
Type 2 Diabetes and Alzheimer’s Disease: The Emerging Role of Cellular Lipotoxicity

 , , and
, , and 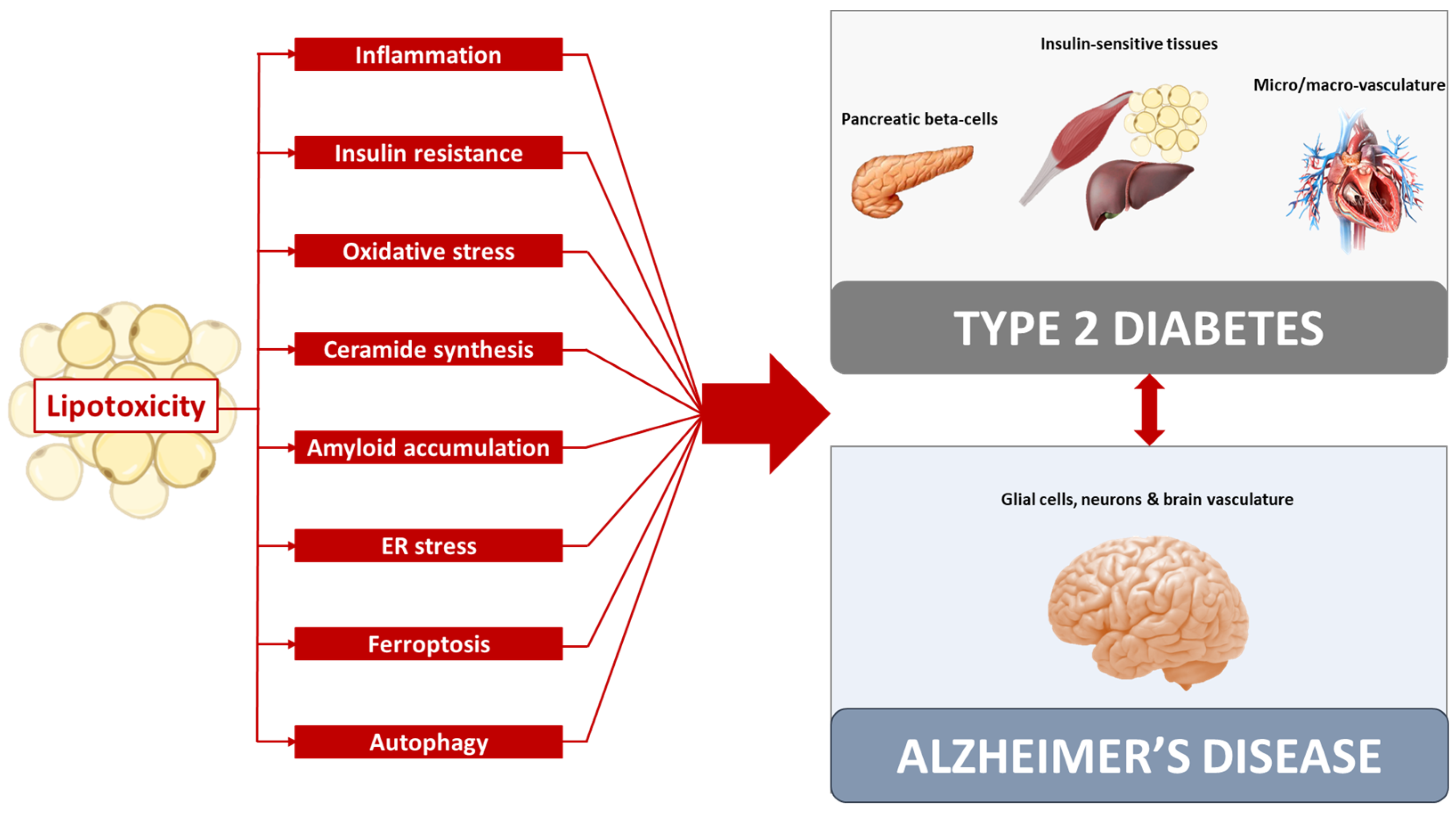{kind=link}
Abstract
:1. Introduction
2. Lipotoxicity Common Mechanism in Type 2 Diabetes and Alzheimer’s Disease
2.1. Inflammation
2.2. Insulin Resistance
2.3. Oxidative Stress
2.4. Ceramides
2.5. Amyloid Accumulation
2.6. ER Stress
2.7. Ferroptosis
2.8. Autophagy
3. Possible Common Therapies
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- International Diabetes Federation (IDF). IDF Diabetes Atlas, 10th ed. 2021. Available online: https://www.diabetesatlas.org/en/ (accessed on 10 December 2022).
- WHO World Health Organization (WHO). Obesity and Overweight. Available online: https://www.who.int/news-room/fact-sheets/detail/obesity-and-overweight#:~:text=Of%20these%20over%20650%20million,overweight%20or%20obese%20in%202020 (accessed on 10 December 2022).
- Biondi, G.; Marrano, N.; Borrelli, A.; Rella, M.; Palma, G.; Calderoni, I.; Siciliano, E.; Lops, P.; Giorgino, F.; Natalicchio, A. Adipose Tissue Secretion Pattern Influences β-Cell Wellness in the Transition from Obesity to Type 2 Diabetes. Int. J. Mol. Sci. 2022, 23, 5522. [Google Scholar] [CrossRef] [PubMed]
- Lytrivi, M.; Castell, A.L.; Poitout, V.; Cnop, M. Recent Insights Into Mechanisms of β-Cell Lipo- and Glucolipotoxicity in Type 2 Diabetes. J. Mol. Biol. 2020, 432, 1514–1534. [Google Scholar] [CrossRef] [PubMed]
- Yazıcı, D.; Sezer, H. Insulin Resistance, Obesity and Lipotoxicity. Adv. Exp. Med. Biol. 2017, 960, 277–304. [Google Scholar] [CrossRef] [PubMed]
- Sletten, A.C.; Peterson, L.R.; Schaffer, J.E. Manifestations and Mechanisms of Myocardial Lipotoxicity in Obesity. J. Intern. Med. 2018, 284, 478–491. [Google Scholar] [CrossRef] [Green Version]
- Nishi, H.; Higashihara, T.; Inagi, R. Lipotoxicity in Kidney, Heart, and Skeletal Muscle Dysfunction. Nutrients 2019, 11, 1664. [Google Scholar] [CrossRef] [Green Version]
- Opazo-Ríos, L.; Mas, S.; Marín-Royo, G.; Mezzano, S.; Gómez-Guerrero, C.; Moreno, J.A.; Egido, J. Lipotoxicity and Diabetic Nephropathy: Novel Mechanistic Insights and Therapeutic Opportunities. Int. J. Mol. Sci. 2020, 21, 2632. [Google Scholar] [CrossRef] [Green Version]
- Patel, V.N.; Chorawala, M.R.; Shah, M.B.; Shah, K.C.; Dave, B.P.; Shah, M.P.; Patel, T.M. Emerging Pathophysiological Mechanisms Linking Diabetes Mellitus and Alzheimer’s Disease: An Old Wine in a New Bottle. J. Alzheimers Dis. Rep. 2022, 6, 349–357. [Google Scholar] [CrossRef]
- El Gaamouch, F.; Jing, P.; Xia, J.; Cai, D. Alzheimer’s Disease Risk Genes and Lipid Regulators. J. Alzheimers Dis. 2016, 53, 15–29. [Google Scholar] [CrossRef] [Green Version]
- Nichols, E.; Szoeke, C.E.I.; Vollset, S.E.; Abbasi, N.; Abd-Allah, F.; Abdela, J.; Aichour, M.T.E.; Akinyemi, R.O.; Alahdab, F.; Asgedom, S.W.; et al. Global, Regional, and National Burden of Alzheimer’s Disease and Other Dementias, 1990-2016: A Systematic Analysis for the Global Burden of Disease Study 2016. Lancet Neurol. 2019, 18, 88–106. [Google Scholar] [CrossRef] [Green Version]
- Ott, A.; Stolk, R.P.; Van Harskamp, F.; Pols, H.A.P.; Hofman, A.; Breteler, M.M.B. Diabetes Mellitus and the Risk of Dementia: The Rotterdam Study. Neurology 1999, 53, 1937–1942. [Google Scholar] [CrossRef]
- Peila, R.; Rodriguez, B.L.; Launer, L.J. Type 2 Diabetes, APOE Gene, and the Risk for Dementia and Related Pathologies: The Honolulu-Asia Aging Study. Diabetes 2002, 51, 1256–1262. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, K.C.; Woung, L.C.; Tsai, M.T.; Liu, C.C.; Su, Y.H.; Li, C.Y. Risk of Alzheimer’s Disease in Relation to Diabetes: A Population-Based Cohort Study. Neuroepidemiology 2012, 38, 237–244. [Google Scholar] [CrossRef] [PubMed]
- Arvanitakis, Z.; Wilson, R.S.; Bienias, J.L.; Evans, D.A.; Bennett, D.A. Diabetes Mellitus and Risk of Alzheimer Disease and Decline in Cognitive Function. Arch. Neurol.. 2004, 61, 661–666. [Google Scholar] [CrossRef] [PubMed]
- Leibson, C.L.; Rocca, W.A.; Hanson, V.A.; Cha, R.; Kokmen, E.; O’Brien, P.C.; Palumbo, P.J. Risk of Dementia among Persons with Diabetes Mellitus: A Population-Based Cohort Study. Am. J. Epidemiol. 1997, 145, 301–308. [Google Scholar] [CrossRef] [Green Version]
- Athanasaki, A.; Melanis, K.; Tsantzali, I.; Stefanou, M.I.; Ntymenou, S.; Paraskevas, S.G.; Kalamatianos, T.; Boutati, E.; Lambadiari, V.; Voumvourakis, K.I.; et al. Type 2 Diabetes Mellitus as a Risk Factor for Alzheimer’s Disease: Review and Meta-Analysis. Biomedicines 2022, 10, 778. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Chen, C.; Hua, S.; Liao, H.; Wang, M.; Xiong, Y.; Cao, F. An Updated Meta-Analysis of Cohort Studies: Diabetes and Risk of Alzheimer’s Disease. Diabetes Res. Clin. Pract. 2017, 124, 41–47. [Google Scholar] [CrossRef]
- Michailidis, M.; Moraitou, D.; Tata, D.A.; Kalinderi, K.; Papamitsou, T.; Papaliagkas, V. Alzheimer’s Disease as Type 3 Diabetes: Common Pathophysiological Mechanisms between Alzheimer’s Disease and Type 2 Diabetes. Int. J. Mol. Sci. 2022, 23, 2687. [Google Scholar] [CrossRef]
- De La Monte, S.M.; Wands, J.R. Alzheimer’s Disease Is Type 3 Diabetes-Evidence Reviewed. J. Diabetes Sci. Technol. 2008, 2, 1101–1113. [Google Scholar] [CrossRef] [Green Version]
- Sebastião, I.; Candeias, E.; Santos, M.S.; de Oliveira, C.R.; Moreira, P.I.; Duarte, A.I. Insulin as a Bridge between Type 2 Diabetes and Alzheimer Disease—How Anti-Diabetics Could Be a Solution for Dementia. Front Endocrinol. 2014, 5, 110. [Google Scholar] [CrossRef] [Green Version]
- Picone, P.; Di Carlo, M.; Nuzzo, D. Obesity and Alzheimer’s Disease: Molecular Bases. Eur. J. Neurosci. 2020, 52, 3944–3950. [Google Scholar] [CrossRef]
- Balasubramanian, P.; Kiss, T.; Tarantini, S.; Nyúl-Toth, Á.; Ahire, C.; Yabluchanskiy, A.; Csipo, T.; Lipecz, A.; Tabak, A.; Institoris, A.; et al. Obesity-Induced Cognitive Impairment in Older Adults: A Microvascular Perspective. Am. J. Physiol. Heart Circ Physiol. 2021, 320, H740–H761. [Google Scholar] [CrossRef] [PubMed]
- Bischof, G.N.; Park, D.C. Obesity and Aging: Consequences for Cognition, Brain Structure, and Brain Function. Psychosom Med. 2015, 77, 697–709. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pugazhenthi, S.; Qin, L.; Reddy, P.H. Common Neurodegenerative Pathways in Obesity, Diabetes, and Alzheimer’s Disease. Biochim. Biophys. Acta Mol. Basis Dis. 2017, 1863, 1037–1045. [Google Scholar] [CrossRef] [PubMed]
- Terzo, S.; Amato, A.; Mulè, F. From Obesity to Alzheimer’s Disease through Insulin Resistance. J. Diabetes Complicat. 2021, 35, 108026. [Google Scholar] [CrossRef] [PubMed]
- O’Brien, P.D.; Hinder, L.M.; Callaghan, B.C.; Feldman, E.L. Neurological Consequences of Obesity. Lancet Neurol. 2017, 16, 465–477. [Google Scholar] [CrossRef]
- Vesga-jiménez, D.J.; Martin, C.; Barreto, G.E.; Aristizábal-pachón, A.F.; Pinzón, A.; González, J. Fatty Acids: An Insight into the Pathogenesis of Neurodegenerative Diseases and Therapeutic Potential. Int. J. Mol. Sci. 2022, 23, 2577. [Google Scholar] [CrossRef]
- Weisberg, S.P.; McCann, D.; Desai, M.; Rosenbaum, M.; Leibel, R.L.; Ferrante, A.W. Obesity Is Associated with Macrophage Accumulation in Adipose Tissue. J. Clin. Investig. 2003, 112, 1796–1808. [Google Scholar] [CrossRef]
- Wensveen, F.M.; Jelenčić, V.; Valentić, S.; Šestan, M.; Wensveen, T.T.; Theurich, S.; Glasner, A.; Mendrila, D.; Štimac, D.; Wunderlich, F.T.; et al. NK Cells Link Obesity-Induced Adipose Stress to Inflammation and Insulin Resistance. Nat. Immunol. 2015, 16, 376–385. [Google Scholar] [CrossRef]
- Saltiel, A.R.; Olefsky, J.M. Inflammatory mechanisms linking obesity and metabolic disease. J. Clin. Investig. 2017, 127, 1–4. [Google Scholar] [CrossRef] [Green Version]
- Boucher, J.; Masri, B.; Daviaud, D.; Gesta, S.; Guigné, C.; Mazzucotelli, A.; Castan-Laurell, I.; Tack, I.; Knibiehler, B.; Carpéné, C.; et al. Apelin, a Newly Identified Adipokine up-Regulated by Insulin and Obesity. Endocrinology 2005, 146, 1764–1771. [Google Scholar] [CrossRef]
- Jialal, I.; Adams-Huet, B.; Duong, F.; Smith, G. Relationship between Retinol-Binding Protein-4/Adiponectin and Leptin/Adiponectin Ratios with Insulin Resistance and Inflammation. Metab. Syndr. Relat. Disord. 2014, 12, 227–230. [Google Scholar] [CrossRef] [PubMed]
- Maury, E.; Brichard, S.M.; Pataky, Z.; Carpentier, A.; Golay, A.; Bobbioni-Harsch, E. Effect of Obesity on Growth-Related Oncogene Factor-Alpha, Thrombopoietin, and Tissue Inhibitor Metalloproteinase-1 Serum Levels. Obesity 2010, 18, 1503–1509. [Google Scholar] [CrossRef] [PubMed]
- Maury, E.; Ehala-Aleksejev, K.; Guiot, Y.; Detry, R.; Vandenhooft, A.; Brichard, S.M. Adipokines Oversecreted by Omental Adipose Tissue in Human Obesity. Am. J. Physiol. Endocrinol. Metab. 2007, 293, E656–E665. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, K.; Kusminski, C.M.; Scherer, P.E. Adipose Tissue Remodeling and Obesity. J. Clin. Investig. 2011, 121, 2094–2101. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shu, C.J.; Benoist, C.; Mathis, D. The Immune System’s Involvement in Obesity-Driven Type 2 Diabetes. Semin. Immunol. 2012, 24, 436–442. [Google Scholar] [CrossRef] [Green Version]
- Igoillo-Esteve, M.; Marselli, L.; Cunha, D.A.; Ladrière, L.; Ortis, F.; Grieco, F.A.; Dotta, F.; Weir, G.C.; Marchetti, P.; Eizirik, D.L.; et al. Palmitate Induces a Pro-Inflammatory Response in Human Pancreatic Islets That Mimics CCL2 Expression by Beta Cells in Type 2 Diabetes. Diabetologia 2010, 53, 1395–1405. [Google Scholar] [CrossRef] [Green Version]
- Maedler, K.; Oberholzer, J.; Bucher, P.; Spinas, G.A.; Donath, M.Y. Monounsaturated Fatty Acids Prevent the Deleterious Effects of Palmitate and High Glucose on Human Pancreatic Beta-Cell Turnover and Function. Diabetes 2003, 52, 726–733. [Google Scholar] [CrossRef] [Green Version]
- Maedler, K.; Sergeev, P.; Ehses, J.A.; Mathe, Z.; Bosco, D.; Berney, T.; Dayer, J.M.; Reinecke, M.; Halban, P.A.; Donath, M.Y. Leptin Modulates Beta Cell Expression of IL-1 Receptor Antagonist and Release of IL-1beta in Human Islets. Proc. Natl. Acad. Sci. USA 2004, 101, 8138–8143. [Google Scholar] [CrossRef] [Green Version]
- Dinarello, C.A. Immunological and Inflammatory Functions of the Interleukin-1 Family. Annu. Rev. Immunol. 2009, 27, 519–550. [Google Scholar] [CrossRef]
- Shi, H.; Kokoeva, M.v.; Inouye, K.; Tzameli, I.; Yin, H.; Flier, J.S. TLR4 Links Innate Immunity and Fatty Acid-Induced Insulin Resistance. J. Clin. Investig. 2006, 116, 3015–3025. [Google Scholar] [CrossRef]
- Hollingworth, P.; Harold, D.; Sims, R.; Gerrish, A.; Lambert, J.C.; Carrasquillo, M.M.; Abraham, R.; Hamshere, M.L.; Pahwa, J.S.; Moskvina, V.; et al. Common Variants at ABCA7, MS4A6A/MS4A4E, EPHA1, CD33 and CD2AP Are Associated with Alzheimer’s Disease. Nat. Genet. 2011, 43, 429–436. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Naj, A.C.; Jun, G.; Beecham, G.W.; Wang, L.S.; Vardarajan, B.N.; Buros, J.; Gallins, P.J.; Buxbaum, J.D.; Jarvik, G.P.; Crane, P.K.; et al. Common Variants at MS4A4/MS4A6E, CD2AP, CD33 and EPHA1 Are Associated with Late-Onset Alzheimer’s Disease. Nat. Genet. 2011, 43, 436–443. [Google Scholar] [CrossRef] [Green Version]
- Zhang, B.; Gaiteri, C.; Bodea, L.G.; Wang, Z.; McElwee, J.; Podtelezhnikov, A.A.; Zhang, C.; Xie, T.; Tran, L.; Dobrin, R.; et al. Integrated Systems Approach Identifies Genetic Nodes and Networks in Late-Onset Alzheimer’s Disease. Cell 2013, 153, 707–720. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Glass, C.K.; Saijo, K.; Winner, B.; Marchetto, M.C.; Gage, F.H. Mechanisms Underlying Inflammation in Neurodegeneration. Cell 2010, 140, 918–934. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hanzel, C.E.; Pichet-Binette, A.; Pimentel, L.S.B.; Iulita, M.F.; Allard, S.; Ducatenzeiler, A.; do Carmo, S.; Cuello, A.C. Neuronal Driven Pre-Plaque Inflammation in a Transgenic Rat Model of Alzheimer’s Disease. Neurobiol. Aging 2014, 35, 2249–2262. [Google Scholar] [CrossRef] [PubMed]
- Di Filippo, M.; Chiasserini, D.; Gardoni, F.; Viviani, B.; Tozzi, A.; Giampà, C.; Costa, C.; Tantucci, M.; Zianni, E.; Boraso, M.; et al. Effects of Central and Peripheral Inflammation on Hippocampal Synaptic Plasticity. Neurobiol. Dis. 2013, 52, 229–236. [Google Scholar] [CrossRef]
- Khan, M.S.H.; Hegde, V. Obesity and Diabetes Mediated Chronic Inflammation: A Potential Biomarker in Alzheimer’s Disease. J. Pers. Med. 2020, 10, 42. [Google Scholar] [CrossRef]
- Banks, W.A.; Kastin, A.J.; Broadwell, R.D. Passage of Cytokines across the Blood-Brain Barrier. Neuroimmunomodulation 1995, 2, 241–248. [Google Scholar] [CrossRef]
- Skelly, D.T.; Hennessy, E.; Dansereau, M.A.; Cunningham, C. A Systematic Analysis of the Peripheral and CNS Effects of Systemic LPS, IL-1Β, TNF-α and IL-6 Challenges in C57BL/6 Mice. PLoS ONE 2013, 8, e69123. [Google Scholar] [CrossRef]
- Nguyen, J.C.D.; Killcross, A.S.; Jenkins, T.A. Obesity and Cognitive Decline: Role of Inflammation and Vascular Changes. Front Neurosci. 2014, 8, 375. [Google Scholar] [CrossRef]
- Zhang, X.; Dong, F.; Ren, J.; Driscoll, M.J.; Culver, B. High Dietary Fat Induces NADPH Oxidase-Associated Oxidative Stress and Inflammation in Rat Cerebral Cortex. Exp. Neurol. 2005, 191, 318–325. [Google Scholar] [CrossRef] [PubMed]
- Thaler, J.P.; Yi, C.X.; Schur, E.A.; Guyenet, S.J.; Hwang, B.H.; Dietrich, M.O.; Zhao, X.; Sarruf, D.A.; Izgur, V.; Maravilla, K.R.; et al. Obesity Is Associated with Hypothalamic Injury in Rodents and Humans. J. Clin. Investig. 2012, 122, 153–162. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Dyken, P.; Lacoste, B. Impact of Metabolic Syndrome on Neuroinflammation and the Blood-Brain Barrier. Front Neurosci. 2018, 12, 930. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rapoport, S.I.; Chang, M.C.J.; Spector, A.A. Delivery and Turnover of Plasma-Derived Essential PUFAs in Mammalian Brain. J. Lipid Res. 2001, 42, 678–685. [Google Scholar] [CrossRef] [PubMed]
- Kumar Jha, M.; Ho Park, D.; Kook, H.; Lee, I.K.; Lee, W.H.; Suk, K. Metabolic Control of Glia-Mediated Neuroinflammation. Curr. Alzheimer Res. 2016, 13, 387–402. [Google Scholar] [CrossRef] [PubMed]
- Tyagi, A.; Mirita, C.; Shah, I.; Reddy, P.H.; Pugazhenthi, S. Effects of Lipotoxicity in Brain Microvascular Endothelial Cells During Sirt3 Deficiency-Potential Role in Comorbid Alzheimer’s Disease. Front Aging Neurosci. 2021, 13, 716616. [Google Scholar] [CrossRef]
- Marwarha, G.; Claycombe-Larson, K.; Lund, J.; Schommer, J.; Ghribi, O. A Diet Enriched in Palmitate and Deficient in Linoleate Exacerbates Oxidative Stress and Amyloid-β Burden in the Hippocampus of 3xTg-AD Mouse Model of Alzheimer’s Disease. J. Alzheimers Dis. 2019, 68, 219–237. [Google Scholar] [CrossRef]
- Cole, G.M.; Ma, Q.L.; Frautschy, S.A. Dietary Fatty Acids and the Aging Brain. Nutr. Rev. 2010, 68, S102–S111. [Google Scholar] [CrossRef] [Green Version]
- Gupta, S.; Knight, A.G.; Gupta, S.; Keller, J.N.; Bruce-Keller, A.J. Saturated Long-Chain Fatty Acids Activate Inflammatory Signaling in Astrocytes. J. Neurochem. 2012, 120, 1060–1071. [Google Scholar] [CrossRef] [Green Version]
- Edwards, G.A.; Gamez, N.; Escobedo, G.; Calderon, O.; Moreno-Gonzalez, I. Modifiable Risk Factors for Alzheimer’s Disease. Front Aging Neurosci. 2019, 11, 146. [Google Scholar] [CrossRef]
- Wang, Z.; Liu, D.; Wang, F.; Liu, S.; Zhao, S.; Ling, E.A.; Hao, A. Saturated Fatty Acids Activate Microglia via Toll-like Receptor 4/NF-ΚB Signalling. Br. J. Nutr. 2012, 107, 229–241. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carroll, R.G.; Zasłona, Z.; Galván-Peña, S.; Koppe, E.L.; Sévin, D.C.; Angiari, S.; Triantafilou, M.; Triantafilou, K.; Modis, L.K.; O’Neill, L.A. An Unexpected Link between Fatty Acid Synthase and Cholesterol Synthesis in Proinflammatory Macrophage Activation. J. Biol. Chem. 2018, 293, 5509–5521. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arnold, S.E.; Lucki, I.; Brookshire, B.R.; Carlson, G.C.; Browne, C.A.; Kazi, H.; Bang, S.; Choi, B.R.; Chen, Y.; McMullen, M.F.; et al. High Fat Diet Produces Brain Insulin Resistance, Synaptodendritic Abnormalities and Altered Behavior in Mice. Neurobiol. Dis. 2014, 67, 79–87. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Desale, S.E.; Chinnathambi, S. Role of Dietary Fatty Acids in Microglial Polarization in Alzheimer’s Disease. J. Neuroinflammation 2020, 17, 93. [Google Scholar] [CrossRef] [Green Version]
- Patil, S.; Chan, C. Palmitic and Stearic Fatty Acids Induce Alzheimer-like Hyperphosphorylation of Tau in Primary Rat Cortical Neurons. Neurosci. Lett. 2005, 384, 288–293. [Google Scholar] [CrossRef]
- Amtul, Z.; Keet, M.; Wang, L.; Merrifield, P.; Westaway, D.; Rozmahel, R.F. DHA Supplemented in Peptamen Diet Offers No Advantage in Pathways to Amyloidosis: Is It Time to Evaluate Composite Lipid Diet? PLoS ONE 2011, 6, e0176644. [Google Scholar] [CrossRef] [Green Version]
- Liu, L.; Chan, C. IPAF Inflammasome Is Involved in Interleukin-1β Production from Astrocytes, Induced by Palmitate; Implications for Alzheimer’s Disease. Neurobiol. Aging 2014, 35, 309–321. [Google Scholar] [CrossRef] [Green Version]
- Kacířová, M.; Zmeškalová, A.; Kořínková, L.; Železná, B.; Kuneš, J.; Maletínská, L. Inflammation: Major Denominator of Obesity, Type 2 Diabetes and Alzheimer’s Disease-like Pathology? Clin. Sci. 2020, 134, 547–570. [Google Scholar] [CrossRef] [Green Version]
- Goldstein, B.J. Insulin Resistance as the Core Defect in Type 2 Diabetes Mellitus. Am. J. Cardiol. 2002, 90, 3–10. [Google Scholar] [CrossRef]
- Biddinger, S.B.; Kahn, C.R. FROM MICE TO MEN: Insights into the Insulin Resistance Syndromes. Annu. Rev. Physiol. 2006, 68, 123–158. [Google Scholar] [CrossRef]
- Lazar, M.A. How Obesity Causes Diabetes: Not a Tall Tale. Science 2005, 307, 373–375. [Google Scholar] [CrossRef] [Green Version]
- Kahn, S.E.; Hull, R.L.; Utzschneider, K.M. Mechanisms Linking Obesity to Insulin Resistance and Type 2 Diabetes. Nature 2006, 444, 840–846. [Google Scholar] [CrossRef]
- Shulman, G.I. Cellular Mechanisms of Insulin Resistance. J. Clin. Investig. 2000, 106, 171–176. [Google Scholar] [CrossRef]
- Ter Horst, K.W.; Gilijamse, P.W.; Versteeg, R.I.; Ackermans, M.T.; Nederveen, A.J.; la Fleur, S.E.; Romijn, J.A.; Nieuwdorp, M.; Zhang, D.; Samuel, V.T.; et al. Hepatic Diacylglycerol-Associated Protein Kinase Cε Translocation Links Hepatic Steatosis to Hepatic Insulin Resistance in Humans. Cell Rep. 2017, 19, 1997–2004. [Google Scholar] [CrossRef] [Green Version]
- Timmers, S.; Schrauwen, P.; de Vogel, J. Muscular Diacylglycerol Metabolism and Insulin Resistance. Physiol. Behav. 2008, 94, 242–251. [Google Scholar] [CrossRef]
- Biondi, G.; Marrano, N.; Dipaola, L.; Borrelli, A.; Rella, M.; D’Oria, R.; Genchi, V.A.; Caccioppoli, C.; Porreca, I.; Cignarelli, A.; et al. The P66Shc Protein Mediates Insulin Resistance and Secretory Dysfunction in Pancreatic β-Cells Under Lipotoxic Conditions. Diabetes 2022, 71, 1763–1771. [Google Scholar] [CrossRef]
- Ho, L.; Qin, W.; Pompl, P.N.; Xiang, Z.; Wang, J.; Zhao, Z.; Peng, Y.; Cambareri, G.; Rocher, A.; Mobbs, C.v.; et al. Diet-Induced Insulin Resistance Promotes Amyloidosis in a Transgenic Mouse Model of Alzheimer’s Disease. FASEB J. 2004, 18, 902–904. [Google Scholar] [CrossRef] [Green Version]
- Julien, C.; Tremblay, C.; Phivilay, A.; Berthiaume, L.; Émond, V.; Julien, P.; Calon, F. High-Fat Diet Aggravates Amyloid-Beta and Tau Pathologies in the 3xTg-AD Mouse Model. Neurobiol. Aging 2010, 31, 1516–1531. [Google Scholar] [CrossRef]
- Tschritter, O.; Preissl, H.; Hennige, A.M.; Stumvoll, M.; Porubska, K.; Frost, R.; Marx, H.; Klösel, B.; Lutzenberger, W.; Birbaumer, N.; et al. The Cerebrocortical Response to Hyperinsulinemia Is Reduced in Overweight Humans: A Magnetoencephalographic Study. Proc. Natl. Acad. Sci. USA 2006, 103, 12103–12108. [Google Scholar] [CrossRef] [Green Version]
- Tschritter, O.; Preissl, H.; Yokoyama, Y.; Machicao, F.; Häring, H.U.; Fritsche, A. Variation in the FTO Gene Locus Is Associated with Cerebrocortical Insulin Resistance in Humans. Diabetologia 2007, 50, 2602–2603. [Google Scholar] [CrossRef]
- Xu, J.; Gao, H.; Zhang, L.; Rong, S.; Yang, W.; Ma, C.; Chen, M.; Huang, Q.; Deng, Q.; Huang, F. Melatonin Alleviates Cognition Impairment by Antagonizing Brain Insulin Resistance in Aged Rats Fed a High-Fat Diet. J. Pineal Res. 2019, 67, e12584. [Google Scholar] [CrossRef] [PubMed]
- Hallschmid, M.; Benedict, C.; Schultes, B.; Born, J.; Kern, W. Obese Men Respond to Cognitive but Not to Catabolic Brain Insulin Signaling. Int. J. Obes. 2008, 32, 275–282. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guest, J.; Garg, M.; Bilgin, A.; Grant, R. Relationship between Central and Peripheral Fatty Acids in Humans. Lipids Health Dis. 2013, 12, 79. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nuzzo, D.; Amato, A.; Picone, P.; Terzo, S.; Galizzi, G.; Bonina, F.P.; Mulè, F.; di Carlo, M. A Natural Dietary Supplement with a Combination of Nutrients Prevents Neurodegeneration Induced by a High Fat Diet in Mice. Nutrients 2018, 10, 1130. [Google Scholar] [CrossRef] [Green Version]
- Campana, M.; Bellini, L.; Rouch, C.; Rachdi, L.; Coant, N.; Butin, N.; Bandet, C.L.; Philippe, E.; Meneyrol, K.; Kassis, N.; et al. Inhibition of Central de Novo Ceramide Synthesis Restores Insulin Signaling in Hypothalamus and Enhances β-Cell Function of Obese Zucker Rats. Mol. Metab. 2018, 8, 23–36. [Google Scholar] [CrossRef]
- Maciejczyk, M.; Żebrowska, E.; Chabowski, A. Insulin Resistance and Oxidative Stress in the Brain: What’s New? Int. J. Mol. Sci. 2019, 20, 874. [Google Scholar] [CrossRef] [Green Version]
- Pizzino, G.; Irrera, N.; Cucinotta, M.; Pallio, G.; Mannino, F.; Arcoraci, V.; Squadrito, F.; Altavilla, D.; Bitto, A. Oxidative Stress: Harms and Benefits for Human Health. Oxid Med. Cell Longev. 2017, 2017, 8416763. [Google Scholar] [CrossRef] [Green Version]
- Di Meo, S.; Reed, T.T.; Venditti, P.; Victor, V.M. Role of ROS and RNS Sources in Physiological and Pathological Conditions. Oxid Med. Cell Longev. 2016, 2016, 1245049. [Google Scholar] [CrossRef]
- Newsholme, P.; Morgan, D.; Rebelato, E.; Oliveira-Emilio, H.C.; Procopio, J.; Curi, R.; Carpinelli, A. Insights into the Critical Role of NADPH Oxidase(s) in the Normal and Dysregulated Pancreatic Beta Cell. Diabetologia 2009, 52, 2489–2498. [Google Scholar] [CrossRef] [Green Version]
- Cobley, J.N.; Fiorello, M.L.; Bailey, D.M. 13 Reasons Why the Brain Is Susceptible to Oxidative Stress. Redox Biol. 2018, 15, 490–503. [Google Scholar] [CrossRef]
- Martins, R.N.; Harper, C.G.; Stokes, G.B.; Masters, C.L. Increased Cerebral Glucose-6-Phosphate Dehydrogenase Activity in Alzheimer’s Disease May Reflect Oxidative Stress. J. Neurochem. 1986, 46, 1042–1045. [Google Scholar] [CrossRef]
- Butterfield, D.A.; Poon, H.F.; st. Clair, D.; Keller, J.N.; Pierce, W.M.; Klein, J.B.; Markesbery, W.R. Redox Proteomics Identification of Oxidatively Modified Hippocampal Proteins in Mild Cognitive Impairment: Insights into the Development of Alzheimer’s Disease. Neurobiol. Dis. 2006, 22, 223–232. [Google Scholar] [CrossRef]
- Thériault, P.; ElAli, A.; Rivest, S. High Fat Diet Exacerbates Alzheimer’s Disease-Related Pathology in APPswe/PS1 Mice. Oncotarget 2016, 7, 67808–67827. [Google Scholar] [CrossRef] [Green Version]
- Button, E.B.; Mitchell, A.S.; Domingos, M.M.; Chung, J.H.J.; Bradley, R.M.; Hashemi, A.; Marvyn, P.M.; Patterson, A.C.; Stark, K.D.; Quadrilatero, J.; et al. Microglial Cell Activation Increases Saturated and Decreases Monounsaturated Fatty Acid Content, but Both Lipid Species Are Proinflammatory. Lipids 2014, 49, 305–316. [Google Scholar] [CrossRef]
- Mett, J. The Impact of Medium Chain and Polyunsaturated ω-3-Fatty Acids on Amyloid-β Deposition, Oxidative Stress and Metabolic Dysfunction Associated with Alzheimer’s Disease. Antioxidants 2021, 10, 1991. [Google Scholar] [CrossRef]
- Lenzen, S. Oxidative Stress: The Vulnerable Beta-Cell. Biochem. Soc. Trans. 2008, 36, 343–347. [Google Scholar] [CrossRef]
- Oprescu, A.I.; Bikopoulos, G.; Naassan, A.; Allister, E.M.; Tang, C.; Park, E.; Uchino, H.; Lewis, G.F.; Fantus, I.G.; Rozakis-Adcock, M.; et al. Free Fatty Acid–Induced Reduction in Glucose-Stimulated Insulin Secretion Evidence for a Role of Oxidative Stress In Vitro and In Vivo. Diabetes 2007, 56, 2927–2937. [Google Scholar] [CrossRef] [Green Version]
- Ly, L.D.; Xu, S.; Choi, S.K.; Ha, C.M.; Thoudam, T.; Cha, S.K.; Wiederkehr, A.; Wollheim, C.B.; Lee, I.K.; Park, K.S. Oxidative Stress and Calcium Dysregulation by Palmitate in Type 2 Diabetes. Exp. Mol. Med. 2017, 49, e291. [Google Scholar] [CrossRef] [Green Version]
- Piro, S.; Anello, M.; di Pietro, C.; Lizzio, M.N.; Patanè, G.; Rabuazzo, A.M.; Vigneri, R.; Purrello, M.; Purrello, F. Chronic Exposure to Free Fatty Acids or High Glucose Induces Apoptosis in Rat Pancreatic Islets: Possible Role of Oxidative Stress. Metabolism 2002, 51, 1340–1347. [Google Scholar] [CrossRef]
- Hasnain, S.Z.; Prins, J.B.; McGuckin, M.A. Oxidative and Endoplasmic Reticulum Stress in β-Cell Dysfunction in Diabetes. J. Mol. Endocrinol. 2016, 56, R33–R54. [Google Scholar] [CrossRef]
- Plötz, T.; von Hanstein, A.S.; Krümmel, B.; Laporte, A.; Mehmeti, I.; Lenzen, S. Structure-Toxicity Relationships of Saturated and Unsaturated Free Fatty Acids for Elucidating the Lipotoxic Effects in Human EndoC-ΒH1 Beta-Cells. Biochim. Biophys. Acta Mol. Basis Dis. 2019, 1865, 165525. [Google Scholar] [CrossRef]
- Natalicchio, A.; Tortosa, F.; Labarbuta, R.; Biondi, G.; Marrano, N.; Carchia, E.; Leonardini, A.; Cignarelli, A.; Bugliani, M.; Marchetti, P.; et al. The P66(Shc) Redox Adaptor Protein Is Induced by Saturated Fatty Acids and Mediates Lipotoxicity-Induced Apoptosis in Pancreatic Beta Cells. Diabetologia 2015, 58, 1260–1271. [Google Scholar] [CrossRef]
- Zamora, M.; Villena, J. Targeting Mitochondrial Biogenesis to Treat Insulin Resistance. Curr. Pharm. Des. 2014, 20, 5527–5557. [Google Scholar] [CrossRef]
- Liu, J.; Shen, W.; Zhao, B.; Wang, Y.; Wertz, K.; Weber, P.; Zhang, P. Targeting Mitochondrial Biogenesis for Preventing and Treating Insulin Resistance in Diabetes and Obesity: Hope from Natural Mitochondrial Nutrients. Adv. Drug Deliv. Rev. 2009, 61, 1343–1352. [Google Scholar] [CrossRef]
- Zhu, M.; Liu, X.; Liu, W.; Lu, Y.; Cheng, J.; Chen, Y. β Cell Aging and Age-Related Diabetes. Aging 2021, 13, 7691–7706. [Google Scholar] [CrossRef]
- Kuzmenko, D.I.; Klimentyeva, T.K. Role of Ceramide in Apoptosis and Development of Insulin Resistance. Biochemistry 2016, 81, 913–927. [Google Scholar] [CrossRef]
- Rial, E.; Rodríguez-Sánchez, L.; Gallardo-Vara, E.; Zaragoza, P.; Moyano, E.; González-Barroso, M.M. Lipotoxicity, Fatty Acid Uncoupling and Mitochondrial Carrier Function. Biochim. Biophys. Acta (BBA)-Bioenerg. 2010, 1797, 800–806. [Google Scholar] [CrossRef] [Green Version]
- Grishko, V.; Rachek, L.; Musiyenko, S.; LeDoux, S.P.; Wilson, G.L. Involvement of MtDNA Damage in Free Fatty Acid-Induced Apoptosis. Free Radic. Biol. Med. 2005, 38, 755–762. [Google Scholar] [CrossRef]
- Wright, M.M.; Schopfer, F.J.; Baker, P.R.S.; Vidyasagar, V.; Powell, P.; Chumley, P.; Iles, K.E.; Freeman, B.A.; Agarwal, A. Fatty Acid Transduction of Nitric Oxide Signaling: Nitrolinoleic Acid Potently Activates Endothelial Heme Oxygenase 1 Expression. Proc. Natl. Acad. Sci. USA 2006, 103, 4299–4304. [Google Scholar] [CrossRef] [Green Version]
- Carmona-Montesinos, E.; Velazquez-Perez, R.; Pichardo Aguirre, E.; Rivas-Arancibia, S. Obesity, Oxidative Stress, and Their Effect on Serum Heme Oxygenase-1 Concentrations and Insulin in Children Aged 3 to 5 Years in a Pediatric Hospital of the Ministry of Health CDMX. Child Obes. 2016, 12, 474–481. [Google Scholar] [CrossRef]
- Butterfield, D.A.; Di Domenico, F.; Barone, E. Elevated Risk of Type 2 Diabetes for Development of Alzheimer Disease: A Key Role for Oxidative Stress in Brain. Biochim Biophys Acta 2014, 1842, 1693–1706. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barone, E.; Di Domenico, F.; Mancuso, C.; Butterfield, D.A. The Janus Face of the Heme Oxygenase/Biliverdin Reductase System in Alzheimer Disease: It’s Time for Reconciliation. Neurobiol Dis 2014, 62, 144–159. [Google Scholar] [CrossRef] [Green Version]
- Barone, E.; Butterfield, D.A. Insulin Resistance in Alzheimer Disease: Is Heme Oxygenase-1 an Achille’s Heel? Neurobiol. Dis. 2015, 84, 69–77. [Google Scholar] [CrossRef] [PubMed]
- Morgan, D.; Oliveira-Emilio, H.R.; Keane, D.; Hirata, A.E.; Santos da Rocha, M.; Bordin, S.; Curi, R.; Newsholme, P.; Carpinelli, A.R. Glucose, Palmitate and pro-Inflammatory Cytokines Modulate Production and Activity of a Phagocyte-like NADPH Oxidase in Rat Pancreatic Islets and a Clonal Beta Cell Line. Diabetologia 2007, 50, 359–369. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Michalska, M.; Wolf, G.; Walther, R.; Newsholme, P. Effects of Pharmacological Inhibition of NADPH Oxidase or INOS on Pro-Inflammatory Cytokine, Palmitic Acid or H2O2-Induced Mouse Islet or Clonal Pancreatic β-Cell Dysfunction. Biosci. Rep. 2010, 30, 445–453. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yuan, H.; Zhang, X.; Huang, X.; Lu, Y.; Tang, W.; Man, Y.; Wang, S.; Xi, J.; Li, J. NADPH Oxidase 2-Derived Reactive Oxygen Species Mediate FFAs-Induced Dysfunction and Apoptosis of β-Cells via JNK, P38 MAPK and P53 Pathways. PLoS ONE 2010, 5, e15726. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vilas-Boas, E.A.; Nalbach, L.; Ampofo, E.; Lucena, C.F.; Naudet, L.; Ortis, F.; Carpinelli, A.R.; Morgan, B.; Roma, L.P. Transient NADPH Oxidase 2-Dependent H 2 O 2 Production Drives Early Palmitate-Induced Lipotoxicity in Pancreatic Islets. Free Radic. Biol. Med. 2021, 162, 1–13. [Google Scholar] [CrossRef]
- Inoguchi, T.; Nawata, H. NAD(P)H Oxidase Activation: A Potential Target Mechanism for Diabetic Vascular Complications, Progressive Beta-Cell Dysfunction and Metabolic Syndrome. Curr. Drug Targets 2005, 6, 495–501. [Google Scholar] [CrossRef]
- Guichard, C.; Moreau, R.; Pessayre, D.; Epperson, T.K.; Krause, K.H. NOX Family NADPH Oxidases in Liver and in Pancreatic Islets: A Role in the Metabolic Syndrome and Diabetes? Biochem. Soc. Trans. 2008, 36, 920–929. [Google Scholar] [CrossRef] [Green Version]
- Anvari, E.; Wikström, P.; Walum, E.; Welsh, N. The Novel NADPH Oxidase 4 Inhibitor GLX351322 Counteracts Glucose Intolerance in High-Fat Diet-Treated C57BL/6 Mice. Free Radic. Res. 2015, 49, 1308–1318. [Google Scholar] [CrossRef]
- Lynch, C.M.; Kinzenbaw, D.A.; Chen, X.; Zhan, S.; Mezzetti, E.; Filosa, J.; Ergul, A.; Faulkner, J.L.; Faraci, F.M.; Didion, S.P. Nox2-Derived Superoxide Contributes to Cerebral Vascular Dysfunction in Diet-Induced Obesity. Stroke 2013, 44, 3195–3201. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jeong, J.H.; Koo, J.H.; Cho, J.Y.; Kang, E.B. Neuroprotective Effect of Treadmill Exercise against Blunted Brain Insulin Signaling, NADPH Oxidase, and Tau Hyperphosphorylation in Rats Fed a High-Fat Diet. Brain Res. Bull 2018, 142, 374–383. [Google Scholar] [CrossRef] [PubMed]
- Hammerschmidt, P.; Brüning, J.C. Contribution of Specific Ceramides to Obesity-Associated Metabolic Diseases. Cell. Mol. Life Sci. 2022, 79, 395. [Google Scholar] [CrossRef]
- Hannun, Y.A.; Obeid, L.M. Sphingolipids and Their Metabolism in Physiology and Disease. Nat. Rev. Mol. Cell Biol. 2018, 19, 175. [Google Scholar] [CrossRef]
- Boslem, E.; Meikle, P.J.; Biden, T.J. Roles of Ceramide and Sphingolipids in Pancreatic β-Cell Function and Dysfunction. Islets 2012, 4, 177–187. [Google Scholar] [CrossRef] [Green Version]
- Paumen, M.B.; Ishida, Y.; Muramatsu, M.; Yamamoto, M.; Honjo, T. Inhibition of Carnitine Palmitoyltransferase I Augments Sphingolipid Synthesis and Palmitate-Induced Apoptosis. J. Biol. Chem. 1997, 272, 3324–3329. [Google Scholar] [CrossRef] [Green Version]
- Ji, R.; Akashi, H.; Drosatos, K.; Liao, X.; Jiang, H.; Kennel, P.J.; Brunjes, D.L.; Castillero, E.; Zhang, X.; Deng, L.Y.; et al. Increased de Novo Ceramide Synthesis and Accumulation in Failing Myocardium. JCI Insight 2017, 2, e82922. [Google Scholar] [CrossRef] [Green Version]
- Shimabukuro, M.; Higa, M.; Zhou, Y.T.; Wang, M.Y.; Newgard, C.B.; Unger, R.H. Lipoapoptosis in Beta-Cells of Obese Prediabetic Fa/Fa Rats. Role of Serine Palmitoyltransferase Overexpression. J. Biol. Chem. 1998, 273, 32487–32490. [Google Scholar] [CrossRef] [Green Version]
- Lupi, R.; Dotta, F.; Marselli, L.; del Guerra, S.; Masini, M.; Santangelo, C.; Patané, G.; Boggi, U.; Piro, S.; Anello, M.; et al. Prolonged Exposure to Free Fatty Acids Has Cytostatic and Pro-Apoptotic Effects on Human Pancreatic IsletsEvidence That β-Cell Death Is Caspase Mediated, Partially Dependent on Ceramide Pathway, and Bcl-2 Regulated. Diabetes 2002, 51, 1437–1442. [Google Scholar] [CrossRef] [Green Version]
- Boslem, E.; Weir, J.M.; MacIntosh, G.; Sue, N.; Cantley, J.; Meikle, P.J.; Biden, T.J. Alteration of Endoplasmic Reticulum Lipid Rafts Contributes to Lipotoxicity in Pancreatic β-Cells. J. Biol. Chem. 2013, 288, 26569–26582. [Google Scholar] [CrossRef]
- Straczkowski, M.; Kowalska, I.; Baranowski, M.; Nikolajuk, A.; Otziomek, E.; Zabielski, P.; Adamska, A.; Blachnio, A.; Gorski, J.; Gorska, M. Increased Skeletal Muscle Ceramide Level in Men at Risk of Developing Type 2 Diabetes. Diabetologia 2007, 50, 2366–2373. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- de la Maza, M.P.; Rodriguez, J.M.; Hirsch, S.; Leiva, L.; Barrera, G.; Bunout, D. Skeletal Muscle Ceramide Species in Men with Abdominal Obesity. J. Nutr. Health Aging 2015, 19, 389–396. [Google Scholar] [CrossRef] [PubMed]
- Kolak, M.; Westerbacka, J.; Velagapudi, V.R.; Wågsäter, D.; Yetukuri, L.; Makkonen, J.; Rissanen, A.; Häkkinen, A.M.; Lindell, M.; Bergholm, R.; et al. Adipose Tissue Inflammation and Increased Ceramide Content Characterize Subjects with High Liver Fat Content Independent of Obesity. Diabetes 2007, 56, 1960–1968. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jazvinšćak Jembrek, M.; Hof, P.R.; Šimić, G. Ceramides in Alzheimer’s Disease: Key Mediators of Neuronal Apoptosis Induced by Oxidative Stress and Aβ Accumulation. Oxid. Med. Cell Longev. 2015, 2015, 346783. [Google Scholar] [CrossRef] [Green Version]
- Patil, S.; Melrose, J.; Chan, C. Involvement of Astroglial Ceramide in Palmitic Acid-Induced Alzheimer-like Changes in Primary Neurons. Eur. J. Neurosci. 2007, 26, 2131–2141. [Google Scholar] [CrossRef]
- Filippov, V.; Song, M.A.; Zhang, K.; Vinters, H.v.; Tung, S.; Kirsch, W.M.; Yang, J.; Duerksen-Hughes, P.J. Increased Ceramide in Brains with Alzheimer’s and Other Neurodegenerative Diseases. J. Alzheimer’s Dis. 2012, 29, 537–547. [Google Scholar] [CrossRef] [Green Version]
- Mielke, M.M.; Bandaru, V.V.R.; Haughey, N.J.; Xia, J.; Fried, L.P.; Yasar, S.; Albert, M.; Varma, V.; Harris, G.; Schneider, E.B.; et al. Serum Ceramides Increase the Risk of Alzheimer Disease: The Women’s Health and Aging Study II. Neurology 2012, 79, 633–641. [Google Scholar] [CrossRef] [Green Version]
- de la Monte, S.M.; Tong, M.; Nguyen, V.; Setshedi, M.; Longato, L.; Wands, J.R. Ceramide-Mediated Insulin Resistance and Impairment of Cognitive-Motor Functions. J. Alzheimers Dis. 2010, 21, 967. [Google Scholar] [CrossRef] [Green Version]
- Tong, M.; de La Monte, S.M. Mechanisms of Ceramide-Mediated Neurodegeneration. J. Alzheimers Dis. 2009, 16, 705–714. [Google Scholar] [CrossRef] [Green Version]
- Stoica, B.A.; Movsesyan, V.A.; Lea IV, P.M.; Faden, A.I. Ceramide-Induced Neuronal Apoptosis Is Associated with Dephosphorylation of Akt, BAD, FKHR, GSK-3β, and Induction of the Mitochondrial-Dependent Intrinsic Caspase Pathway. Mol. Cell. Neurosci. 2003, 22, 365–382. [Google Scholar] [CrossRef]
- Kahn, S.E.; D’Alessio, D.A.; Schwartz, M.W.; Fujimoto, W.Y.; Ensinck, J.W.; Taborsky, G.J.; Porte, D. Evidence of Cosecretion of Islet Amyloid Polypeptide and Insulin by β-Cells. Diabetes 1990, 39, 634–638. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, S.; Liu, J.; MacGibbon, G.; Dragunow, M.; Cooper, G.J.S. Increased Expression and Activation of C-Jun Contributes to Human Amylin-Induced Apoptosis in Pancreatic Islet β-Cells. J. Mol. Biol. 2002, 324, 271–285. [Google Scholar] [CrossRef]
- Oshima, M.; Pechberty, S.; Bellini, L.; Göpel, S.O.; Campana, M.; Rouch, C.; Dairou, J.; Cosentino, C.; Fantuzzi, F.; Toivonen, S.; et al. Stearoyl CoA Desaturase Is a Gatekeeper That Protects Human Beta Cells against Lipotoxicity and Maintains Their Identity. Diabetologia 2020, 63, 395–409. [Google Scholar] [CrossRef] [Green Version]
- Krizhanovskii, C.; Fred, R.G.; Oskarsson, M.E.; Westermark, G.T.; Welsh, N. Addition of Exogenous Sodium Palmitate Increases the IAPP/Insulin MRNA Ratio via GPR40 in Human EndoC-ΒH1 Cells. Ups. J. Med. Sci. 2017, 122, 149–159. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hull, R.L.; Andrikopoulos, S.; Verchere, C.B.; Vidal, J.; Wang, F.; Cnop, M.; Prigeon, R.L.; Kahn, S.E. Increased Dietary Fat Promotes Islet Amyloid Formation and β-Cell Secretory Dysfunction in a Transgenic Mouse Model of Islet Amyloid. Diabetes 2003, 52, 372–379. [Google Scholar] [CrossRef]
- Casas, S.; Gomis, R.; Gribble, F.M.; Altirriba, J.; Knuutila, S.; Novials, A. Impairment of the Ubiquitin-Proteasome Pathway Is a Downstream Endoplasmic Reticulum Stress Response Induced by Extracellular Human Islet Amyloid Polypeptide and Contributes to Pancreatic β-Cell Apoptosis. Diabetes 2007, 56, 2284–2294. [Google Scholar] [CrossRef] [Green Version]
- Konarkowska, B.; Aitken, J.F.; Kistler, J.; Zhang, S.; Cooper, G.J.S. The Aggregation Potential of Human Amylin Determines Its Cytotoxicity towards Islet β-Cells. FEBS J. 2006, 273, 3614–3624. [Google Scholar] [CrossRef]
- Zhang, S.; Liu, J.; Dragunow, M.; Cooper, G.J.S. Fibrillogenic Amylin Evokes Islet β-Cell Apoptosis through Linked Activation of a Caspase Cascade and JNK1. J. Biol. Chem. 2003, 278, 52810–52819. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Raleigh, D.; Zhang, X.; Hastoy, B.; Clark, A. The β-Cell Assassin: IAPP Cytotoxicity. J. Mol. Endocrinol. 2017, 59, R121–R140. [Google Scholar] [CrossRef]
- Zhang, X.; Fu, Z.; Meng, L.; He, M.; Zhang, Z. The Early Events That Initiate β-Amyloid Aggregation in Alzheimer’s Disease. Front Aging Neurosci. 2018, 10, 359. [Google Scholar] [CrossRef]
- Xiong, H.; Callaghan, D.; Jones, A.; Walker, D.G.; Lue, L.F.; Beach, T.G.; Sue, L.I.; Woulfe, J.; Xu, H.; Stanimirovic, D.B.; et al. Cholesterol Retention in Alzheimer’s Brain Is Responsible for High β- and γ-Secretase Activities and Aβ Production. Neurobiol. Dis. 2008, 29, 422–437. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rudge, J.D. A New Hypothesis for Alzheimer’s Disease: The Lipid Invasion Model. J. Alzheimers Dis. Rep. 2022, 6, 129–161. [Google Scholar] [CrossRef] [PubMed]
- Cutler, R.G.; Kelly, J.; Storie, K.; Pedersen, W.A.; Tammara, A.; Hatanpaa, K.; Troncoso, J.C.; Mattson, M.P. Involvement of Oxidative Stress-Induced Abnormalities in Ceramide and Cholesterol Metabolism in Brain Aging and Alzheimer’s Disease. Proc. Natl. Acad. Sci. USA 2004, 101, 2070–2075. [Google Scholar] [CrossRef] [Green Version]
- Li, H.; Kim, W.S.; Guillemin, G.J.; Hill, A.F.; Evin, G.; Garner, B. Modulation of Amyloid Precursor Protein Processing by Synthetic Ceramide Analogues. Biochim. Biophys. Acta (BBA)-Mol. Cell Biol. Lipids 2010, 1801, 887–895. [Google Scholar] [CrossRef] [Green Version]
- Takasugi, N.; Sasaki, T.; Shinohara, M.; Iwatsubo, T.; Tomita, T. Synthetic Ceramide Analogues Increase Amyloid-β 42 Production by Modulating γ-Secretase Activity. Biochem. Biophys. Res. Commun. 2015, 457, 194–199. [Google Scholar] [CrossRef] [PubMed]
- Grimm, M.O.W.; Grimm, H.S.; Pätzold, A.J.; Zinser, E.G.; Halonen, R.; Duering, M.; Tschäpe, J.A.; de Strooper, B.; Müller, U.; Shen, J.; et al. Regulation of Cholesterol and Sphingomyelin Metabolism by Amyloid-β and Presenilin. Nat. Cell Biol. 2005, 7, 1118–1123. [Google Scholar] [CrossRef]
- Wang, X.; Ching, Y.P.; Lam, W.H.; Qi, Z.; Zhang, M.; Wang, J.H. Identification of a Common Protein Association Region in the Neuronal Cdk5 Activator. J. Biol. Chem. 2000, 275, 31763–31769. [Google Scholar] [CrossRef] [Green Version]
- Malaplate-Armand, C.; Florent-Béchard, S.; Youssef, I.; Koziel, V.; Sponne, I.; Kriem, B.; Leininger-Muller, B.; Olivier, J.L.; Oster, T.; Pillot, T. Soluble Oligomers of Amyloid-β Peptide Induce Neuronal Apoptosis by Activating a CPLA2-Dependent Sphingomyelinase-Ceramide Pathway. Neurobiol. Dis. 2006, 23, 178–189. [Google Scholar] [CrossRef]
- Kook, S.Y.; Hong, H.S.; Moon, M.; Ha, C.M.; Chang, S.; Mook-Jung, I. A 1-42-RAGE Interaction Disrupts Tight Junctions of the Blood-Brain Barrier Via Ca2+-Calcineurin Signaling. J. Neurosci. 2012, 32, 8845–8854. [Google Scholar] [CrossRef]
- Takechi, R.; Galloway, S.; Pallebage-Gamarallage, M.M.S.; Lam, V.; Mamo, J.C.L. Dietary Fats, Cerebrovasculature Integrity and Alzheimer’s Disease Risk. Prog. Lipid Res. 2010, 49, 159–170. [Google Scholar] [CrossRef]
- Galloway, S.; Takechi, R.; Nesbit, M.; Pallebage-Gamarallage, M.M.; Lam, V.; Mamo, J.C.L. The Differential Effects of Fatty Acids on Enterocytic Abundance of Amyloid-Beta. Lipids Health Dis. 2019, 18, 209. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Walter, P.; Ron, D. The Unfolded Protein Response: From Stress Pathway to Homeostatic Regulation. Science 2011, 334, 1081–1086. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Karaskov, E.; Scott, C.; Zhang, L.; Teodoro, T.; Ravazzola, M.; Volchuk, A. Chronic Palmitate But Not Oleate Exposure Induces Endoplasmic Reticulum Stress, Which May Contribute to INS-1 Pancreatic β-Cell Apoptosis. Endocrinology 2006, 147, 3398–3407. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cunha, D.A.; Hekerman, P.; Ladrière, L.; Bazarra-Castro, A.; Ortis, F.; Wakeham, M.C.; Moore, F.; Rasschaert, J.; Cardozo, A.K.; Bellomo, E.; et al. Initiation and Execution of Lipotoxic ER Stress in Pancreatic β-Cells. J. Cell Sci. 2008, 121, 2308–2318. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Laybutt, D.R.; Preston, A.M.; Åkerfeldt, M.C.; Kench, J.G.; Busch, A.K.; Biankin, A.v.; Biden, T.J. Endoplasmic Reticulum Stress Contributes to Beta Cell Apoptosis in Type 2 Diabetes. Diabetologia 2007, 50, 752–763. [Google Scholar] [CrossRef] [Green Version]
- Gwiazda, K.S.; Yang, T.L.B.; Lin, Y.; Johnson, J.D. Effects of Palmitate on ER and Cytosolic Ca 2+ Homeostasis in β-Cells. Am. J. Physiol. Endocrinol. Metab. 2009, 296, E690–E701. [Google Scholar] [CrossRef]
- Lei, X.; Zhang, S.; Bohrer, A.; Ramanadham, S. Calcium-Independent Phospholipase A2 (IPLA2 Beta)-Mediated Ceramide Generation Plays a Key Role in the Cross-Talk between the Endoplasmic Reticulum (ER) and Mitochondria during ER Stress-Induced Insulin-Secreting Cell Apoptosis. J. Biol. Chem. 2008, 283, 34819–34832. [Google Scholar] [CrossRef] [Green Version]
- Özcan, U.; Cao, Q.; Yilmaz, E.; Lee, A.H.; Iwakoshi, N.N.; Özdelen, E.; Tuncman, G.; Görgün, C.; Glimcher, L.H.; Hotamisligil, G.S. Endoplasmic Reticulum Stress Links Obesity, Insulin Action, and Type 2 Diabetes. Science 2004, 306, 457–461. [Google Scholar] [CrossRef] [Green Version]
- Koh, H.J.; Toyoda, T.; Didesch, M.M.; Lee, M.Y.; Sleeman, M.W.; Kulkarni, R.N.; Musi, N.; Hirshman, M.F.; Goodyear, L.J. Tribbles 3 Mediates Endoplasmic Reticulum Stress-Induced Insulin Resistance in Skeletal Muscle. Nat. Commun. 2013, 4, 1871. [Google Scholar] [CrossRef] [Green Version]
- Ajoolabady, A.; Lindholm, D.; Ren, J.; Pratico, D. ER Stress and UPR in Alzheimer’s Disease: Mechanisms, Pathogenesis, Treatments. Cell Death Dis. 2022, 13, 706. [Google Scholar] [CrossRef]
- Gerakis, Y.; Hetz, C. Emerging Roles of ER Stress in the Etiology and Pathogenesis of Alzheimer’s Disease. FEBS J. 2018, 285, 995–1011. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, J.; Park, Y.J.; Jang, Y.; Kwon, Y.H. AMPK Activation Inhibits Apoptosis and Tau Hyperphosphorylation Mediated by Palmitate in SH-SY5Y Cells. Brain Res. 2011, 1418, 42–51. [Google Scholar] [CrossRef] [PubMed]
- Hsiao, Y.H.; Lin, C.I.; Liao, H.; Chen, Y.H.; Lin, S.H. Palmitic Acid-Induced Neuron Cell Cycle G2/M Arrest and Endoplasmic Reticular Stress through Protein Palmitoylation in SH-SY5Y Human Neuroblastoma Cells. Int. J. Mol. Sci. 2014, 15, 20876–20899. [Google Scholar] [CrossRef] [Green Version]
- Marwarha, G.; Claycombe, K.; Schommer, J.; Collins, D.; Ghribi, O. Palmitate-Induced Endoplasmic Reticulum Stress and Subsequent C/EBPα Homologous Protein Activation Attenuates Leptin and Insulin-like Growth Factor 1 Expression in the Brain. Cell Signal 2016, 28, 1789–1805. [Google Scholar] [CrossRef] [Green Version]
- Hitomi, J.; Katayama, T.; Eguchi, Y.; Kudo, T.; Taniguchi, M.; Koyama, Y.; Manabe, T.; Yamagishi, S.; Bando, Y.; Imaizumi, K.; et al. Involvement of Caspase-4 in Endoplasmic Reticulum Stress-Induced Apoptosis and Aβ-Induced Cell Death. J. Cell Biol. 2004, 165, 347–356. [Google Scholar] [CrossRef] [PubMed]
- Yukioka, F.; Matsuzaki, S.; Kawamoto, K.; Koyama, Y.; Hitomi, J.; Katayama, T.; Tohyama, M. Presenilin-1 Mutation Activates the Signaling Pathway of Caspase-4 in Endoplasmic Reticulum Stress-Induced Apoptosis. Neurochem. Int. 2008, 52, 683–687. [Google Scholar] [CrossRef]
- Alberdi, E.; Wyssenbach, A.; Alberdi, M.; Sánchez-Gómez, M.v.; Cavaliere, F.; Rodríguez, J.J.; Verkhratsky, A.; Matute, C. Ca 2+ -Dependent Endoplasmic Reticulum Stress Correlates with Astrogliosis in Oligomeric Amyloid β-Treated Astrocytes and in a Model of Alzheimer’s Disease. Aging Cell 2013, 12, 292–302. [Google Scholar] [CrossRef] [Green Version]
- Seyb, K.I.; Ansar, S.; Bean, J.; Michaelis, M.L. β-Amyloid and Endoplasmic Reticulum Stress Reponses in Primary Neurons: Effects of Drugs That Interact with the Cytoskeleton. J. Mol. Neurosci. 2006, 28, 111–124. [Google Scholar] [CrossRef]
- Costa, R.O.; Ferreiro, E.; Martins, I.; Santana, I.; Cardoso, S.M.; Oliveira, C.R.; Pereira, C.M.F. Amyloid β-Induced ER Stress Is Enhanced under Mitochondrial Dysfunction Conditions. Neurobiol. Aging 2012, 33, 824.e5–824.e16. [Google Scholar] [CrossRef]
- Resende, R.; Ferreiro, E.; Pereira, C.; Resende de Oliveira, C. Neurotoxic Effect of Oligomeric and Fibrillar Species of Amyloid-Beta Peptide 1-42: Involvement of Endoplasmic Reticulum Calcium Release in Oligomer-Induced Cell Death. Neuroscience 2008, 155, 725–737. [Google Scholar] [CrossRef]
- O’Connor, T.; Sadleir, K.R.; Maus, E.; Velliquette, R.A.; Zhao, J.; Cole, S.L.; Eimer, W.A.; Hitt, B.; Bembinster, L.A.; Lammich, S.; et al. Phosphorylation of the Translation Initiation Factor EIF2α Increases BACE1 Levels and Promotes Amyloidogenesis. Neuron 2008, 60, 988–1009. [Google Scholar] [CrossRef] [Green Version]
- Yoon, S.O.; Park, D.J.; Ryu, J.C.; Ozer, H.G.; Tep, C.; Shin, Y.J.; Lim, T.H.; Pastorino, L.; Kunwar, A.J.; Walton, J.C.; et al. JNK3 Perpetuates Metabolic Stress Induced by Aβ Peptides. Neuron 2012, 75, 824–837. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mouton-Liger, F.; Paquet, C.; Dumurgier, J.; Bouras, C.; Pradier, L.; Gray, F.; Hugon, J. Oxidative Stress Increases BACE1 Protein Levels through Activation of the PKR-EIF2α Pathway. Biochimica et Biophysica Acta (BBA)-Molecular Basis of Disease 2012, 1822, 885–896. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sims-Robinson, C.; Bakeman, A.; Glasser, R.; Boggs, J.; Pacut, C.; Feldman, E.L. The Role of Endoplasmic Reticulum Stress in Hippocampal Insulin Resistance. Exp. Neurol. 2016, 277, 261–267. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Z.; Chen, L.; Chen, C.; Zhou, Y.; Hu, D.; Yang, J.; Chen, Y.; Zhuo, W.; Mao, M.; Zhang, X.; et al. Targeting Ferroptosis in Breast Cancer. Biomark Res. 2020, 8, 58. [Google Scholar] [CrossRef] [PubMed]
- Dixon, S.J.; Lemberg, K.M.; Lamprecht, M.R.; Skouta, R.; Zaitsev, E.M.; Gleason, C.E.; Patel, D.N.; Bauer, A.J.; Cantley, A.M.; Yang, W.S.; et al. Ferroptosis: An Iron-Dependent Form of Nonapoptotic Cell Death. Cell 2012, 149, 1060–1072. [Google Scholar] [CrossRef] [Green Version]
- Römer, A.; Linn, T.; Petry, S.F. Lipotoxic Impairment of Mitochondrial Function in β-Cells: A Review. Antioxidants 2021, 10, 293. [Google Scholar] [CrossRef]
- Krümmel, B.; von Hanstein, A.S.; Plötz, T.; Lenzen, S.; Mehmeti, I. Differential Effects of Saturated and Unsaturated Free Fatty Acids on Ferroptosis in Rat β-Cells. J. Nutr. Biochem. 2022, 106, 109013. [Google Scholar] [CrossRef]
- Krümmel, B.; Plötz, T.; Jörns, A.; Lenzen, S.; Mehmeti, I. The Central Role of Glutathione Peroxidase 4 in the Regulation of Ferroptosis and Its Implications for Pro-Inflammatory Cytokine-Mediated Beta-Cell Death. Biochim. Et Biophys. Acta (BBA)-Mol. Basis Dis. 2021, 1867, 166114. [Google Scholar] [CrossRef]
- Yang, X.D.; Yang, Y.Y. Ferroptosis as a Novel Therapeutic Target for Diabetes and Its Complications. Front Endocrinol (Lausanne) 2022, 13, 853822. [Google Scholar] [CrossRef]
- Yan, N.; Zhang, J.J. Iron Metabolism, Ferroptosis, and the Links with Alzheimer’s Disease. Front Neurosci. 2020, 13, 1443. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weiland, A.; Wang, Y.; Wu, W.; Lan, X.; Han, X.; Li, Q.; Wang, J. Ferroptosis and Its Role in Diverse Brain Diseases. Mol. Neurobiol. 2019, 56, 4880–4893. [Google Scholar] [CrossRef] [PubMed]
- Jin, W.; Fan, M.; Zhang, Y.; Zhang, Q.; Jing, C.; Jiang, R.; Piao, C.; Sun, L. Polydatin Prevents Lipotoxicity-Induced Dysfunction in Pancreatic β-Cells by Inhibiting Endoplasmic Reticulum Stress and Excessive Autophagy. Phytomedicine 2022, 106, 154410. [Google Scholar] [CrossRef] [PubMed]
- Šrámek, J.; Němcová-Fürstová, V.; Kovář, J. Molecular Mechanisms of Apoptosis Induction and Its Regulation by Fatty Acids in Pancreatic β-Cells. Int. J. Mol. Sci. 2021, 22, 4285. [Google Scholar] [CrossRef]
- Li, Q.; Liu, Y.; Sun, M. Autophagy and Alzheimer’s Disease. Cell Mol. Neurobiol. 2017, 37, 377–388. [Google Scholar] [CrossRef]
- Las, G.; Shirihai, O.S. The Role of Autophagy in β-Cell Lipotoxicity and Type 2 Diabetes. Diabetes Obes. Metab. 2010, 12, 15–19. [Google Scholar] [CrossRef]
- Chen, Y.Y.; Sun, L.Q.; Wang, B.A.; Zou, X.M.; Mu, Y.M.; Lu, J.M. Palmitate Induces Autophagy in Pancreatic β-Cells via Endoplasmic Reticulum Stress and Its Downstream JNK Pathway. Int. J. Mol. Med. 2013, 32, 1401–1406. [Google Scholar] [CrossRef] [Green Version]
- Ebato, C.; Uchida, T.; Arakawa, M.; Komatsu, M.; Ueno, T.; Komiya, K.; Azuma, K.; Hirose, T.; Tanaka, K.; Kominami, E.; et al. Autophagy Is Important in Islet Homeostasis and Compensatory Increase of Beta Cell Mass in Response to High-Fat Diet. Cell Metab. 2008, 8, 325–332. [Google Scholar] [CrossRef] [Green Version]
- Doria, M.; Nury, T.; Delmas, D.; Moreau, T.; Lizard, G.; Vejux, A. Protective Function of Autophagy during VLCFA-Induced Cytotoxicity in a Neurodegenerative Cell Model. Free Radic. Biol. Med. 2019, 137, 46–58. [Google Scholar] [CrossRef]
- Michailidis, M.; Tata, D.A.; Moraitou, D.; Kavvadas, D.; Karachrysafi, S.; Papamitsou, T.; Vareltzis, P.; Papaliagkas, V. Antidiabetic Drugs in the Treatment of Alzheimer’s Disease. Int. J. Mol. Sci. 2022, 23, 4641. [Google Scholar] [CrossRef]
- Femminella, G.D.; Bencivenga, L.; Petraglia, L.; Visaggi, L.; Gioia, L.; Grieco, F.V.; De Lucia, C.; Komici, K.; Corbi, G.; Edison, P.; et al. Antidiabetic Drugs in Alzheimer’s Disease: Mechanisms of Action and Future Perspectives. J. Diabetes Res. 2017, 2017, 7420796. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Logroscino, G.; Kang, J.H.; Grodstein, F. Prospective Study of Type 2 Diabetes and Cognitive Decline in Women Aged 70–81 Years. BMJ 2004, 328, 548–551. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scherer, T.; Sakamoto, K.; Buettner, C. Brain Insulin Signalling in Metabolic Homeostasis and Disease. Nat. Rev. Endocrinol. 2021, 17, 468–483. [Google Scholar] [CrossRef] [PubMed]
- Freiherr, J.; Hallschmid, M.; Frey, W.H.; Brünner, Y.F.; Chapman, C.D.; Hölscher, C.; Craft, S.; De Felice, F.G.; Benedict, C. Intranasal Insulin as a Treatment for Alzheimer’s Disease: A Review of Basic Research and Clinical Evidence. CNS Drugs 2013, 27, 505–514. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, Y.; Zhang, J.; Zhang, B.; Gong, C.X. Targeting Insulin Signaling for the Treatment of Alzheimer’s Disease. Curr. Top Med. Chem. 2016, 16, 485–492. [Google Scholar] [CrossRef]
- Craft, S.; Asthana, S.; Newcomer, J.W.; Wilkinson, C.W.; Tio Matos, I.; Baker, L.D.; Cherrier, M.; Lofgreen, C.; Latendresse, S.; Petrova, A.; et al. Enhancement of Memory in Alzheimer Disease with Insulin and Somatostatin, but Not Glucose. Arch Gen Psychiatry 1999, 56, 1135–1140. [Google Scholar] [CrossRef] [Green Version]
- Reger, M.A.; Watson, G.S.; Green, P.S.; Baker, L.D.; Cholerton, B.; Fishel, M.A.; Plymate, S.R.; Cherrier, M.M.; Schellenberg, G.D.; Frey, W.H.; et al. Intranasal Insulin Administration Dose-Dependently Modulates Verbal Memory and Plasma Amyloid-Beta in Memory-Impaired Older Adults. J. Alzheimers Dis. 2008, 13, 323–331. [Google Scholar] [CrossRef]
- Craft, S.; Baker, L.D.; Montine, T.J.; Minoshima, S.; Watson, G.S.; Claxton, A.; Arbuckle, M.; Callaghan, M.; Tsai, E.; Plymate, S.R.; et al. Intranasal Insulin Therapy for Alzheimer Disease and Amnestic Mild Cognitive Impairment: A Pilot Clinical Trial. Arch. Neurol. 2012, 69, 29–38. [Google Scholar] [CrossRef] [Green Version]
- De La Monte, S.M. Intranasal Insulin Therapy for Cognitive Impairment and Neurodegeneration: Current State of the Art. Expert Opin. Drug Deliv. 2013, 10, 1699–1709. [Google Scholar] [CrossRef] [Green Version]
- Li, H.Q.; Wang, B.P.; Deng, X.L.; Zhang, J.Y.; Wang, Y.B.; Zheng, J.; Xia, W.F.; Zeng, T.S.; Chen, L.L. Insulin Improves β-Cell Function in Glucose-Intolerant Rat Models Induced by Feeding a High-Fat Diet. Metabolism 2011, 60, 1566–1574. [Google Scholar] [CrossRef]
- Tang, S.; Wu, W.; Tang, W.; Ge, Z.; Wang, H.; Hong, T.; Zhu, D.; Bi, Y. Suppression of Rho-Kinase 1 Is Responsible for Insulin Regulation of the AMPK/SREBP-1c Pathway in Skeletal Muscle Cells Exposed to Palmitate. Acta Diabetol. 2017, 54, 635–644. [Google Scholar] [CrossRef] [PubMed]
- Cork, S.C.; Richards, J.E.; Holt, M.K.; Gribble, F.M.; Reimann, F.; Trapp, S. Distribution and Characterisation of Glucagon-like Peptide-1 Receptor Expressing Cells in the Mouse Brain. Mol. Metab. 2015, 4, 718–731. [Google Scholar] [CrossRef] [Green Version]
- Liu, X.Y.; Zhang, N.; Zhang, S.X.; Xu, P. Potential New Therapeutic Target for Alzheimer’s Disease: Glucagon-like Peptide-1. Eur. J. Neurosci. 2021, 54, 7749–7769. [Google Scholar] [CrossRef] [PubMed]
- Cheng, D.; Yang, S.; Zhao, X.; Wang, G. The Role of Glucagon-Like Peptide-1 Receptor Agonists (GLP-1 RA) in Diabetes-Related Neurodegenerative Diseases. Drug Des. Devel Ther. 2022, 16, 665–684. [Google Scholar] [CrossRef] [PubMed]
- Mullins, R.J.; Mustapic, M.; Chia, C.W.; Carlson, O.; Gulyani, S.; Tran, J.; Li, Y.; Mattson, M.P.; Resnick, S.; Egan, J.M.; et al. A Pilot Study of Exenatide Actions in Alzheimer’s Disease. Curr. Alzheimer Res. 2019, 16, 741–752. [Google Scholar] [CrossRef] [PubMed]
- Gejl, M.; Brock, B.; Egefjord, L.; Vang, K.; Rungby, J.; Gjedde, A. Blood-Brain Glucose Transfer in Alzheimer’s Disease: Effect of GLP-1 Analog Treatment. Sci. Rep. 2017, 7, 17490. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Watson, K.T.; Wroolie, T.E.; Tong, G.; Foland-Ross, L.C.; Frangou, S.; Singh, M.; McIntyre, R.S.; Roat-Shumway, S.; Myoraku, A.; Reiss, A.L.; et al. Neural Correlates of Liraglutide Effects in Persons at Risk for Alzheimer’s Disease. Behav. Brain Res. 2019, 356, 271–278. [Google Scholar] [CrossRef] [PubMed]
- Gejl, M.; Gjedde, A.; Egefjord, L.; Møller, A.; Hansen, S.B.; Vang, K.; Rodell, A.; Brændgaard, H.; Gottrup, H.; Schacht, A.; et al. In Alzheimer’s Disease, 6-Month Treatment with GLP-1 Analog Prevents Decline of Brain Glucose Metabolism: Randomized, Placebo-Controlled, Double-Blind Clinical Trial. Front Aging Neurosci. 2016, 8, 108. [Google Scholar] [CrossRef]
- Marrano, N.; Biondi, G.; Borrelli, A.; Cignarelli, A.; Perrini, S.; Laviola, L.; Giorgino, F.; Natalicchio, A. Irisin and Incretin Hormones: Similarities, Differences, and Implications in Type 2 Diabetes and Obesity. Biomolecules 2021, 11, 286. [Google Scholar] [CrossRef]
- Marrano, N.; Biondi, G.; Cignarelli, A.; Perrini, S.; Laviola, L.; Giorgino, F.; Natalicchio, A. Functional Loss of Pancreatic Islets in Type 2 Diabetes: How Can We Halt It? Metabolism 2020, 110, 154304. [Google Scholar] [CrossRef]
- Jo, D.; Yoon, G.; Song, J. Role of Exendin-4 in Brain Insulin Resistance, Mitochondrial Function, and Neurite Outgrowth in Neurons under Palmitic Acid-Induced Oxidative Stress. Antioxidants 2021, 10, 78. [Google Scholar] [CrossRef] [PubMed]
- Almaguel, F.G.; Liu, J.W.; Pacheco, F.J.; Casiano, C.A.; De Leon, M. Activation and Reversal of Lipotoxicity in PC12 and Rat Cortical Cells Following Exposure to Palmitic Acid. J. Neurosci. Res. 2009, 87, 1207–1218. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bomba, M.; Granzotto, A.; Castelli, V.; Onofrj, M.; Lattanzio, R.; Cimini, A.; Sensi, S.L. Exenatide Reverts the High-Fat-Diet-Induced Impairment of BDNF Signaling and Inflammatory Response in an Animal Model of Alzheimer’s Disease. J. Alzheimers Dis. 2019, 70, 793–810. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.; Yoon, G.; Song, J.; Jo, J. Exendin-4 Improves Long-Term Potentiation and Neuronal Dendritic Growth in Vivo and in Vitro Obesity Condition. Sci. Rep. 2021, 11, 8326. [Google Scholar] [CrossRef]
- Zhu, C.G.; Luo, Y.; Wang, H.; Li, J.Y.; Yang, J.; Liu, Y.X.; Qu, H.Q.; Wang, B.L.; Zhu, M. Liraglutide Ameliorates Lipotoxicity-Induced Oxidative Stress by Activating the NRF2 Pathway in HepG2 Cells. Horm. Metab. Res. 2020, 52, 532–539. [Google Scholar] [CrossRef]
- Ao, N.; Ma, Z.; Yang, J.; Jin, S.; Zhang, K.; Luo, E.; Du, J. Liraglutide Ameliorates Lipotoxicity-Induced Inflammation through the MTORC1 Signalling Pathway. Peptides 2020, 133, 170375. [Google Scholar] [CrossRef]
- Somm, E.; Montandon, S.A.; Loizides-Mangold, U.; Gaïa, N.; Lazarevic, V.; de Vito, C.; Perroud, E.; Bochaton-Piallat, M.L.; Dibner, C.; Schrenzel, J.; et al. The GLP-1R Agonist Liraglutide Limits Hepatic Lipotoxicity and Inflammatory Response in Mice Fed a Methionine-Choline Deficient Diet. Transl. Res. 2021, 227, 75–88. [Google Scholar] [CrossRef]
- Leonardini, A.; D’Oria, R.; Incalza, M.A.; Caccioppoli, C.; Buccheri, V.A.; Cignarelli, A.; Paparella, D.; Margari, V.; Natalicchio, A.; Perrini, S.; et al. GLP-1 Receptor Activation Inhibits Palmitate-Induced Apoptosis via Ceramide in Human Cardiac Progenitor Cells. J. Clin. Endocrinol. Metab. 2017, 102, 4136–4147. [Google Scholar] [CrossRef]
- Costes, S.; Bertrand, G.; Ravier, M.A. Mechanisms of Beta-Cell Apoptosis in Type 2 Diabetes-Prone Situations and Potential Protection by GLP-1-Based Therapies. Int. J. Mol. Sci. 2021, 22, 5303. [Google Scholar] [CrossRef]
- Draznin, B.; Aroda, V.R.; Bakris, G.; Benson, G.; Brown, F.M.; Freeman, R.; Green, J.; Huang, E.; Isaacs, D.; American Diabetes Association Professional Practice Committee. 9. Pharmacologic Approaches to Glycemic Treatment: Standards of Medical Care in Diabetes-2022. Diabetes Care 2022, 45, S125–S143. [Google Scholar] [CrossRef]
- Bhatti, G.K.; Reddy, A.P.; Reddy, P.H.; Bhatti, J.S. Lifestyle Modifications and Nutritional Interventions in Aging-Associated Cognitive Decline and Alzheimer’s Disease. Front Aging Neurosci. 2020, 11, 369. [Google Scholar] [CrossRef] [PubMed]
- Boström, P.; Wu, J.; Jedrychowski, M.P.; Korde, A.; Ye, L.; Lo, J.C.; Rasbach, K.A.; Boström, E.A.; Choi, J.H.; Long, J.Z.; et al. A PGC1-α-Dependent Myokine That Drives Brown-Fat-like Development of White Fat and Thermogenesis. Nature 2012, 481, 463–468. [Google Scholar] [CrossRef] [Green Version]
- Natalicchio, A.; Marrano, N.; Biondi, G.; Spagnuolo, R.; Labarbuta, R.; Porreca, I.; Cignarelli, A.; Bugliani, M.; Marchetti, P.; Perrini, S.; et al. The Myokine Irisin Is Released in Response to Saturated Fatty Acids and Promotes Pancreatic β-Cell Survival and Insulin Secretion. Diabetes 2017, 66, 2849–2856. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Young, M.F.; Valaris, S.; Wrann, C.D. A Role for FNDC5/Irisin in the Beneficial Effects of Exercise on the Brain and in Neurodegenerative Diseases. Prog Cardiovasc. Dis. 2019, 62, 172–178. [Google Scholar] [CrossRef] [PubMed]
- Natalicchio, A.; Marrano, N.; Biondi, G.; Dipaola, L.; Spagnuolo, R.; Cignarelli, A.; Perrini, S.; Laviola, L.; Giorgino, F. Irisin Increases the Expression of Anorexigenic and Neurotrophic Genes in Mouse Brain. Diabetes Metab. Res. Rev. 2019, 36, e3238. [Google Scholar] [CrossRef] [PubMed]
- Kim, O.Y.; Song, J. The Role of Irisin in Alzheimer’s Disease. J. Clin. Med. 2018, 7, 407. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lourenco, M.V.; Frozza, R.L.; de Freitas, G.B.; Zhang, H.; Kincheski, G.C.; Ribeiro, F.C.; Gonçalves, R.A.; Clarke, J.R.; Beckman, D.; Staniszewski, A.; et al. Exercise-Linked FNDC5/Irisin Rescues Synaptic Plasticity and Memory Defects in Alzheimer’s Models. Nat. Med. 2019, 25, 165–175. [Google Scholar] [CrossRef]
- Islam, M.R.; Valaris, S.; Young, M.F.; Haley, E.B.; Luo, R.; Bond, S.F.; Mazuera, S.; Kitchen, R.R.; Caldarone, B.J.; Bettio, L.E.B.; et al. Exercise Hormone Irisin Is a Critical Regulator of Cognitive Function. Nat. Metab. 2021, 3, 1058–1070. [Google Scholar] [CrossRef]
- Madhu, L.N.; Somayaji, Y.; Shetty, A.K. Promise of Irisin to Attenuate Cognitive Dysfunction in Aging and Alzheimer’s Disease. Ageing Res. Rev. 2022, 78, 101637. [Google Scholar] [CrossRef]
- Song, R.; Zhao, X.; Zhang, D.Q.; Wang, R.; Feng, Y. Lower Levels of Irisin in Patients with Type 2 Diabetes Mellitus: A Meta-Analysis. Diabetes Res. Clin. Pract 2021, 175, 108788. [Google Scholar] [CrossRef]
- Toi, P.L.; Anothaisintawee, T.; Chaikledkaew, U.; Briones, J.R.; Reutrakul, S.; Thakkinstian, A. Preventive Role of Diet Interventions and Dietary Factors in Type 2 Diabetes Mellitus: An Umbrella Review. Nutrients 2020, 12, 2722. [Google Scholar] [CrossRef] [PubMed]
- Coelho, O.G.L.; da Silva, B.P.; Rocha, D.M.U.P.; Lopes, L.L.; Alfenas, R.C.G. Polyunsaturated Fatty Acids and Type 2 Diabetes: Impact on the Glycemic Control Mechanism. Crit. Rev. Food Sci. Nutr. 2017, 57, 3614–3619. [Google Scholar] [CrossRef] [PubMed]
- Salas-Salvadó, J.; Bulló, M.; Estruch, R.; Ros, E.; Covas, M.I.; Ibarrola-Jurado, N.; Corella, D.; Arós, F.; Gómez-Gracia, E.; Ruiz-Gutiérrez, V.; et al. Prevention of Diabetes with Mediterranean Diets: A Subgroup Analysis of a Randomized Trial. Ann. Intern. Med. 2014, 160, 1–10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pivari, F.; Mingione, A.; Brasacchio, C.; Soldati, L. Curcumin and Type 2 Diabetes Mellitus: Prevention and Treatment. Nutrients 2019, 11, 1837. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marrano, N.; Spagnuolo, R.; Biondi, G.; Cignarelli, A.; Perrini, S.; Vincenti, L.; Laviola, L.; Giorgino, F.; Natalicchio, A. Effects of Extra Virgin Olive Oil Polyphenols on Beta-Cell Function and Survival. Plants 2021, 10, 286. [Google Scholar] [CrossRef] [PubMed]
- Blahova, J.; Martiniakova, M.; Babikova, M.; Kovacova, V.; Mondockova, V.; Omelka, R. Pharmaceutical Drugs and Natural Therapeutic Products for the Treatment of Type 2 Diabetes Mellitus. Pharmaceuticals 2021, 14, 806. [Google Scholar] [CrossRef] [PubMed]
- Román, G.C.; Jackson, R.E.; Gadhia, R.; Román, A.N.; Reis, J. Mediterranean Diet: The Role of Long-Chain ω-3 Fatty Acids in Fish; Polyphenols in Fruits, Vegetables, Cereals, Coffee, Tea, Cacao and Wine; Probiotics and Vitamins in Prevention of Stroke, Age-Related Cognitive Decline, and Alzheimer Disease. Rev. Neurol. 2019, 175, 724–741. [Google Scholar] [CrossRef]
- Chainoglou, E.; Hadjipavlou-Litina, D. Curcumin in Health and Diseases: Alzheimer’s Disease and Curcumin Analogues, Derivatives, and Hybrids. Int. J. Mol. Sci. 2020, 21, 1975. [Google Scholar] [CrossRef] [Green Version]
- Salehi, B.; Venditti, A.; Sharifi-Rad, M.; Kręgiel, D.; Sharifi-Rad, J.; Durazzo, A.; Lucarini, M.; Santini, A.; Souto, E.B.; Novellino, E.; et al. The Therapeutic Potential of Apigenin. Int. J. Mol. Sci. 2019, 20, 1305. [Google Scholar] [CrossRef] [Green Version]
- Yan, Y.; Yang, H.; Xie, Y.; Ding, Y.; Kong, D.; Yu, H. Research Progress on Alzheimer’s Disease and Resveratrol. Neurochem Res. 2020, 45, 989–1006. [Google Scholar] [CrossRef]
- Khan, H.; Ullah, H.; Aschner, M.; Cheang, W.S.; Akkol, E.K. Neuroprotective Effects of Quercetin in Alzheimer’s Disease. Biomolecules 2019, 10, 59. [Google Scholar] [CrossRef] [PubMed]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Marrano, N.; Biondi, G.; Borrelli, A.; Rella, M.; Zambetta, T.; Di Gioia, L.; Caporusso, M.; Logroscino, G.; Perrini, S.; Giorgino, F.; et al. Type 2 Diabetes and Alzheimer’s Disease: The Emerging Role of Cellular Lipotoxicity. Biomolecules 2023, 13, 183. https://doi.org/10.3390/biom13010183
Marrano N, Biondi G, Borrelli A, Rella M, Zambetta T, Di Gioia L, Caporusso M, Logroscino G, Perrini S, Giorgino F, et al. Type 2 Diabetes and Alzheimer’s Disease: The Emerging Role of Cellular Lipotoxicity. Biomolecules. 2023; 13(1):183. https://doi.org/10.3390/biom13010183
Chicago/Turabian StyleMarrano, Nicola, Giuseppina Biondi, Anna Borrelli, Martina Rella, Tommaso Zambetta, Ludovico Di Gioia, Mariangela Caporusso, Giancarlo Logroscino, Sebastio Perrini, Francesco Giorgino, and et al. 2023. "Type 2 Diabetes and Alzheimer’s Disease: The Emerging Role of Cellular Lipotoxicity" Biomolecules 13, no. 1: 183. https://doi.org/10.3390/biom13010183