
Single-Cell Profiling Comparisons of Tumor Microenvironment between Primary Advanced Lung Adenocarcinomas and Brain Metastases and Machine Learning Algorithms in Predicting Immunotherapeutic Responses

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Methods
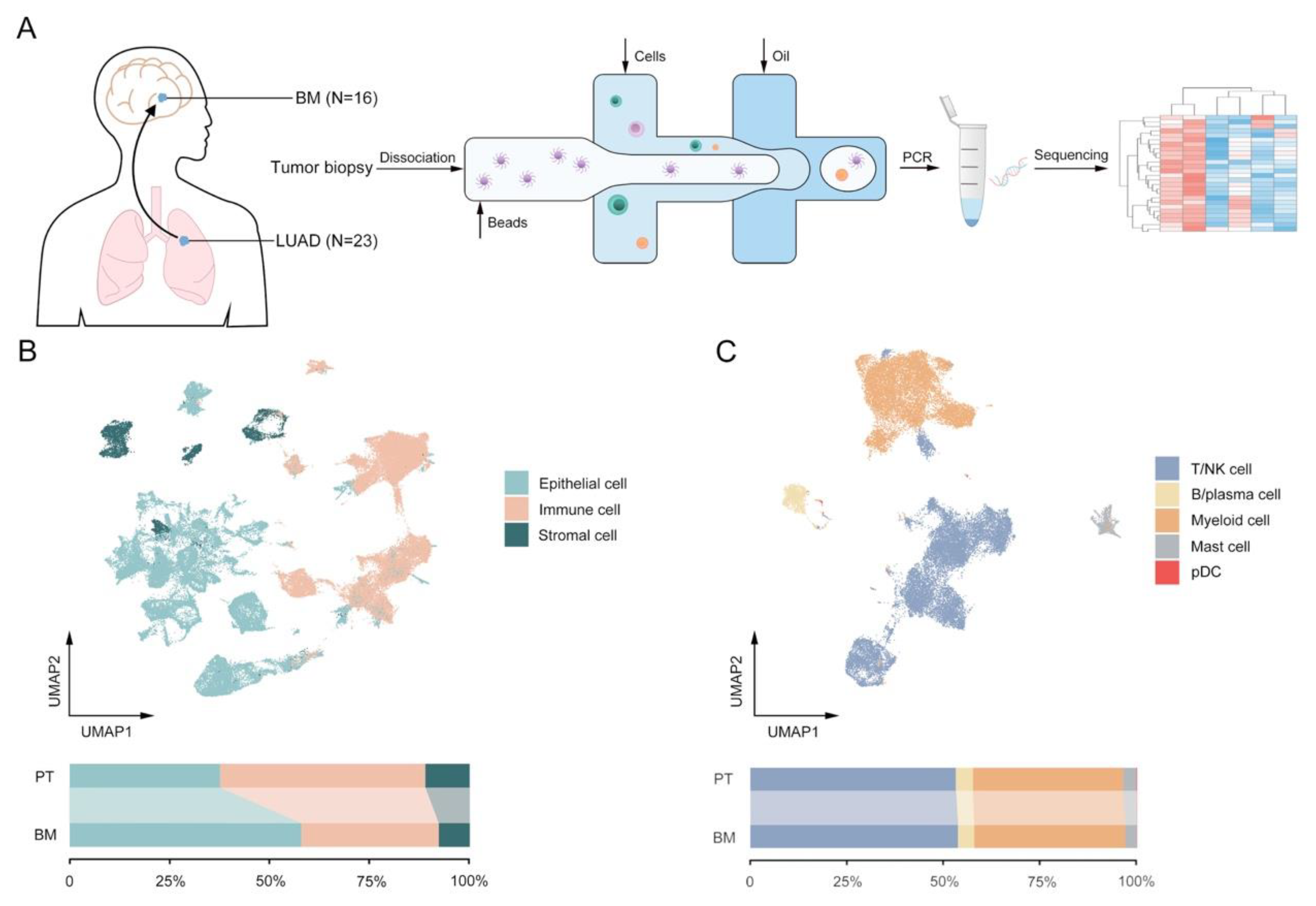
2.1. Collection of Human Specimens for LUAD and BM
2.2. Data Integration, Processing and Quality Control
2.3. Trajectory Analysis
2.4. Functional Enrichment Analysis
2.5. Gene Signature Scores
2.6. Copy Number Variations and Tumor Heterogeneity
2.7. Transcription Factor Analysis
2.8. External Validation in Bulk RNA Sequencing
2.9. Cell-Cell Interaction Analysis
2.10. Statistical Analysis
3. Results
3.1. The Global Single-Cell Landscape of Primary LUAD and Brain Metastases
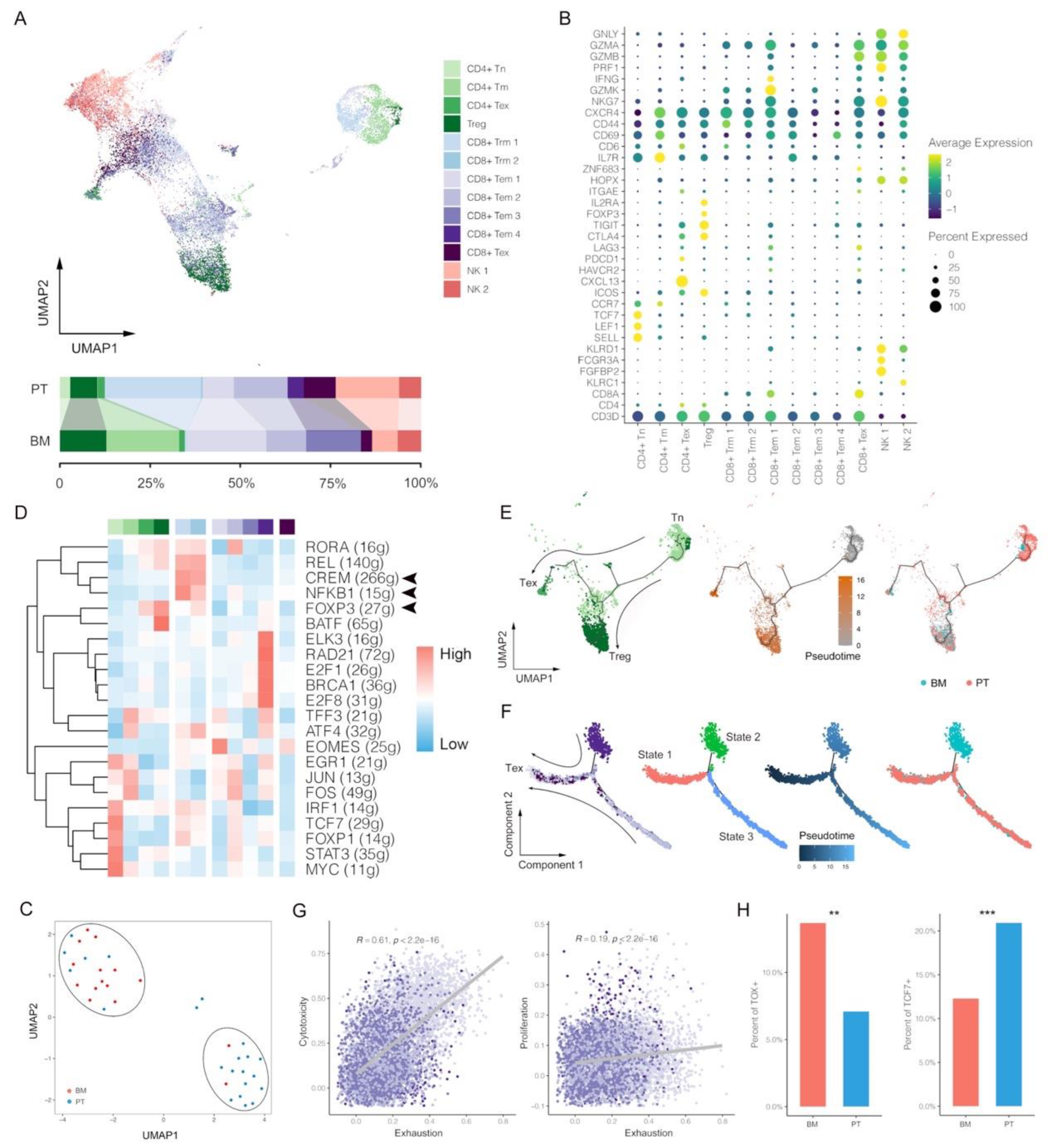
3.2. Deletion of CD8+ Trm Cells and More Dysfunctional CD8+ Tem Cells within BM Tumors
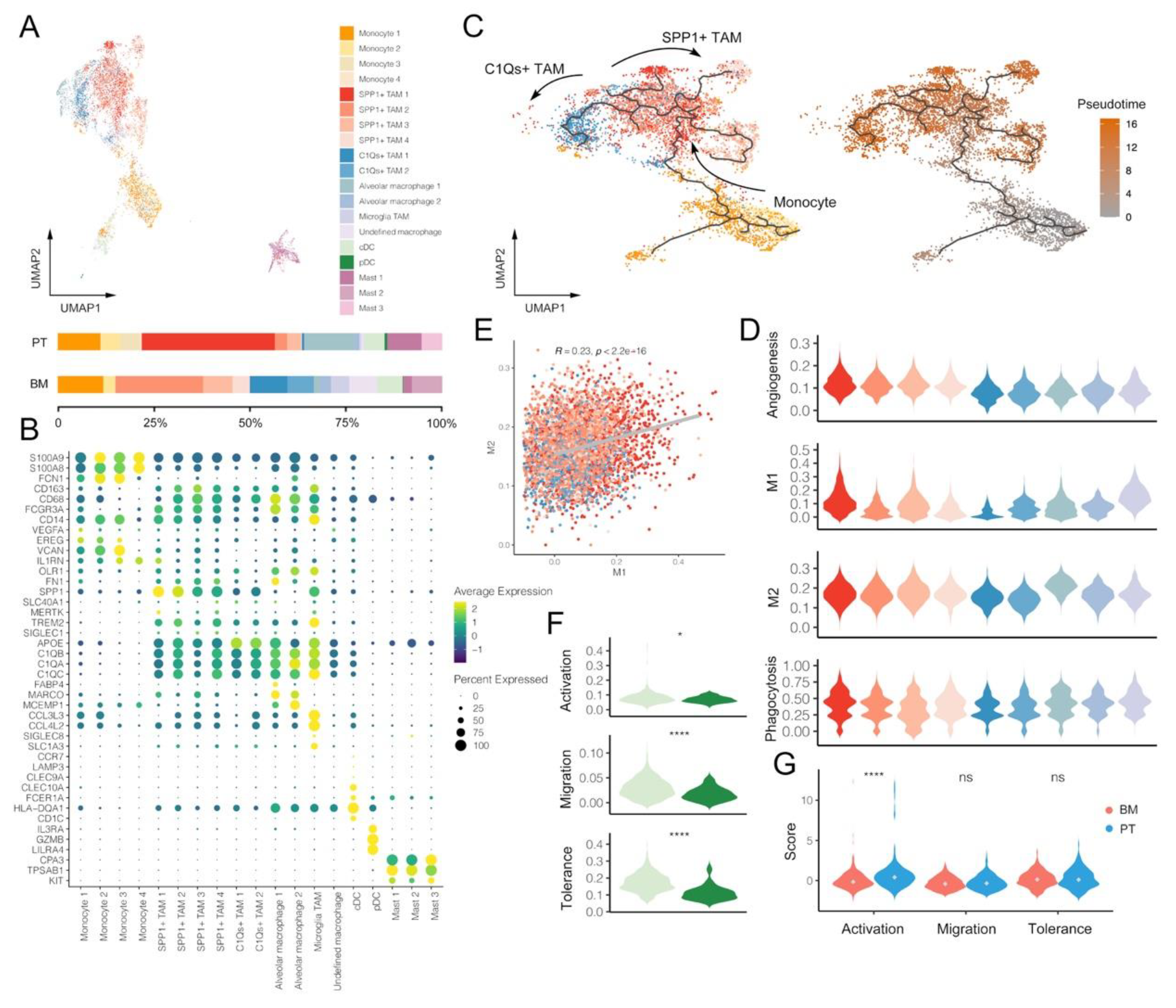
3.3. Macrophages and DCs within BM Tumors Reveal More Pro-Tumoral and Anti-Inflammatory Effects
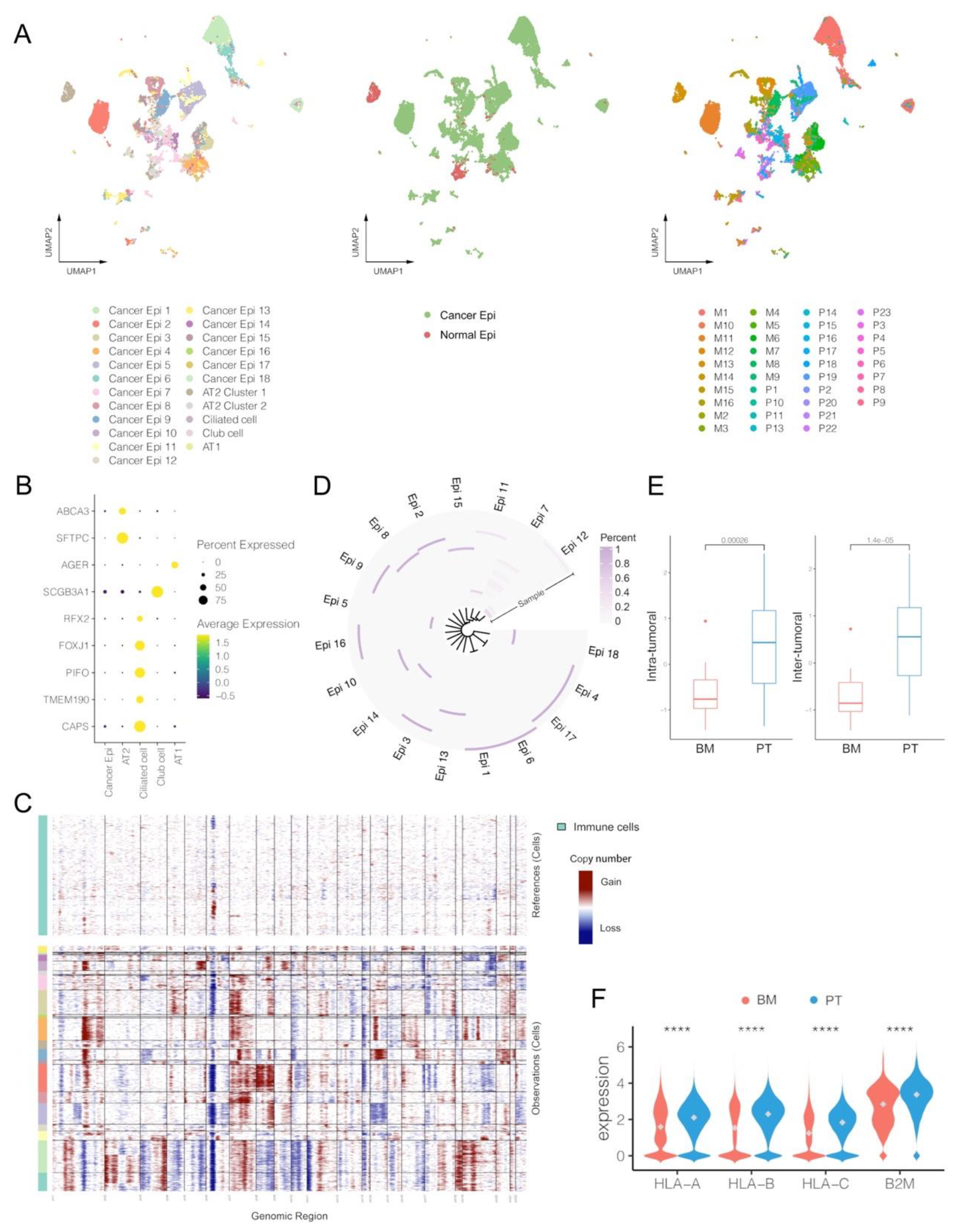
3.4. Cancer Epithelial Cells in BM Demonstrate Limited Heterogeneity and Lower Expression Levels of HLA-I-Associated Genes
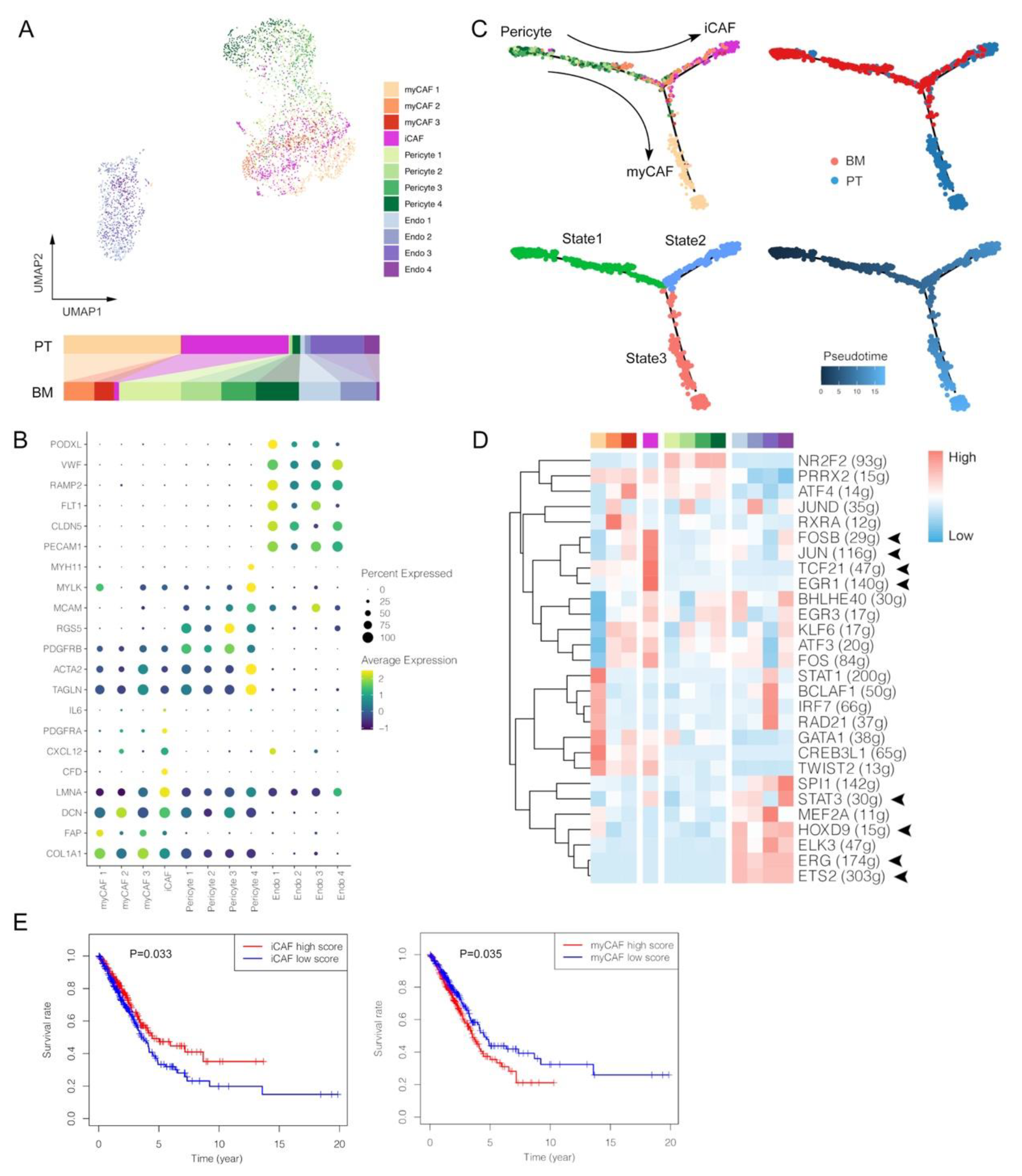
3.5. Lack of Inflammatory-like CAFs and Enrichment of Pericytes with Ambiguous Phenotypes within BM Tumors
3.6. Cell-Cell Interactions Indicate the Critical Role of Pericytes in Shaping TME of Brain Metastasis
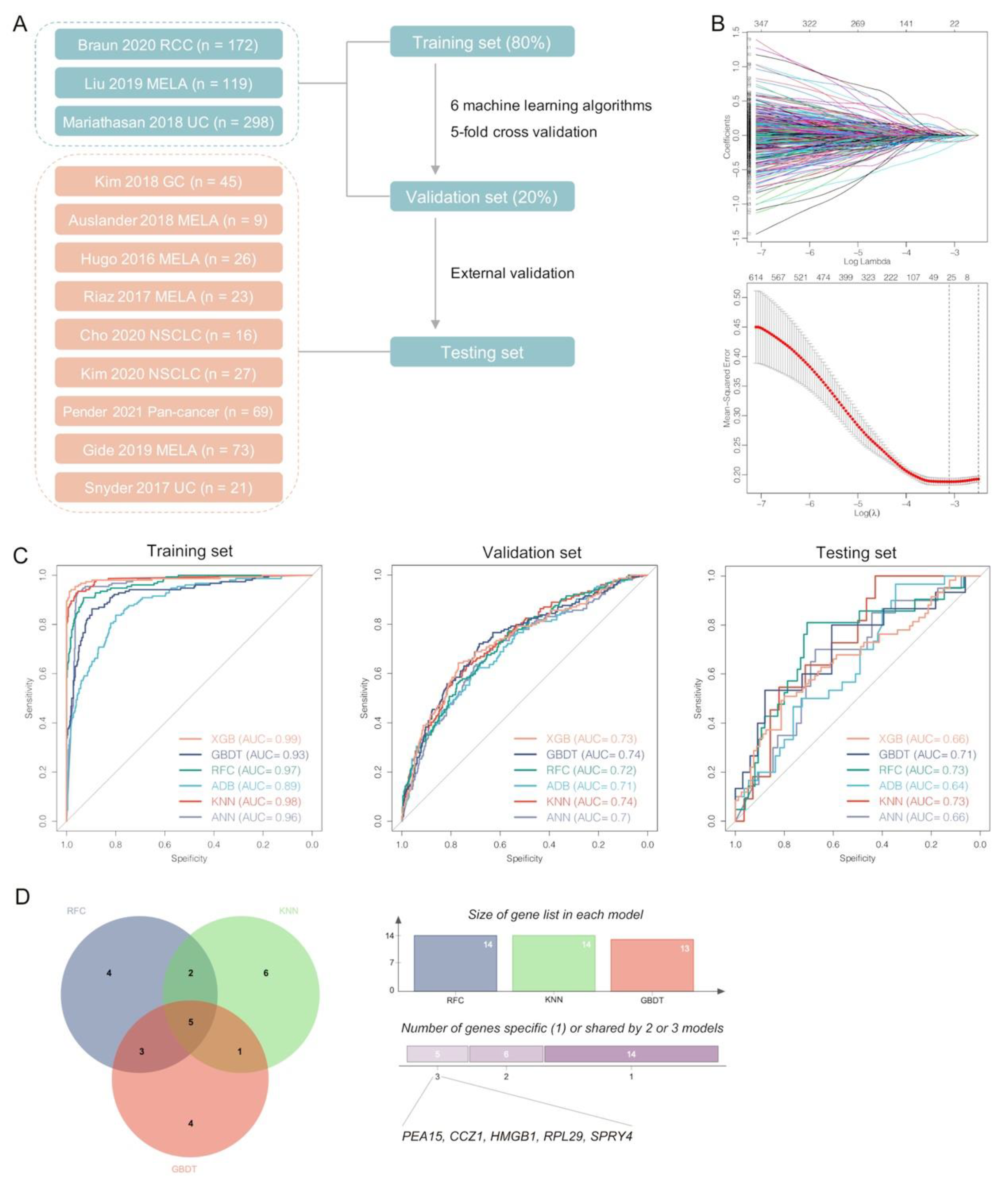
3.7. Pericyte-Related Machine Learning Models Predicting Responses to ICI Therapy
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Sung, H.; Ferlay, J.; Siegel, R.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef] [PubMed]
- Sher, T.; Dy, G.; Adjei, A. Small cell lung cancer. Mayo Clin. Proc. 2008, 83, 355–367. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alberg, A.; Brock, M.; Samet, J. Epidemiology of lung cancer: Looking to the future. Am. J. Clin. Oncol. 2005, 23, 3175–3185. [Google Scholar] [CrossRef] [PubMed]
- Barnholtz-Sloan, J.; Sloan, A.; Davis, F.; Vigneau, F.; Lai, P.; Sawaya, R. Incidence proportions of brain metastases in patients diagnosed (1973 to 2001) in the Metropolitan Detroit Cancer Surveillance System. Am. J. Clin. Oncol. 2004, 22, 2865–2872. [Google Scholar] [CrossRef] [PubMed]
- Schouten, L.; Rutten, J.; Huveneers, H.; Twijnstra, A. Incidence of brain metastases in a cohort of patients with carcinoma of the breast, colon, kidney, and lung and melanoma. Cancer 2002, 94, 2698–2705. [Google Scholar] [CrossRef]
- Yamaguchi, H.; Wakuda, K.; Fukuda, M.; Kenmotsu, H.; Mukae, H.; Ito, K.; Chibana, K.; Inoue, K.; Miura, S.; Tanaka, K.; et al. A Phase II Study of Osimertinib for Radiotherapy-Naive Central Nervous System Metastasis From NSCLC: Results for the T790M Cohort of the OCEAN Study (LOGIK1603/WJOG9116L). J. Thorac. Oncol. 2021, 16, 2121–2132. [Google Scholar] [CrossRef]
- Bischoff, P.; Trinks, A.; Obermayer, B.; Pett, J.; Wiederspahn, J.; Uhlitz, F.; Liang, X.; Lehmann, A.; Jurmeister, P.; Elsner, A.; et al. Single-cell RNA sequencing reveals distinct tumor microenvironmental patterns in lung adenocarcinoma. Oncogene 2021, 40, 6748–6758. [Google Scholar] [CrossRef]
- Kim, N.; Kim, H.; Lee, K.; Hong, Y.; Cho, J.; Choi, J.; Lee, J.; Suh, Y.; Ku, B.; Eum, H.; et al. Single-cell RNA sequencing demonstrates the molecular and cellular reprogramming of metastatic lung adenocarcinoma. Nat. Commun. 2020, 11, 2285. [Google Scholar] [CrossRef]
- Wu, F.; Fan, J.; He, Y.; Xiong, A.; Yu, J.; Li, Y.; Zhang, Y.; Zhao, W.; Zhou, F.; Li, W.; et al. Single-cell profiling of tumor heterogeneity and the microenvironment in advanced non-small cell lung cancer. Nat. Commun. 2021, 12, 2540. [Google Scholar] [CrossRef]
- Yang, L.; He, Y.; Dong, S.; Wei, X.; Chen, Z.; Zhang, B.; Chen, W.; Yang, X.; Wang, F.; Shang, X.; et al. Single-cell transcriptome analysis revealed a suppressive tumor immune microenvironment in EGFR mutant lung adenocarcinoma. J. Immunother. Cancer 2022, 10, e003534. [Google Scholar] [CrossRef]
- Hao, Y.; Hao, S.; Andersen-Nissen, E.; Mauck, W.; Zheng, S.; Butler, A.; Lee, M.; Wilk, A.; Darby, C.; Zager, M.; et al. Integrated analysis of multimodal single-cell data. Cell 2021, 184, 3573–3587. [Google Scholar] [CrossRef]
- Hafemeister, C.; Satija, R. Normalization and variance stabilization of single-cell RNA-seq data using regularized negative binomial regression. Genome Biol. 2019, 20, 296. [Google Scholar] [CrossRef] [Green Version]
- Korsunsky, I.; Millard, N.; Fan, J.; Slowikowski, K.; Zhang, F.; Wei, K.; Baglaenko, Y.; Brenner, M.; Loh, P.; Raychaudhuri, S. Fast, sensitive and accurate integration of single-cell data with Harmony. Nat. Methods 2019, 16, 1289–1296. [Google Scholar] [CrossRef]
- Cao, J.; Spielmann, M.; Qiu, X.; Huang, X.; Ibrahim, D.; Hill, A.; Zhang, F.; Mundlos, S.; Christiansen, L.; Steemers, F.; et al. The single-cell transcriptional landscape of mammalian organogenesis. Nature 2019, 566, 496–502. [Google Scholar] [CrossRef]
- Trapnell, C.; Cacchiarelli, D.; Grimsby, J.; Pokharel, P.; Li, S.; Morse, M.; Lennon, N.; Livak, K.; Mikkelsen, T.; Rinn, J. The dynamics and regulators of cell fate decisions are revealed by pseudotemporal ordering of single cells. Nat. Biotechnol. 2014, 32, 381–386. [Google Scholar] [CrossRef] [Green Version]
- Haghverdi, L.; Lun, A.; Morgan, M.; Marioni, J. Batch effects in single-cell RNA-sequencing data are corrected by matching mutual nearest neighbors. Nat. Biotechnol. 2018, 36, 421–427. [Google Scholar] [CrossRef]
- Finak, G.; McDavid, A.; Yajima, M.; Deng, J.; Gersuk, V.; Shalek, A.K.; Slichter, C.K.; Miller, H.W.; McElrath, M.J.; Prlic, M.; et al. MAST: A flexible statistical framework for assessing transcriptional changes and characterizing heterogeneity in single-cell RNA sequencing data. Genome Biol. 2015, 16, 278. [Google Scholar] [CrossRef] [Green Version]
- Wu, T.; Hu, E.; Xu, S.; Chen, M.; Guo, P.; Dai, Z.; Feng, T.; Zhou, L.; Tang, W.; Zhan, L.; et al. clusterProfiler 4.0: A universal enrichment tool for interpreting omics data. Innovation 2021, 2, 100141. [Google Scholar] [CrossRef]
- Charoentong, P.; Finotello, F.; Angelova, M.; Mayer, C.; Efremova, M.; Rieder, D.; Hackl, H.; Trajanoski, Z. Pan-cancer Immunogenomic Analyses Reveal Genotype-Immunophenotype Relationships and Predictors of Response to Checkpoint Blockade. Cell Rep. 2017, 18, 248–262. [Google Scholar] [CrossRef] [Green Version]
- Guo, X.; Zhang, Y.; Zheng, L.; Zheng, C.; Song, J.; Zhang, Q.; Kang, B.; Liu, Z.; Jin, L.; Xing, R.; et al. Global characterization of T cells in non-small-cell lung cancer by single-cell sequencing. Nat. Med. 2018, 24, 978–985. [Google Scholar] [CrossRef]
- Zheng, L.; Qin, S.; Si, W.; Wang, A.; Xing, B.; Gao, R.; Ren, X.; Wang, L.; Wu, X.; Zhang, J.; et al. Pan-cancer single-cell landscape of tumor-infiltrating T. cells. Science 2021, 374, abe6474. [Google Scholar] [CrossRef] [PubMed]
- Cheng, S.; Li, Z.; Gao, R.; Xing, B.; Gao, Y.; Yang, Y.; Qin, S.; Zhang, L.; Ouyang, H.; Du, P.; et al. A pan-cancer single-cell transcriptional atlas of tumor infiltrating myeloid cells. Cell 2021, 184, 792–809. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Y.; Chen, Z.; Han, Y.; Han, L.; Zou, X.; Zhou, B.; Hu, R.; Hao, J.; Bai, S.; Xiao, H.; et al. Immune suppressive landscape in the human esophageal squamous cell carcinoma microenvironment. Nat. Commun. 2020, 11, 6268. [Google Scholar] [CrossRef] [PubMed]
- Aibar, S.; Gonzalez-Blas, C.B.; Moerman, T.; Huynh-Thu, V.A.; Imrichova, H.; Hulselmans, G.; Rambow, F.; Marine, J.C.; Geurts, P.; Aerts, J.; et al. SCENIC: Single-cell regulatory network inference and clustering. Nat. Methods 2017, 14, 1083–1086. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tickle, T.; Tirosh, I.; Georgescu, C.; Brown, M.; Haas, B. inferCNV of the Trinity CTAT Project. In Klarman Cell Observatory; Broad Institute of MIT and Harvard: Cambridge, MA, USA, 2019. [Google Scholar]
- Hänzelmann, S.; Castelo, R.; Guinney, J. GSVA: Gene set variation analysis for microarray and RNA-seq data. BMC Bioinform. 2013, 14, 7. [Google Scholar] [CrossRef] [Green Version]
- Tirosh, I.; Izar, B.; Prakadan, S.; Wadsworth, M.; Treacy, D.; Trombetta, J.; Rotem, A.; Rodman, C.; Lian, C.; Murphy, G.; et al. Dissecting the multicellular ecosystem of metastatic melanoma by single-cell RNA-seq. Science 2016, 352, 189–196. [Google Scholar] [CrossRef] [Green Version]
- Byrne, A.; Savas, P.; Sant, S.; Li, R.; Virassamy, B.; Luen, S.; Beavis, P.; Mackay, L.; Neeson, P.; Loi, S. Tissue-resident memory T cells in breast cancer control and immunotherapy responses. Nat. Rev. Clin. Oncol. 2020, 17, 341–348. [Google Scholar] [CrossRef]
- Edwards, J.; Wilmott, J.; Madore, J.; Gide, T.; Quek, C.; Tasker, A.; Ferguson, A.; Chen, J.; Hewavisenti, R.; Hersey, P.; et al. CD103 Tumor-Resident CD8 T Cells Are Associated with Improved Survival in Immunotherapy-Naïve Melanoma Patients and Expand Significantly During Anti-PD-1 Treatment. Am. J. Cancer Res. 2018, 24, 3036–3045. [Google Scholar] [CrossRef] [Green Version]
- Yu, K.; Kuang, L.; Fu, T.; Zhang, C.; Zhou, Y.; Zhu, C.; Zhang, Q.; Zhang, Z.; Le, A. CREM Is Correlated With Immune-Suppressive Microenvironment and Predicts Poor Prognosis in Gastric Adenocarcinoma. Front. Cell Dev. Biol. 2021, 9, 697748. [Google Scholar] [CrossRef]
- Ren, X.; Zhang, L.; Zhang, Y.; Li, Z.; Siemers, N.; Zhang, Z. Insights Gained from Single-Cell Analysis of Immune Cells in the Tumor Microenvironment. Annu. Rev. Immunol. 2021, 39, 583–609. [Google Scholar] [CrossRef]
- Khan, O.; Giles, J.; McDonald, S.; Manne, S.; Ngiow, S.; Patel, K.; Werner, M.; Huang, A.; Alexander, K.; Wu, J.; et al. TOX transcriptionally and epigenetically programs CD8 T cell exhaustion. Nature 2019, 571, 211–218. [Google Scholar] [CrossRef]
- Wolf, N.; Kissiov, D.; Raulet, D. Roles of natural killer cells in immunity to cancer, and applications to immunotherapy. Nat. Rev. Immunol. 2022. [Google Scholar] [CrossRef]
- Benoit, M.; Clarke, E.; Morgado, P.; Fraser, D.; Tenner, A. Complement protein C1q directs macrophage polarization and limits inflammasome activity during the uptake of apoptotic cells. J. Immunol. 2012, 188, 5682–5693. [Google Scholar] [CrossRef] [Green Version]
- Hu, Z.; Li, Z.; Ma, Z.; Curtis, C. Multi-cancer analysis of clonality and the timing of systemic spread in paired primary tumors and metastases. Nat. Genet. 2020, 52, 701–708. [Google Scholar] [CrossRef]
- Belle, J.; DeNardo, D. A Single-Cell Window into Pancreas Cancer Fibroblast Heterogeneity. Cancer Discov. 2019, 9, 1001–1002. [Google Scholar] [CrossRef] [Green Version]
- Bhowmick, N.; Neilson, E.; Moses, H. Stromal fibroblasts in cancer initiation and progression. Nature 2004, 432, 332–337. [Google Scholar] [CrossRef] [Green Version]
- Kieffer, Y.; Hocine, H.; Gentric, G.; Pelon, F.; Bernard, C.; Bourachot, B.; Lameiras, S.; Albergante, L.; Bonneau, C.; Guyard, A.; et al. Single-Cell Analysis Reveals Fibroblast Clusters Linked to Immunotherapy Resistance in Cancer. Cancer Discov. 2020, 10, 1330–1351. [Google Scholar] [CrossRef]
- Dong, X.; Yang, Y.; Xu, G.; Tian, Z.; Yang, Q.; Gong, Y.; Wu, G. The initial expression alterations occurring to transcription factors during the formation of breast cancer: Evidence from bioinformatics. Cancer Med. 2022, 11, 1371–1395. [Google Scholar] [CrossRef]
- Hussain, A.; Voisin, V.; Poon, S.; Karamboulas, C.; Bui, N.; Means, J.; Dmytryshyn, J.; Ho, V.; Tang, K.; Paterson, J.; et al. Distinct fibroblast functional states drive clinical outcomes in ovarian cancer and are regulated by TCF21. J. Exp. Med. 2020, 217, e20191094. [Google Scholar] [CrossRef]
- Wu, S.Z.; Roden, D.L.; Wang, C.; Holliday, H.; Harvey, K.; Cazet, A.S.; Murphy, K.J.; Pereira, B.; Al-Eryani, G.; Bartonicek, N.; et al. Stromal cell diversity associated with immune evasion in human triple-negative breast cancer. EMBO J. 2020, 39, e104063. [Google Scholar] [CrossRef]
- Molnár, K.; Mészáros, Á.; Fazakas, C.; Kozma, M.; Győri, F.; Reisz, Z.; Tiszlavicz, L.; Farkas, A.; Nyúl-Tóth, Á.; Haskó, J.; et al. Pericyte-secreted IGF2 promotes breast cancer brain metastasis formation. Mol. Oncol. 2020, 14, 2040–2057. [Google Scholar] [CrossRef] [PubMed]
- Ujifuku, K.; Fujimoto, T.; Sato, K.; Morofuji, Y.; Muto, H.; Masumoto, H.; Nakagawa, S.; Niwa, M.; Matsuo, T. Exploration of Pericyte-Derived Factors Implicated in Lung Cancer Brain Metastasis Protection: A Pilot Messenger RNA Sequencing Using the Blood-Brain Barrier In Vitro Model. Cell. Mol. Neurobiol. 2022, 42, 997–1004. [Google Scholar] [CrossRef] [PubMed]
- Le Tran, N.; Wang, Y.; Nie, G. Podocalyxin in Normal Tissue and Epithelial Cancer. Cancers 2021, 13, 2863. [Google Scholar] [CrossRef] [PubMed]
- Moh-Moh-Aung, A.; Fujisawa, M.; Ito, S.; Katayama, H.; Ohara, T.; Ota, Y.; Yoshimura, T.; Matsukawa, A. Decreased miR-200b-3p in cancer cells leads to angiogenesis in HCC by enhancing endothelial ERG expression. Sci. Rep. 2020, 10, 10418. [Google Scholar] [CrossRef] [PubMed]
- Parakh, S.; Ernst, M.; Poh, A. Multicellular Effects of STAT3 in Non-small Cell Lung Cancer: Mechanistic Insights and Therapeutic Opportunities. Cancers 2021, 13, 6228. [Google Scholar] [CrossRef]
- Toshner, M.; Dunmore, B.; McKinney, E.; Southwood, M.; Caruso, P.; Upton, P.; Waters, J.; Ormiston, M.; Skepper, J.; Nash, G.; et al. Transcript analysis reveals a specific HOX signature associated with positional identity of human endothelial cells. PLoS ONE 2014, 9, e91334. [Google Scholar] [CrossRef] [Green Version]
- Wallace, J.; Li, F.; Balakrishnan, S.; Cantemir-Stone, C.; Pecot, T.; Martin, C.; Kladney, R.; Sharma, S.; Trimboli, A.; Fernandez, S.; et al. Ets2 in tumor fibroblasts promotes angiogenesis in breast cancer. PLoS ONE 2013, 8, e71533. [Google Scholar] [CrossRef]
- Fujimoto, T.; Nakagawa, S.; Morofuji, Y.; Watanabe, D.; Ujifuku, K.; Horie, N.; Izumo, T.; Niwa, M.; Banks, W.A.; Deli, M.A.; et al. Pericytes Suppress Brain Metastasis from Lung Cancer In Vitro. Cell. Mol. Neurobiol. 2020, 40, 113–121. [Google Scholar] [CrossRef]
- Kürten, C.H.L.; Kulkarni, A.; Cillo, A.R.; Santos, P.M.; Roble, A.K.; Onkar, S.; Reeder, C.; Lang, S.; Chen, X.; Duvvuri, U.; et al. Investigating immune and non-immune cell interactions in head and neck tumors by single-cell RNA sequencing. Nat. Commun. 2021, 12, 7338. [Google Scholar] [CrossRef]
- Zhang, X.N.; Yang, K.D.; Chen, C.; He, Z.C.; Wang, Q.H.; Feng, H.; Lv, S.Q.; Wang, Y.; Mao, M.; Liu, Q.; et al. Pericytes augment glioblastoma cell resistance to temozolomide through CCL5-CCR5 paracrine signaling. Cell Res. 2021, 31, 1072–1087. [Google Scholar] [CrossRef]
- Biktasova, A.K.; Dudimah, D.F.; Uzhachenko, R.V.; Park, K.; Akhter, A.; Arasada, R.R.; Evans, J.V.; Novitskiy, S.V.; Tchekneva, E.E.; Carbone, D.P.; et al. Multivalent Forms of the Notch Ligand DLL-1 Enhance Antitumor T-cell Immunity in Lung Cancer and Improve Efficacy of EGFR-Targeted Therapy. Cancer Res. 2015, 75, 4728–4741. [Google Scholar] [CrossRef] [Green Version]
- Huang, Y.; Lin, L.; Shanker, A.; Malhotra, A.; Yang, L.; Dikov, M.M.; Carbone, D.P. Resuscitating cancer immunosurveillance: Selective stimulation of DLL1-Notch signaling in T cells rescues T-cell function and inhibits tumor growth. Cancer Res. 2011, 71, 6122–6131. [Google Scholar] [CrossRef] [Green Version]
- Tchekneva, E.E.; Goruganthu, M.U.L.; Uzhachenko, R.V.; Thomas, P.L.; Antonucci, A.; Chekneva, I.; Koenig, M.; Piao, L.; Akhter, A.; de Aquino, M.T.P.; et al. Determinant roles of dendritic cell-expressed Notch Delta-like and Jagged ligands on anti-tumor T cell immunity. J. Immunother. Cancer 2019, 7, 95. [Google Scholar] [CrossRef] [Green Version]
- Huang, C.C.; Cheng, S.H.; Wu, C.H.; Li, W.Y.; Wang, J.S.; Kung, M.L.; Chu, T.H.; Huang, S.T.; Feng, C.T.; Huang, S.C.; et al. Delta-like 1 homologue promotes tumorigenesis and epithelial-mesenchymal transition of ovarian high-grade serous carcinoma through activation of Notch signaling. Oncogene 2019, 38, 3201–3215. [Google Scholar] [CrossRef]
- Jin, Z.H.; Yang, R.J.; Dong, B.; Xing, B.C. Progenitor gene DLK1 might be an independent prognostic factor of liver cancer. Expert. Opin. Biol. Ther. 2008, 8, 371–377. [Google Scholar] [CrossRef]
- Takagi, H.; Zhao, S.; Muto, S.; Yokouchi, H.; Nishihara, H.; Harada, T.; Yamaguchi, H.; Mine, H.; Watanabe, M.; Ozaki, Y.; et al. Delta-like 1 homolog (DLK1) as a possible therapeutic target and its application to radioimmunotherapy using (125)I-labelled anti-DLK1 antibody in lung cancer models (HOT1801 and FIGHT004). Lung Cancer 2021, 153, 134–142. [Google Scholar] [CrossRef]
- Pittaway, J.F.H.; Lipsos, C.; Mariniello, K.; Guasti, L. The role of delta-like non-canonical Notch ligand 1 (DLK1) in cancer. Endocr. Relat. Cancer 2021, 28, R271–R287. [Google Scholar] [CrossRef]
- Traustadóttir, G.; Lagoni, L.V.; Ankerstjerne, L.B.S.; Bisgaard, H.C.; Jensen, C.H.; Andersen, D.C. The imprinted gene Delta like non-canonical Notch ligand 1 (Dlk1) is conserved in mammals, and serves a growth modulatory role during tissue development and regeneration through Notch dependent and independent mechanisms. Cytokine Growth Factor Rev. 2019, 46, 17–27. [Google Scholar] [CrossRef]
- Liebner, S.; Dijkhuizen, R.M.; Reiss, Y.; Plate, K.H.; Agalliu, D.; Constantin, G. Functional morphology of the blood-brain barrier in health and disease. Acta. Neuropathol. 2018, 135, 311–336. [Google Scholar] [CrossRef] [Green Version]
- Caspani, E.M.; Crossley, P.H.; Redondo-Garcia, C.; Martinez, S. Glioblastoma: A pathogenic crosstalk between tumor cells and pericytes. PLoS ONE 2014, 9, e101402. [Google Scholar] [CrossRef]
- Ochs, K.; Sahm, F.; Opitz, C.A.; Lanz, T.V.; Oezen, I.; Couraud, P.O.; von Deimling, A.; Wick, W.; Platten, M. Immature mesenchymal stem cell-like pericytes as mediators of immunosuppression in human malignant glioma. J. Neuroimmunol. 2013, 265, 106–116. [Google Scholar] [CrossRef] [PubMed]
- Wirsik, N.M.; Ehlers, J.; Mäder, L.; Ilina, E.I.; Blank, A.E.; Grote, A.; Feuerhake, F.; Baumgarten, P.; Devraj, K.; Harter, P.N.; et al. TGF-β activates pericytes via induction of the epithelial-to-mesenchymal transition protein SLUG in glioblastoma. Neuropathol. Appl. Neurobiol. 2021, 47, 768–780. [Google Scholar] [CrossRef] [PubMed]
- Zonneville, J.; Safina, A.; Truskinovsky, A.M.; Arteaga, C.L.; Bakin, A.V. TGF-β signaling promotes tumor vasculature by enhancing the pericyte-endothelium association. BMC Cancer 2018, 18, 670. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- van Tellingen, O.; Yetkin-Arik, B.; de Gooijer, M.C.; Wesseling, P.; Wurdinger, T.; de Vries, H.E. Overcoming the blood-brain tumor barrier for effective glioblastoma treatment. Drug Resist. Updat. 2015, 19, 1–12. [Google Scholar] [CrossRef]
- Hubert, P.; Roncarati, P.; Demoulin, S.; Pilard, C.; Ancion, M.; Reynders, C.; Lerho, T.; Bruyere, D.; Lebeau, A.; Radermecker, C.; et al. Extracellular HMGB1 blockade inhibits tumor growth through profoundly remodeling immune microenvironment and enhances checkpoint inhibitor-based immunotherapy. J. Immunother. Cancer 2021, 9, e001966. [Google Scholar] [CrossRef]







Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wu, Y.; Kang, K.; Han, C.; Wang, L.; Wang, Z.; Zhao, A. Single-Cell Profiling Comparisons of Tumor Microenvironment between Primary Advanced Lung Adenocarcinomas and Brain Metastases and Machine Learning Algorithms in Predicting Immunotherapeutic Responses. Biomolecules 2023, 13, 185. https://doi.org/10.3390/biom13010185
Wu Y, Kang K, Han C, Wang L, Wang Z, Zhao A. Single-Cell Profiling Comparisons of Tumor Microenvironment between Primary Advanced Lung Adenocarcinomas and Brain Metastases and Machine Learning Algorithms in Predicting Immunotherapeutic Responses. Biomolecules. 2023; 13(1):185. https://doi.org/10.3390/biom13010185
Chicago/Turabian StyleWu, Yijun, Kai Kang, Chang Han, Li Wang, Zhile Wang, and Ailin Zhao. 2023. "Single-Cell Profiling Comparisons of Tumor Microenvironment between Primary Advanced Lung Adenocarcinomas and Brain Metastases and Machine Learning Algorithms in Predicting Immunotherapeutic Responses" Biomolecules 13, no. 1: 185. https://doi.org/10.3390/biom13010185