Cholangiocarcinoma: Current knowledge and future perspectives consensus statement from the European Network for the Study of Cholangiocarcinoma (ENS-CCA). Nat. Rev. Gastroenterol. Hepatol. 2016, 13, 261–280. [Google Scholar] [CrossRef]
- Andersen, J.B.; Spee, B.; Blechacz, B.R.; Avital, I.; Komuta, M.; Barbour, A.; Conner, E.A.; Gillen, M.C.; Roskams, T.; Roberts, L.; et al. Genomic and Genetic Characterization of Cholangiocarcinoma Identifies Therapeutic Targets for Tyrosine Kinase Inhibitors. Gastroenterology 2012, 142, 1021–1031.e15. [Google Scholar] [CrossRef] [PubMed]
- Lindnér, P.; Rizell, M.; Hafström, L. The Impact of Changed Strategies for Patients with Cholangiocarcinoma in This Millenium. HPB Surg. 2015, 2015, 736049. [Google Scholar] [CrossRef] [PubMed]
- Kamsa-Ard, S.; Luvira, V.; Suwanrungruang, K.; Kamsa-Ard, S.; Luvira, V.; Santong, C.; Srisuk, T.; Pugkhem, A.; Bhudhisawasdi, V.; Pairojkul, C. Cholangiocarcinoma Trends, Incidence, and Relative Survival in Khon Kaen, Thailand From 1989 Through 2013: A Population-Based Cancer Registry Study. J. Epidemiol. 2019, 29, 197–204. [Google Scholar] [CrossRef]
- Strijker, M.; Belkouz, A.; van der Geest, L.G.; van Gulik, T.M.; van Hooft, J.E.; de Meijer, V.E.; Haj Mohammad, N.; de Reuver, P.R.; Verheij, J.; de Vos-Geelen, J.; et al. Treatment and survival of resected and unresected distal cholangiocarcinoma: A nationwide study. Acta Oncol. (Madr.) 2019, 58, 1048–1055. [Google Scholar] [CrossRef] [PubMed]
- Alabraba, E.; Joshi, H.; Bird, N.; Griffin, R.; Sturgess, R.; Stern, N.; Sieberhagen, C.; Cross, T.; Camenzuli, A.; Davis, R.; et al. Increased multimodality treatment options has improved survival for Hepatocellular carcinoma but poor survival for biliary tract cancers remains unchanged. Eur. J. Surg. Oncol. (EJSO) 2019, 45, 1660–1667. [Google Scholar] [CrossRef] [PubMed]
- Groot Koerkamp, B.; Wiggers, J.K.; Allen, P.J.; Besselink, M.G.; Blumgart, L.H.; Busch, O.R.C.; Coelen, R.J.; D’Angelica, M.I.; DeMatteo, R.P.; Gouma, D.J.; et al. Recurrence Rate and Pattern of Perihilar Cholangiocarcinoma after Curative Intent Resection Presented at the 11th Congress of the European-African Hepa-to-Pancreato-Biliary Association, Manchester, UK, April 2015. J. Am. Coll. Surg. 2015, 221, 1041–1049. [Google Scholar]
- Komaya, K.; Ebata, T.; Yokoyama, Y.; Igami, T.; Sugawara, G.; Mizuno, T.; Yamaguchi, J.; Nagino, M. Recurrence after curative-intent resection of perihilar cholangiocarcinoma: Analysis of a large cohort with a close postoperative follow-up approach. Surgery 2018, 163, 732–738. [Google Scholar] [CrossRef] [PubMed]
- Cambridge, W.A.; Fairfield, C.; Powell, J.J.; Harrison, E.M.; Søreide, K.; Wigmore, S.J.; Guest, R.V. Meta-analysis and Meta-regression of Survival After Liver Transplantation for Unresectable Perihilar Cholangiocarcinoma. Ann. Surg. 2020, 273, 240–250. [Google Scholar] [CrossRef] [PubMed]
- Spolverato, G.; Kim, Y.; Alexandrescu, S.; Marques, H.P.; Lamelas, J.; Aldrighetti, L.; Clark Gamblin, T.; Maithel, S.K.; Pulitano, C.; Bauer, T.W.; et al. Management and Outcomes of Patients with Recurrent Intrahepatic Cholan-giocarcinoma Following Previous Curative-Intent Surgical Resection. Ann. Surg. Oncol. 2016, 23, 235–243. [Google Scholar] [CrossRef]
- Greene, B.L.; Kang, G.; Cui, C.; Bennati, M.; Nocera, D.G.; Drennan, C.L.; Stubbe, J. Ribonucleotide Reductases: Structure, Chemistry, and Metabolism Suggest New Therapeutic Targets. Annu. Rev. Biochem. 2020, 89, 45–75. [Google Scholar] [CrossRef]
- Brown, N.C.; Canellakis, Z.N.; Lundin, B.; Reichard, P.; Thelander, L. Ribonucleoside Diphosphate Reductase. Purification of the two Subunits, Proteins B1 and B2. Eur. J. Biochem. 1969, 9, 561–573. [Google Scholar] [CrossRef] [PubMed]
- Shao, J.; Liu, X.; Zhu, L.; Yen, Y. Targeting ribonucleotide reductase for cancer therapy. Expert Opin. Ther. Targets 2013, 17, 1423–1437. [Google Scholar] [CrossRef] [PubMed]
- Nordlund, P.; Reichard, P. Ribonucleotide reductases. Annu. Rev. Biochem. 2006, 75, 681–706. [Google Scholar] [CrossRef] [PubMed]
- Reichard, P. Interactions between deoxyribonucleotide and DNA synthesis. Annu. Rev. Biochem. 1988, 57, 349–374. [Google Scholar] [CrossRef] [PubMed]
- Bester, A.C.; Roniger, M.; Oren, Y.S.; Im, M.M.; Sarni, D.; Chaoat, M.; Bensimon, A.; Zamir, G.; Shewach, D.S.; Kerem, B. Nucleotide Deficiency Promotes Genomic Instability in Early Stages of Cancer Development. Cell 2011, 145, 435–446. [Google Scholar] [CrossRef]
- Burrell, R.A.; McClelland, S.E.; Endesfelder, D.; Groth, P.; Weller, M.C.; Shaikh, N.; Domingo, E.; Kanu, N.; Dewhurst, S.M.; Gronroos, E.; et al. Replication stress links structural and numerical cancer chromosomal instability. Nature 2013, 494, 492–496. [Google Scholar] [CrossRef]
- Wheeler, L.J.; Rajagopal, I.; Mathews, C.K. Stimulation of mutagenesis by proportional deoxyribonucleoside tri-phosphate accumulation in Escherichia coli. DNA Repair 2005, 4, 1450–1456. [Google Scholar] [CrossRef]
- Chabes, A.; Georgieva, B.; Domkin, V.; Zhao, X.; Rothstein, R.; Thelander, L. Survival of DNA Damage in Yeast Directly Depends on Increased dNTP Levels Allowed by Relaxed Feedback Inhibition of Ribonucleotide Reductase. Cell 2003, 112, 391–401. [Google Scholar] [CrossRef]
- Fan, H.; Villegas, C.; Huang, A.; Wright, J.A. The mammalian ribonucleotide reductase R2 component cooperates with a variety of oncogenes in mechanisms of cellular transformation. Cancer Res. 1998, 58, 1650–1653. [Google Scholar]
- Cory, J.G.; Cory, A.H.; Rappa, G.; Lorico, A.; Mao-Chin, L.; Tai-Shun, L.; Sartorelli, A.C. Inhibitors of ribonucleo-tide reductase. Comparative effects of amino- and hydroxy-substituted pyri-dine-2-carboxaldehydethiosemicarbazones. Biochem. Pharmacol. 1994, 48, 335–344. [Google Scholar]
- Liu, M.C.; Lin, T.S.; Cory, J.G.; Cory, A.H.; Sartorelli, A.C. Synthesis and biological activity of 3- and 5-amino derivatives of pyridine-2-carboxaldehyde thiosemicarbazone. J. Med. Chem. 1996, 39, 2586–2593. [Google Scholar] [CrossRef] [PubMed]
- Licht, S.; Gerfen, G.J.; Stubbe, J. Thiyl Radicals in Ribonucleotide Reductases. Science 1996, 271, 477–481. [Google Scholar] [CrossRef] [PubMed]
- Fairman, J.W.; Wijerathna, S.R.; Ahmad, F.; Xu, H.; Nakano, R.; Jha, S.; Prendergast, J.; Welin, R.M.; Flodin, S.; Roos, A.; et al. Structural basis for allosteric regulation of human ribonucleotide reductase by nucleotide-induced oligomerization. Nat. Struct. Mol. Biol. 2011, 18, 316–322. [Google Scholar] [CrossRef] [Green Version]
- Misko, T.A.; Liu, Y.-T.; Harris, M.E.; Oleinick, N.L.; Pink, J.; Lee, H.-Y.; Dealwis, C.G. Structure-guided design of anti-cancer ribonucleotide reductase inhibitors. J. Enzym. Inhib. Med. Chem. 2019, 34, 438–450. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Lohman, G.J.S.; Stubbe, J. Enhanced subunit interactions with gemcitabine-5′-diphosphate inhibit ribonucleotide reductases. Proc. Natl. Acad. Sci. USA 2007, 104, 14324–14329. [Google Scholar] [CrossRef]
- Aye, Y.; Stubbe, J. Clofarabine 5′-di and -triphosphates inhibit human ribonucleotide reductase by altering the quaternary structure of its large subunit. Proc. Natl. Acad. Sci. USA 2011, 108, 9815–9820. [Google Scholar] [CrossRef] [PubMed]
- Aye, Y.; Brignole, E.J.; Long, M.J.; Chittuluru, J.; Drennan, C.L.; Asturias, F.J.; Stubbe, J. Clofarabine Targets the Large Subunit (α) of Human Ribonucleotide Reductase in Live Cells by Assembly into Persistent Hexamers. Chem. Biol. 2012, 19, 799–805. [Google Scholar] [CrossRef] [PubMed]
- Xie, C.; Plunkett, W. Metabolism and Actions of 2-Chloro-9-(2-deoxy-2-fluoro-β-D-arabinofuranosyl)-adenine in Human Lymphoblastoid Cells. Cancer Res. 1995, 55, 2847–2852. [Google Scholar] [PubMed]
- Zhang, C.; Liu, G.; Huang, M. Ribonucleotide reductase metallocofactor: Assembly, maintenance and inhibition. Front. Biol. 2014, 9, 104–113. [Google Scholar] [CrossRef]
- Loehre, P.J.L.; Feng, Y.; Cardenes, H.; Wagner, L.; Brell, J.M.; Cella, D.; Flynn, P.; Ramanathan, R.K.; Crane, C.H.; Alberts, S.R.; et al. Gemcitabine Alone Versus Gemcitabine Plus Radiotherapy in Patients With Locally Advanced Pancreatic Cancer: An Eastern Cooperative Oncology Group Trial. J. Clin. Oncol. 2011, 29, 4105–4112. [Google Scholar] [CrossRef]
- Burris, H.A., 3rd; Moore, M.J.; Andersen, J.; Green, M.R.; Rothenberg, M.L.; Modiano, M.R.; Cripps, M.C.; Portenoy, R.K.; Storniolo, A.M.; Tarassoff, P.; et al. Improvements in survival and clinical benefit with gemcitabine as first-line therapy for patients with advanced pancreas cancer: A randomized trial. J. Clin. Oncol. 1997, 15, 2403–2413. [Google Scholar] [CrossRef]
- Oettle, H.; Post, S.; Neuhaus, P.; Gellert, K.; Langrehr, J.; Ridwelski, K.; Schramm, H.; Fahlke, J.; Zuelke, C.; Burkart, C.; et al. Adjuvant chemotherapy with gemcitabine vs observation in patients undergoing curative-intent resection of pancreatic cancer: A randomized controlled trial. J. Am. Med. Assoc. 2007, 297, 267–277. [Google Scholar] [CrossRef] [PubMed]
- Raderer, M.; Hejna, M.H.L.; Valencak, J.B.; Kornek, G.V.; Weinländer, G.S.; Bareck, E.; Lenauer, J.; Brodowicz, T.; Lang, F.; Scheithauer, W. Two consecutive phase II studies of 5-fluorouracil/leucovorin/mitomycin C and of gemcitabine in patients with advanced biliary cancer. Oncology 1999, 56, 177–180. [Google Scholar] [CrossRef] [PubMed]
- Gebbia, V.; Giuliani, F.; Maiello, E.; Colucci, G.; Verderame, F.; Borsellino, N.; Mauceri, G.; Caruso, M.; Tirrito, M.L.; Valdesi, M. Treatment of Inoperable and/or Metastatic Biliary Tree Carcinomas with Single-Agent Gemcitabine or in Combination with Levofolinic Acid and Infusional Fluorouracil: Results of a Multicenter Phase II Study. J. Clin. Oncol. 2001, 19, 4089–4091. [Google Scholar] [CrossRef] [PubMed]
- Kubicka, S.; Rudolph, K.L.; Tietze, M.K.; Lorenz, M.; Manns, M. Phase II study of systemic gemcitabine chemo-therapy for advanced unresectable hepatobiliary carcinomas. Hepatogastroenterology 2001, 48, 783–789. [Google Scholar] [PubMed]
- Hong, Y.S.; Lee, J.; Lee, S.C.; Hwang, I.G.; Choi, S.-H.; Heo, J.-S.; Park, J.O.; Park, Y.S.; Lim, H.Y.; Kang, W.K. Phase II study of capecitabine and cisplatin in previously untreated advanced biliary tract cancer. Cancer Chemother. Pharmacol. 2006, 60, 321–328. [Google Scholar] [CrossRef] [PubMed]
- Kim, T.W.; Chang, H.M.; Kang, H.J.; Lee, J.R.; Ryu, M.H.; Ahn, J.H.; Kim, J.H.; Kang, Y.K. Phase II study of capecitabine plus cisplatin as first-line chemotherapy in advanced biliary cancer. Ann. Oncol. 2003, 14, 1115–1120. [Google Scholar] [CrossRef]
- Hezel, A.F.; Zhu, A.X. Systemic Therapy for Biliary Tract Cancers. Oncol. 2008, 13, 415–423. [Google Scholar] [CrossRef] [PubMed]
- Baker, C.H.; Banzon, J.; Bollinger, J.M.; Stubbe, J.; Samano, V.; Robins, M.J.; Lippert, B.; Jarvi, E.; Resvick, R. 2′-Deoxy-2′-methylenecytidine and 2′-Deoxy-2′,2′-difluorocytidine 5′-Diphosphates: Potent Mechanism-Based Inhibitors of Ribonucleotide Reductase. J. Med. Chem. 1991, 34, 1879–1884. [Google Scholar] [CrossRef]
- Gandhi, V.; Plunkett, W. Modulatory Activity of 2′,2′-Difluorodeoxycytidine on the Phosphorylation and Cyto-toxicity of Arabinosyl Nucleosides. Cancer Res. 1990, 50, 3675–3680. [Google Scholar] [PubMed]
- Heinemann, V.; Schulz, L.; Issels, R.; Plunkett, W. Gemcitabine: A modulator of intracellular nucleotide and deoxynucleotide metabolism. Semin. Oncol. 1995, 22, 11–18. [Google Scholar] [PubMed]
- Clark, D.E. What has virtual screening ever done for drug discovery? Expert Opin. Drug Discov. 2008, 3, 841–851. [Google Scholar] [CrossRef] [PubMed]
- Morgan, S.; Grootendorst, P.; Lexchin, J.; Cunningham, C.; Greyson, D. The cost of drug development: A systematic review. Health Policy 2011, 100, 4–17. [Google Scholar] [CrossRef] [PubMed]
- Dickson, M.; Gagnon, J.P. The cost of new drug discovery and development. Discov. Med. 2004, 4, 172–179. [Google Scholar]
- Mullin, R. Drug Development Costs About $1.7 Billion. Chem. Eng. News Arch. 2003, 81, 8. [Google Scholar] [CrossRef]
- Petrova, E. Innovation in the pharmaceutical industry: The process of drug discovery and development. In Innovation and Marketing in the Pharmaceutical Industry; Springer: Berlin/Heidelberg, Germany, 2014; pp. 19–81. [Google Scholar] [CrossRef]
- Kiriiri, G.K.; Njogu, P.; Mwangi, A.N. Exploring different approaches to improve the success of drug discovery and development projects: A review. Futur. J. Pharm. Sci. 2020, 6, 27. [Google Scholar] [CrossRef]
- Rickels, W.; Dovern, J.; Hoffmann, J.; Quaas, M.F.; Schmidt, J.O.; Visbeck, M. Indicators for monitoring sustainable development goals: An application to oceanic development in the European Union. Earth's Futur. 2016, 4, 252–267. [Google Scholar] [CrossRef]
- Mouchlis, V.D.; Afantitis, A.; Serra, A.; Fratello, M.; Papadiamantis, A.G.; Aidinis, V.; Lynch, I.; Greco, D.; Melagraki, G. Advances in de Novo Drug Design: From Conventional to Machine Learning Methods. Int. J. Mol. Sci. 2021, 22, 1676. [Google Scholar] [CrossRef]
- Devi, R.V.; Sathya, S.S.; Coumar, M.S. Evolutionary algorithms for de novo drug design—A survey. Appl. Soft Comput. 2015, 27, 543–552. [Google Scholar] [CrossRef]
- Jokinen, E.M.; Postila, P.; Ahinko, M.; Niinivehmas, S.; Pentikäinen, O.T. Fragment- and negative image-based screening of phosphodiesterase 10A inhibitors. Chem. Biol. Drug Des. 2019, 94, 1799–1812. [Google Scholar] [CrossRef]
- Bender, B.J.; Gahbauer, S.; Luttens, A.; Lyu, J.; Webb, C.M.; Stein, R.M.; Fink, E.A.; Balius, T.E.; Carlsson, J.; Irwin, J.J.; et al. A practical guide to large-scale docking. Nat. Protoc. 2021, 16, 4799–4832. [Google Scholar] [CrossRef]
- Lo, Y.-C.; Rensi, S.E.; Torng, W.; Altman, R.B. Machine learning in chemoinformatics and drug discovery. Drug Discov. Today 2018, 23, 1538–1546. [Google Scholar] [CrossRef] [PubMed]
- Alam, S.; Khan, F. Virtual screening, Docking, ADMET and System Pharmacology studies on Garcinia caged Xanthone derivatives for Anticancer activity. Sci. Rep. 2018, 8, 5524. [Google Scholar] [CrossRef] [PubMed]
- Korb, O.; Stützle, T.; Exner, T.E. PLANTS: Application of ant colony optimization to structure-based drug design. In Proceedings of the Lecture Notes in Computer Science (including Subseries Lecture Notes in Artificial Intelligence and Lecture Notes in Bioinformatics); Springer: Berlin/Heidelberg, Germany, 2006; Volume 4150 LNCS, pp. 247–258. [Google Scholar]
- Jiménez, J.; Škalič, M.; Martínez-Rosell, G.; De Fabritiis, G. KDEEP: Protein-Ligand Absolute Binding Affinity Pre-diction via 3D-Convolutional Neural Networks. J. Chem. Inf. Model. 2018, 58, 287–296. [Google Scholar] [CrossRef] [PubMed]
- Shetve, V.; Bhowmick, S.; Alissa, S.; Alothman, Z.; Wabaidur, S.; Alasmary, F.; Alhajri, H.; Islam, M. Identification of selective Lyn inhibitors from the chemical databases through integrated molecular modelling approaches. SAR QSAR Environ. Res. 2020, 32, 1–27. [Google Scholar] [CrossRef] [PubMed]
- Tsavaris, N.; Kosmas, C.; Gouveris, P.; Gennatas, K.; Polyzos, A.; Mouratidou, D.; Tsipras, H.; Margaris, H.; Papastratis, G.; Tzima, E.; et al. Weekly Gemcitabine for the Treatment of Biliary Tract and Gallbladder Cancer. Investig. New Drugs 2004, 22, 193–198. [Google Scholar] [CrossRef]
- Morizane, C.; Okusaka, T.; Mizusawa, J.; Katayama, H.; Ueno, M.; Ikeda, M.; Ozaka, M.; Okano, N.; Sugimori, K.; Fukutomi, A.; et al. Combination gemcitabine plus S-1 versus gemcitabine plus cisplatin for advanced/recurrent biliary tract cancer: The FUGA-BT (JCOG1113) randomized phase III clinical trial. Ann. Oncol. 2019, 30, 1950–1958. [Google Scholar] [CrossRef]
- Ahmad, F.; Alam, I.; Huff, S.E.; Pink, J.; Flanagan, S.A.; Shewach, D.; Misko, T.A.; Oleinick, N.L.; Harte, W.E.; Viswanathan, R.; et al. Potent competitive inhibition of human ribonucleotide reductase by a nonnucleoside small molecule. Proc. Natl. Acad. Sci. USA 2017, 114, 8241–8246. [Google Scholar] [CrossRef]
- Berman, H.M.; Westbrook, J.; Feng, Z.; Gilliland, G.; Bhat, T.N.; Weissig, H.; Shindyalov, I.N.; Bourne, P.E. The Protein Data Bank. Nucleic Acids Res. 2000, 28, 235–242. [Google Scholar] [CrossRef]
- Jo, S.; Kim, T.; Iyer, V.G.; Im, W. CHARMM-GUI: A web-based graphical user interface for CHARMM. J. Comput. Chem. 2008, 29, 1859–1865. [Google Scholar] [CrossRef]
- Ahn, Y.; Jun, Y. Measurement of pain-like response to various NICU stimulants for high-risk infants. Early Hum. Dev. 2007, 83, 255–262. [Google Scholar] [CrossRef] [PubMed]
- Trott, O.; Olson, A.J. AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem. 2010, 31, 455–461. [Google Scholar] [CrossRef] [PubMed]
- Yuan, Y.; Pei, J.; Lai, L. LigBuilder V3: A Multi-Target de novo Drug Design Approach. Front. Chem. 2020, 8, 142. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dias, R.; de Azevedo Jr., W. Molecular Docking Algorithms. Curr. Drug Targets 2008, 9, 1040–1047. [Google Scholar] [CrossRef]
- O’Boyle, N.M.; Banck, M.; James, C.A.; Morley, C.; Vandermeersch, T.; Hutchison, G.R. Open babel: An open chemical toolbox. J. Cheminform. 2011, 3, 33. [Google Scholar] [CrossRef]
- Landrum, G. RDKit: Open-Source Cheminformatics Software. 2021. Available online: http://www.Rdkit.Org/ (accessed on 1 September 2022).
- Gasteiger, J.; Marsili, M. A new model for calculating atomic charges in molecules. Tetrahedron Lett. 1978, 19, 3181–3184. [Google Scholar] [CrossRef]
- Hartigan, J.A.; Wong, M.A. Algorithm AS 136: A K-Means Clustering Algorithm. Appl. Stat. 1979, 28, 100. [Google Scholar] [CrossRef]
- Niinivehmas, S.P.; Salokas, K.; Lätti, S.; Raunio, H.; Pentikäinen, O.T. Ultrafast protein structure-based virtual screening with Panther. J. Comput. Aided. Mol. Des. 2015, 29, 989–1006. [Google Scholar] [CrossRef]
- Daina, A.; Michielin, O.; Zoete, V. SwissADME: A free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules. Sci. Rep. 2017, 7, 42717. [Google Scholar] [CrossRef]
- Glassman, P.M.; Muzykantov, V.R. Pharmacokinetic and Pharmacodynamic Properties of Drug Delivery Systems. J. Pharmacol. Exp. Ther. 2019, 370, 570–580. [Google Scholar] [CrossRef]
- Lipinski, C.A.; Lombardo, F.; Dominy, B.W.; Feeney, P.J. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv. Drug Deliv. Rev. 2012, 64, 4–17. [Google Scholar] [CrossRef]
- Veber, D.F.; Johnson, S.R.; Cheng, H.-Y.; Smith, B.R.; Ward, K.W.; Kopple, K.D. Molecular Properties That Influence the Oral Bioavailability of Drug Candidates. J. Med. Chem. 2002, 45, 2615–2623. [Google Scholar] [CrossRef] [PubMed]
- Pires, D.E.V.; Blundell, T.L.; Ascher, D.B. pkCSM: Predicting small-molecule pharmacokinetic and toxicity properties using graph-based signatures. J. Med. Chem. 2015, 58, 4066–4072. [Google Scholar] [CrossRef] [PubMed]
- Zeiger, E. The test that changed the world: The Ames test and the regulation of chemicals. Mutat. Res. Mol. Mech. Mutagen. 2019, 841, 43–48. [Google Scholar] [CrossRef] [PubMed]
- Korb, O.; Stützle, T.; Exner, T.E. An ant colony optimization approach to flexible protein–ligand docking. Swarm Intell. 2007, 1, 115–134. [Google Scholar] [CrossRef]
- Nanda, B.K.; Das, G. Ant Colony Optimization. A Computational Intelligence Technique. Int. J. Comput. Commun. Technol. 2013, 2, 58–63. [Google Scholar] [CrossRef]
- Stützle, T.; Hoos, H.H. MAX-MIN Ant System. Futur. Gener. Comput. Syst. 2000, 16, 889–914. [Google Scholar] [CrossRef]
- Korb, O.; Stützle, T.; Exner, T.E. Empirical Scoring Functions for Advanced Protein−Ligand Docking with PLANTS. J. Chem. Inf. Model. 2009, 49, 84–96. [Google Scholar] [CrossRef]
- Abraham, M.J.; Murtola, T.; Schulz, R.; Páll, S.; Smith, J.C.; Hess, B.; Lindah, E. Gromacs: High performance molecular simulations through multi-level parallelism from laptops to supercomputers. SoftwareX 2015, 1–2, 19–25. [Google Scholar] [CrossRef]
- Lindahl; Abraham; Hess; van der Spoel GROMACS 2021.3 Source code 2021.
- Huang, J.; Rauscher, S.; Nawrocki, G.; Ran, T.; Feig, M.; de Groot, B.L.; Grubmüller, H.; MacKerell, A.D., Jr. CHARMM36m: An improved force field for folded and intrinsically disordered proteins. Nat. Methods 2017, 14, 71–73. [Google Scholar] [CrossRef]
- Zoete, V.; Cuendet, M.A.; Grosdidier, A.; Michielin, O. SwissParam: A fast force field generation tool for small organic molecules. J. Comput. Chem. 2011, 32, 2359–2368. [Google Scholar] [CrossRef] [PubMed]
- Mark, P.; Nilsson, L. Structure and Dynamics of the TIP3P, SPC, and SPC/E Water Models at 298 K. J. Phys. Chem. A 2001, 105, 9954–9960. [Google Scholar] [CrossRef]
- Valdés-Tresanco, M.S.; Valiente, P.A.; Moreno, E. gmx_MMPBSA: A New Tool to Perform End-State Free Energy Calculations with GROMACS. J. Chem. Theory Comput. 2021, 17, 6281–6291. [Google Scholar] [CrossRef] [PubMed]
- Wang, E.; Sun, H.; Wang, J.; Wang, Z.; Liu, H.; Zhang, J.Z.H.; Hou, T. End-Point Binding Free Energy Calculation with MM/PBSA and MM/GBSA: Strategies and Applications in Drug Design. Chem. Rev. 2019, 119, 9478–9508. [Google Scholar] [CrossRef]
- Sitkoff, D.; Sharp, K.A.; Honig, B. Accurate Calculation of Hydration Free Energies Using Macroscopic Solvent Models. J. Phys. Chem. 1994, 98, 1978–1988. [Google Scholar] [CrossRef]
- Tan, C.; Tan, Y.-H.; Luo, R. Implicit Nonpolar Solvent Models. J. Phys. Chem. B 2007, 111, 12263–12274. [Google Scholar] [CrossRef]
- Gohlke, H.; Kiel, C.; Case, D.A. Insights into protein-protein binding by binding free energy calculation and free energy decomposition for the Ras-Raf and Ras-RalGDS complexes. J. Mol. Biol. 2003, 330, 891–913. [Google Scholar] [CrossRef]














{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Molecules | Log (IC50) in µM | Model I | Model II | Model III |
|---|---|---|---|---|
| Gemcitabine | 2.622 | 0.451 | 0.513 | 0.645 |
| M40128 | 2.396 | 0.530 | 0.529 | 0.454 |
| M777989 | 2.879 | 0.307 | 0.720 | 0.548 |
| M951562 | 2.807 | 0.489 | 0.681 | 0.568 |
| NSAH | 2.495 | 0.389 | 0.524 | 0.495 |
| M859980 | 3.420 | 0.654 | 0.653 | 0.597 |
| Correlation coefficient (R) | 0.480 | 0.660 | 0.535 | |
| Mols | PLANTS Score (kcal/mol) | SwissDock ΔG (kcal/mol) | KDEEP ΔG (kcal/mol) |
|---|---|---|---|
| Gemcitabine | −67.106 | −7.46 | −5.819 |
| NSAH | −80.290 | −7.19 | −5.520 |
| BD_1 | −83.636 | −7.70 | −6.961 |
| BD_2 | −76.806 | −7.52 | −8.999 |
| BD_3 | −75.577 | −7.70 | −5.986 |
| BD_4 | −73.217 | −7.22 | −7.777 |
| BD_5 | −75.217 | −7.42 | −8.313 |
| BD_6 | −89.560 | −8.56 | −8.092 |
| BD_7 | −92.362 | −8.16 | −8.854 |
| BD_8 | −82.278 | −7.92 | −8.612 |
| BD_9 | −83.276 | −7.81 | −8.109 |
| BD_10 | −79.143 | −7.74 | −7.821 |
| BD_11 | −71.442 | −7.96 | −7.512 |
| BD_12 | −87.175 | −7.97 | −9.222 |
| BD_13 | −61.565 | −7.50 | −7.382 |
| BD_14 | −84.934 | −8.41 | −7.711 |
| BD_15 | −78.741 | −7.65 | −8.140 |
| BD_16 | −72.471 | −7.59 | −9.358 |
| BD_17 | −83.795 | −8.34 | −8.129 |
| Molecules | 1 HBD | 2 HBA | 3 HY | 4 RA |
|---|---|---|---|---|
| cPharmFrac | ||||
| Gemcitabine + | 0.273 | 0.364 | 0.182 | 0.182 |
| NSAH | ||||
| PharmFrac | ||||
| BD_1 | 0.182 | 0.364 | 0.364 | 0.091 |
| BD_2 | 0.200 | 0.300 | 0.300 | 0.200 |
| BD_3 | 0.300 | 0.200 | 0.400 | 0.100 |
| BD_4 | 0.214 | 0.357 | 0.286 | 0.143 |
| BD_5 | 0.133 | 0.467 | 0.267 | 0.133 |
| BD_6 | 0.167 | 0.417 | 0.333 | 0.083 |
| BD_7 | 0.214 | 0.357 | 0.357 | 0.071 |
| BD_8 | 0.143 | 0.357 | 0.357 | 0.143 |
| BD_9 | 0.286 | 0.286 | 0.357 | 0.071 |
| BD_10 | 0.231 | 0.231 | 0.462 | 0.077 |
| BD_11 | 0.231 | 0.308 | 0.385 | 0.077 |
| BD_12 | 0.267 | 0.200 | 0.400 | 0.133 |
| BD_13 | 0.231 | 0.462 | 0.231 | 0.077 |
| BD_14 | 0.286 | 0.286 | 0.357 | 0.071 |
| BD_15 | 0.214 | 0.286 | 0.357 | 0.143 |
| BD_16 | 0.200 | 0.333 | 0.333 | 0.133 |
| BD_17 | 0.231 | 0.308 | 0.385 | 0.077 |
| Molecule | ΔGbind kcal/mol | Standard Deviation |
|---|---|---|
| Gemcitabine | −36.65 | 3.15 |
| NSAH | −19.53 | 2.91 |
| BD_1 | −24.62 | 3.74 |
| BD_2 | −23.75 | 2.81 |
| BD_3 | −17.79 | 4.11 |
| BD_4 | −12.59 | 3.94 |
| BD_5 | −12.45 | 3.84 |
| BD_6 | −20.74 | 5.22 |
| BD_7 | −48.27 | 4.68 |
| BD_8 | −39.72 | 3.41 |
| BD_9 | −18.27 | 4.12 |
| BD_10 | −27.02 | 3.15 |
| BD_11 | −14.56 | 5.82 |
| BD_12 | −15.04 | 4.91 |
| BD_13 | −17.89 | 5.01 |
| BD_14 | −29.75 | 4.75 |
| BD_15 | −9.74 | 6.16 |
| BD_16 | −14.65 | 3.14 |
| BD_17 | −7.08 | 4.22 |
| Parameters | Gemcitabine | NSAH | BD_1 | BD_7 | BD_8 | BD_10 | BD_14 | |
|---|---|---|---|---|---|---|---|---|
| Backbone RMSD (nm) | Min. | 0.001 | 0.001 | 0.001 | 0.000 | 0.001 | 0.000 | 0.000 |
| Max | 1.263 | 0.681 | 0.947 | 1.106 | 1.599 | 0.875 | 1.250 | |
| Avg | 1.057 | 0.442 | 0.806 | 0.884 | 1.092 | 0.785 | 0.611 | |
| Ligand RMSD (nm) | Min | 0.000 | 0.000 | 0.000 | 0.000 | 0.000 | 0.000 | 0.000 |
| Max | 0.114 | 0.187 | 0.214 | 0.109 | 0.165 | 0.313 | 0.209 | |
| Avg | 0.044 | 0.072 | 0.107 | 0.053 | 0.099 | 0.199 | 0.123 | |
| RMSF (nm) | Min | 0.063 | 0.052 | 0.051 | 0.051 | 0.071 | 0.053 | 0.066 |
| Max | 1.525 | 2.286 | 1.731 | 1.242 | 3.803 | 1.988 | 2.210 | |
| Avg | 0.220 | 0.188 | 0.177 | 0.177 | 0.357 | 0.176 | 0.296 | |
| RoG (nm) | Min | 2.843 | 2.804 | 2.780 | 2.815 | 2.851 | 2.776 | 2.846 |
| Max | 2.999 | 2.991 | 3.028 | 2.976 | 3.205 | 3.035 | 3.156 | |
| Avg | 2.911 | 2.881 | 2.858 | 2.870 | 3.029 | 2.846 | 2.926 |
| Molecule | ΔGbind kcal/mol (Std. Dev.) |
|---|---|
| Gemcitabine | −39.13 (±3.07) |
| NSAH | −17.53 (±3.41) |
| BD_1 | −31.13 (±3.52) |
| BD_7 | −55.27 (±4.75) |
| BD_8 | −47.11 (±2.99) |
| BD_10 | −35.31 (±3.02) |
| BD_14 | −37.39 (±4.65) |
| Binding Interaction Analysis | |||||||
|---|---|---|---|---|---|---|---|
| Molecule | Bonds | Mol. Dock. | Post-MD Simulation | ||||
| 0 ns | 25 ns | 50 ns | 75 ns | 100 ns | |||
| Gemcitabine | HY | ALA201, THR604 | - | - | - | - | - |
| HB | HIS200, SER202, GLU431, SER448, THR607 | SER202, SER217, GLU431, SER448, THR607 | SER202, GLU431, SER448 | SER202, SER448, THR607 | ASN427, GLU431, ALA605, SER606, THR607 | SER202, ASN427, GLU431 | |
| NSAH | HY | GLN288, LEU428 | ALA245, GLN288, LEU428 | PHE206 | ARG153 | PRO203, GLN214, ALA245, ALA296 | PRO203, PHE206, LEU428 |
| HB | ALA245, GLN246, ALA296 | ALA245, ARG293, ARG293, ALA296 | SER202 | SER154 | - | SER217, ALA245 | |
| BD_1 | HY | LEU446, MET602, ALA605 | - | - | - | ALA201, LEU446 | - |
| HB | ALA245, GLY247, ASN427, LEU428, CYS429, SER606, THR607 | ALA245, ASN427, CYS429, SER606, THR607 | SER202, ASN427, LEU428 | SER202, SER217, ASN427, LEU428, THR607 | SER202, SER217, GLY247, ASN427, LEU428, CYS429, THR607 | HIS200, SER202, ASN427, CYS429 | |
| BD_7 | HY | ALA201, LEU446, LEU446, THR604, THR607 | ALA201, LEU446 | ALA201, LEU446 | ALA201, LEU446 | ALA201 | ALA201, LEU446 |
| HB | SER202, ASN427, GLU431, SER448, PRO603, SER606, THR607 | SER202, ASN427, GLU431, PRO603, SER606 | SER202, ASN427, GLU431, PRO603, THR607 | SER202, ASN427, GLU431, PRO603, THR607 | SER202, ASN427, GLU431, PRO603, THR607 | SER202, ASN427, GLU431, PRO603, THR607 | |
| BD_8 | HY | LEU446, ALA605 | LEU446 | LEU446, MET602 | LEU446, ALA605 | LEU446, MET602 | LEU446 |
| HB | SER202, ASN427, GLU431, SER606, THR607 | SER202, ASN427, GLU431, SER606, THR607 | SER202, ASN427, ALA605 | SER202, ASN427, ALA605 | SER202, CYS429, GLU431, ALA447, SER448, THR604, ALA605 | SER202, CYS429, GLU431, THR604, ALA605 | |
| HAL | GLY247 | GLY247 | GLY247 | GLY247 | GLY247 | ||
| BD_10 | HY | ALA201, MET602, PRO603, THR604 | ALA201 | - | LEU428, THR607 | ALA201, LEU428 | ALA296, LEU428, ALA605 |
| HB | TYR155, SER202, GLY247, SER448, THR607 | TYR155, SER202, ALA245, ARG293, SER448, SER606, THR607 | TYR155, SER202, ARG293, SER448, SER606, THR607 | TYR155, SER202, ARG293, ARG293, SER448, THR607 | SER202, SER217, SER448, THR607 | SER448 | |
| pi-Cation | - | - | - | ARG293 | - | - | |
| BD_14 | HY | PRO203, LEU446 | ALA428 | - | PRO203 | PRO203 | - |
| HB | ALA245, ASN427, LEU428, CYS429, SER606, THR607 | SER202, ASN427, SER606, THR607 | SER202, SER217, GLY247, SER606, THR607 | SER202, GLY247, THR607 | SER202, GLY247, ASN427, SER606 | SER202, GLY247, ASN427, SER606, THR607 | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Islam, M.A.; Barshetty, M.M.; Srinivasan, S.; Dudekula, D.B.; Rallabandi, V.P.S.; Mohammed, S.; Natarajan, S.; Park, J. Identification of Novel Ribonucleotide Reductase Inhibitors for Therapeutic Application in Bile Tract Cancer: An Advanced Pharmacoinformatics Study. Biomolecules 2022, 12, 1279. https://doi.org/10.3390/biom12091279
Islam MA, Barshetty MM, Srinivasan S, Dudekula DB, Rallabandi VPS, Mohammed S, Natarajan S, Park J. Identification of Novel Ribonucleotide Reductase Inhibitors for Therapeutic Application in Bile Tract Cancer: An Advanced Pharmacoinformatics Study. Biomolecules. 2022; 12(9):1279. https://doi.org/10.3390/biom12091279
Chicago/Turabian StyleIslam, Md Ataul, Mayuri Makarand Barshetty, Sridhar Srinivasan, Dawood Babu Dudekula, V. P. Subramanyam Rallabandi, Sameer Mohammed, Sathishkumar Natarajan, and Junhyung Park. 2022. "Identification of Novel Ribonucleotide Reductase Inhibitors for Therapeutic Application in Bile Tract Cancer: An Advanced Pharmacoinformatics Study" Biomolecules 12, no. 9: 1279. https://doi.org/10.3390/biom12091279