
Phosphatidylinositol Monophosphates Regulate the Membrane Localization of HSPA1A, a Stress-Inducible 70-kDa Heat Shock Protein

 , ,
, ,  and
and 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
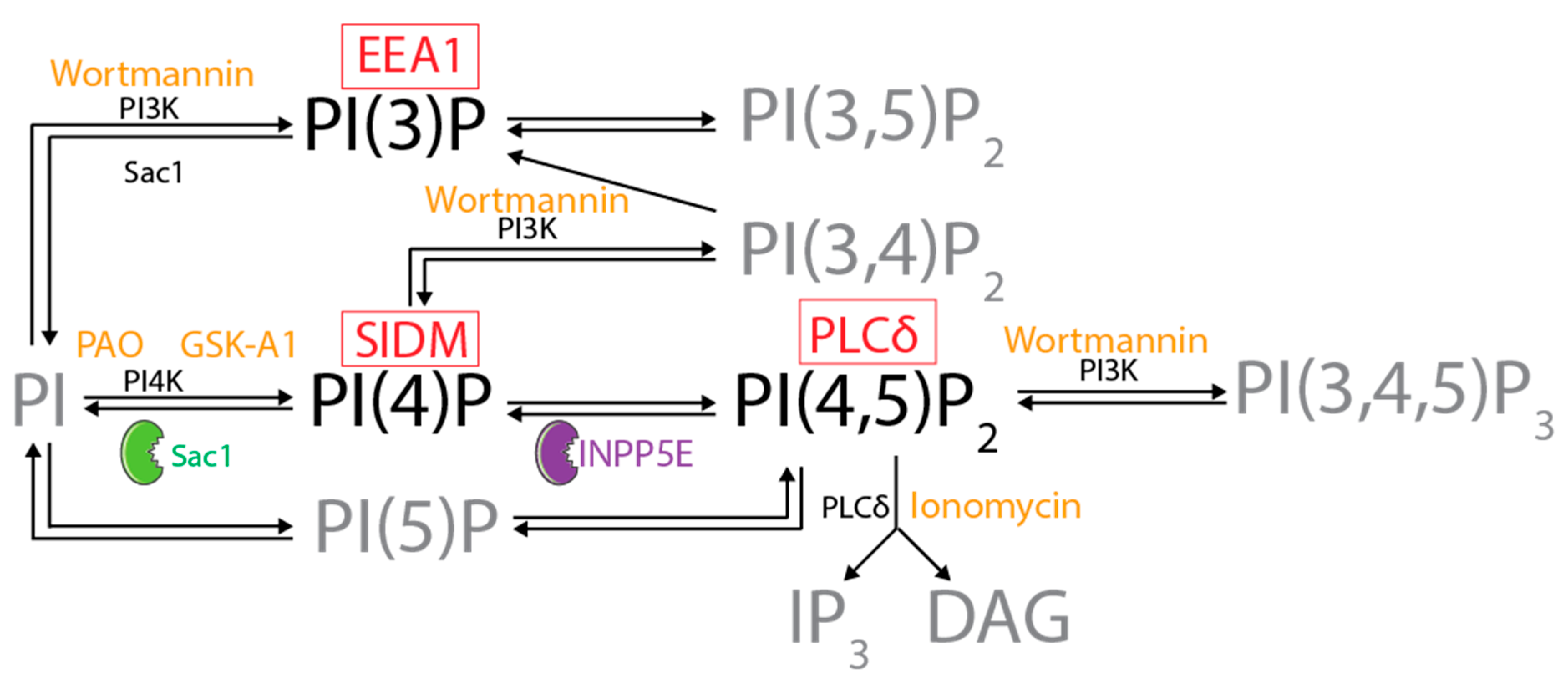
:1. Introduction
2. Materials and Methods
2.1. Plasmids Used in This Study
2.2. Cell Culture, Transfection, and Treatments
2.3. Confocal Microscopy and Image Analysis
2.4. Cell Surface Biotinylation and Total PM Protein Isolation
2.5. Statistical Tests
3. Results
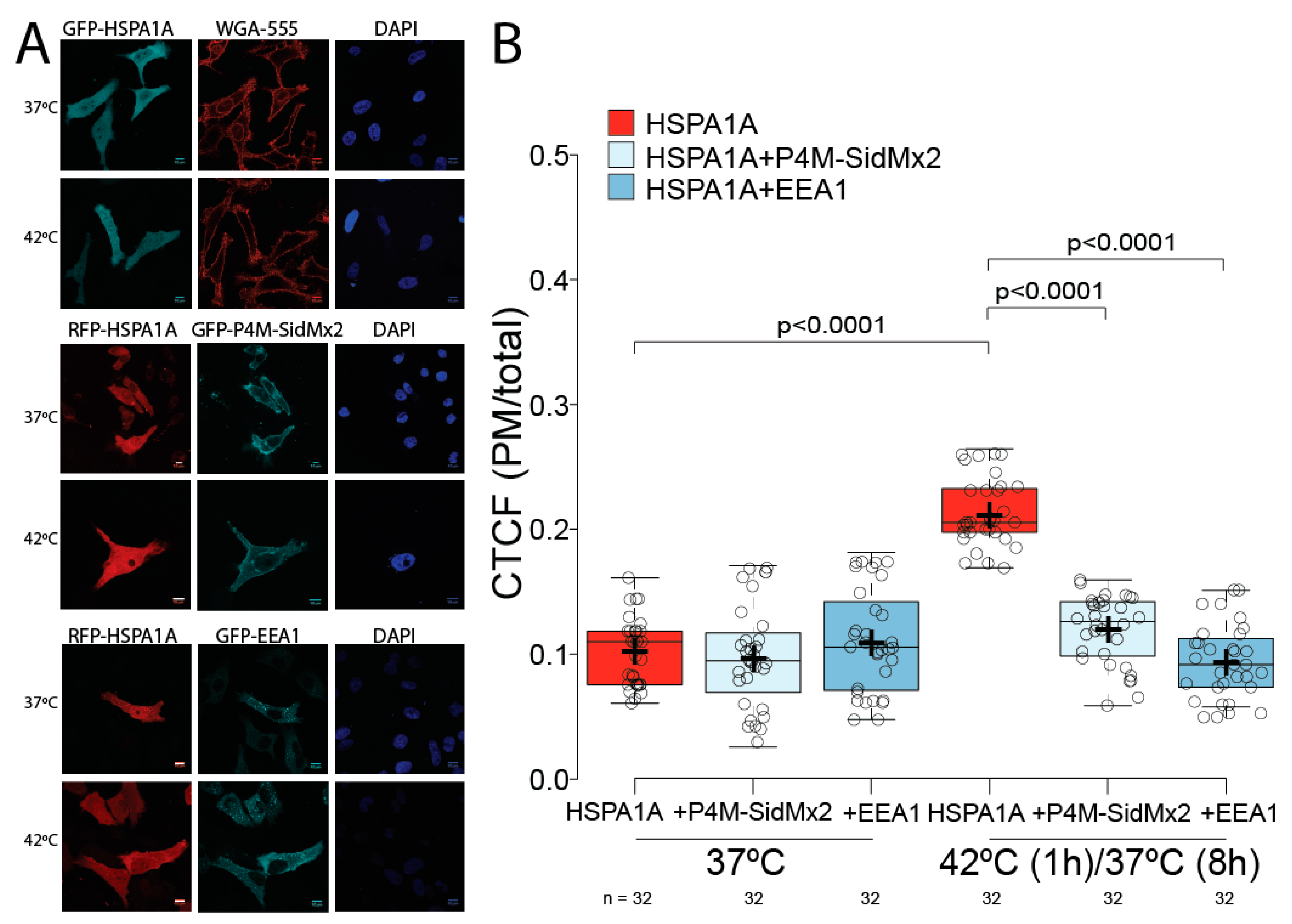
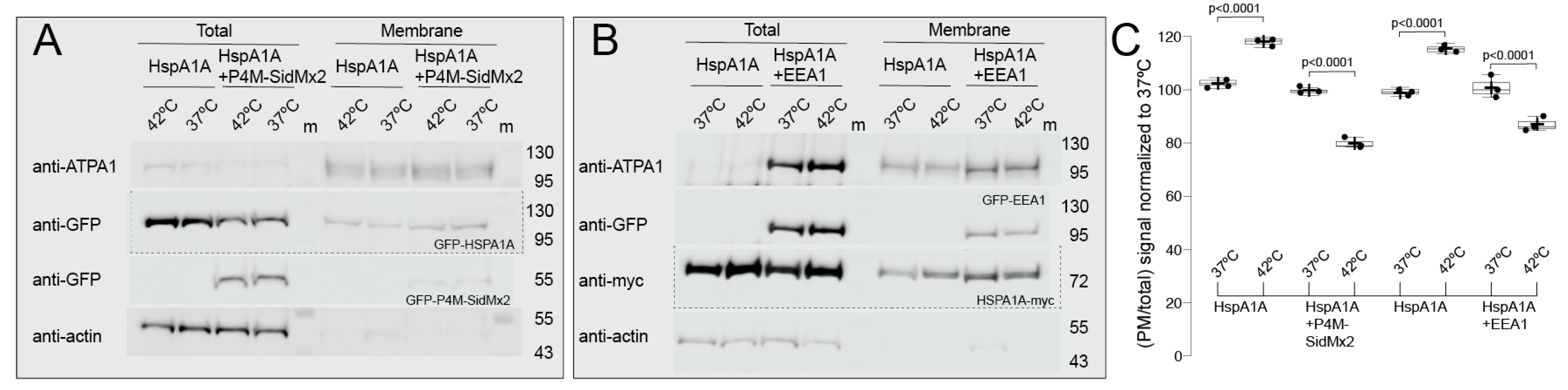
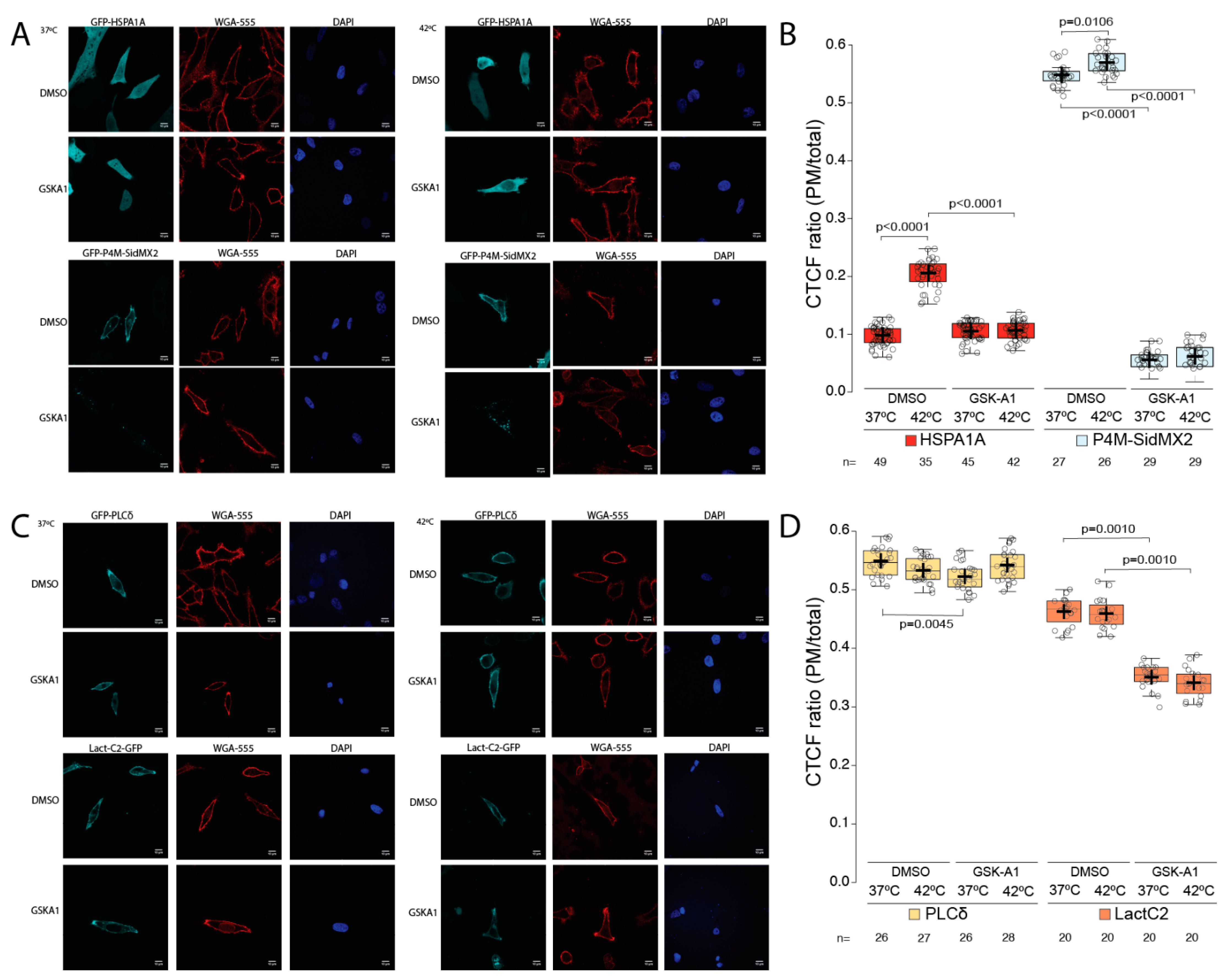
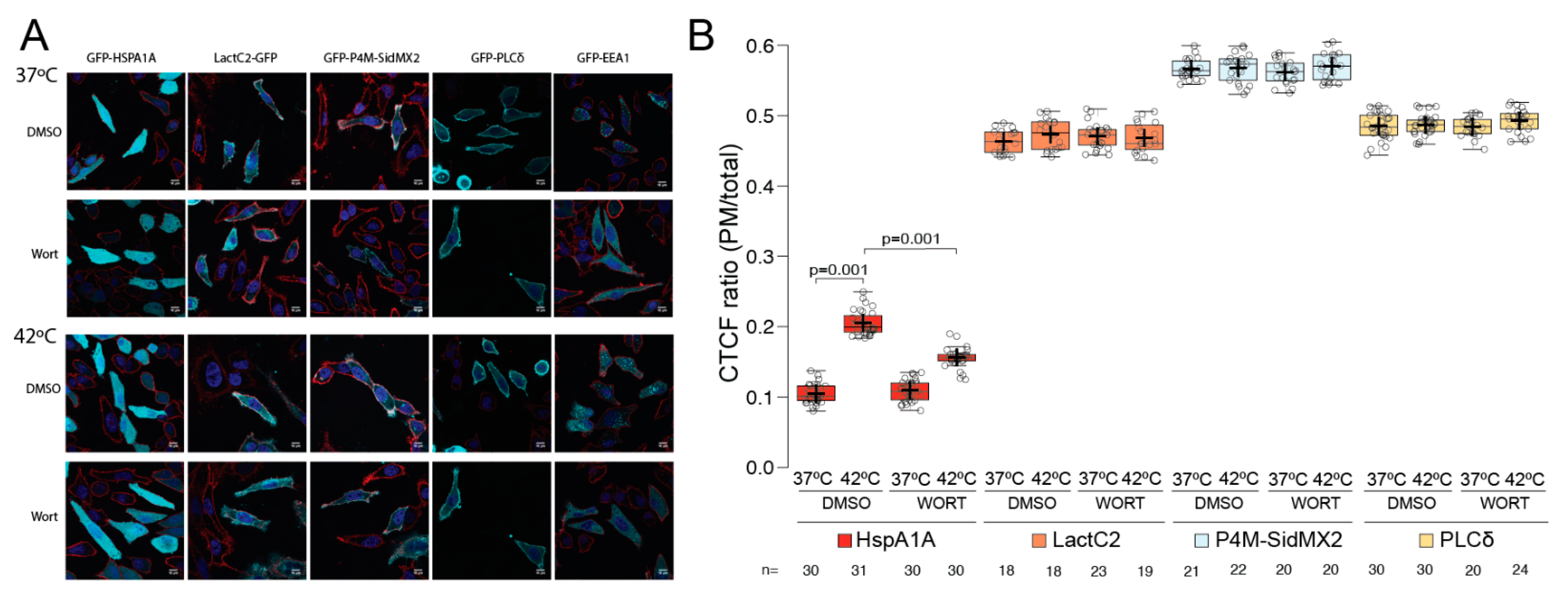
3.1. HSPA1A’s PM Localization Is Significantly Reduced by the Presence of P4M-SidMx2 and EEA1
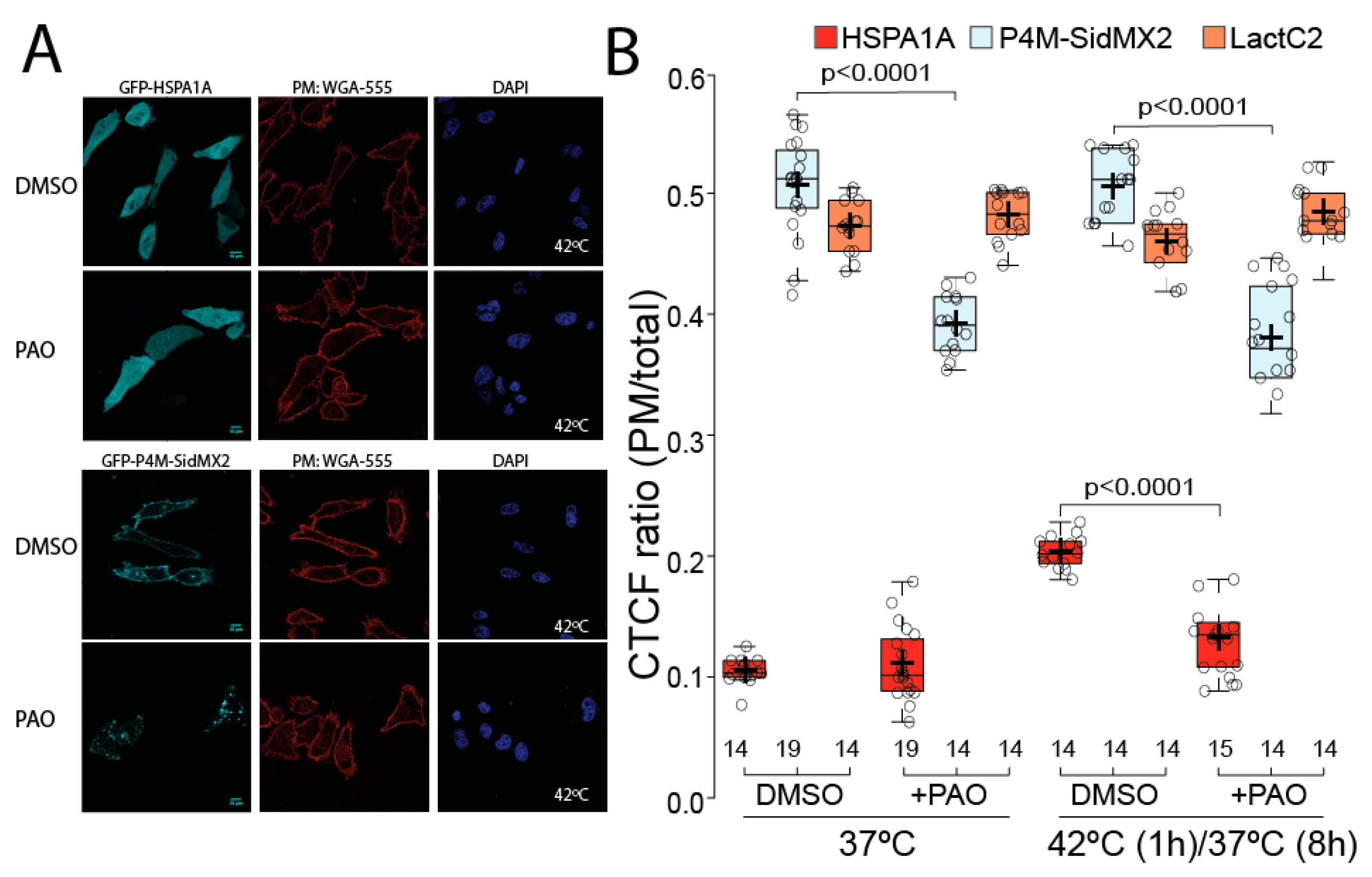
3.2. Pharmacological PIP Depletion Reduces HSPA1A’s PM Localization
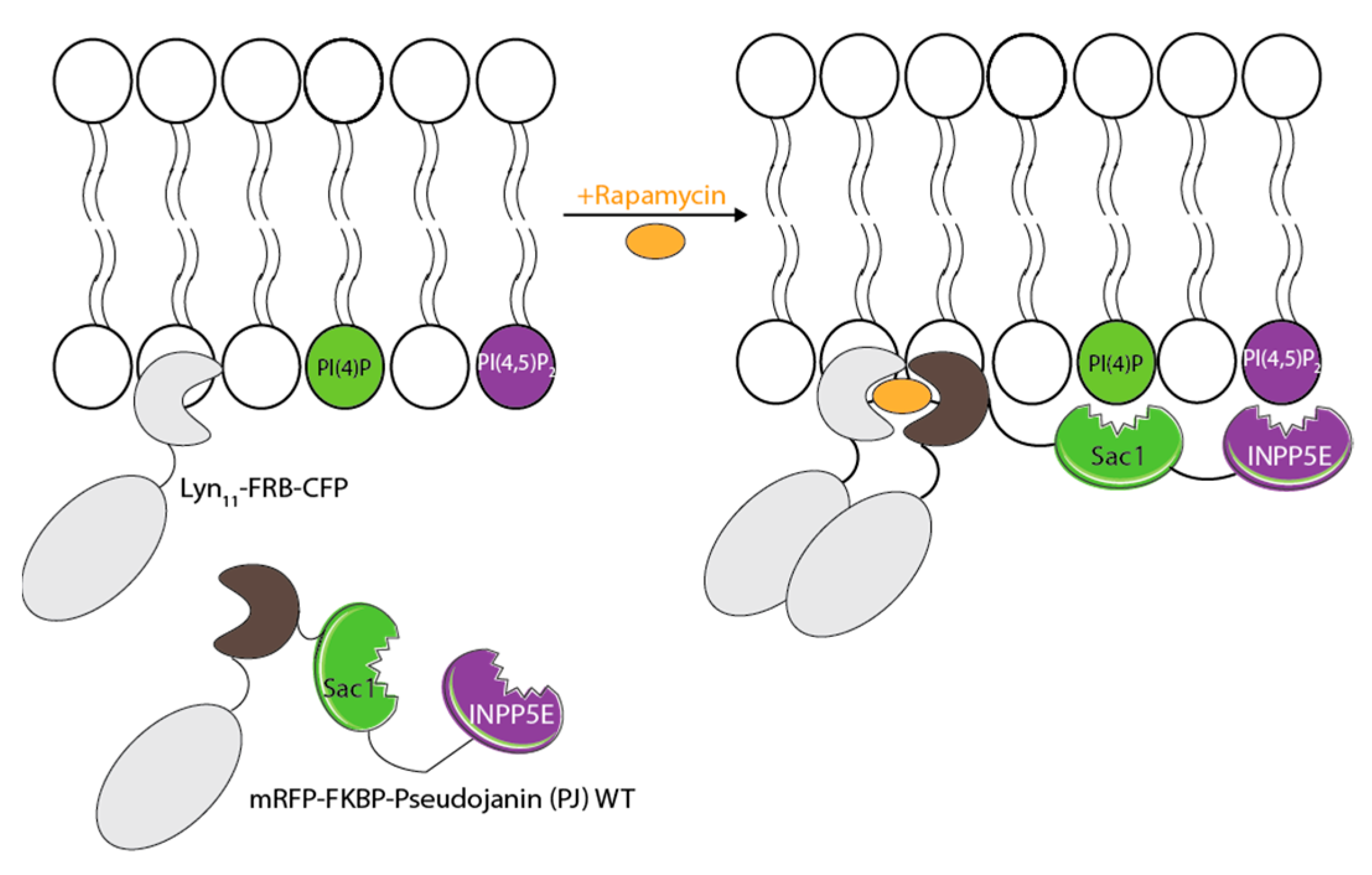
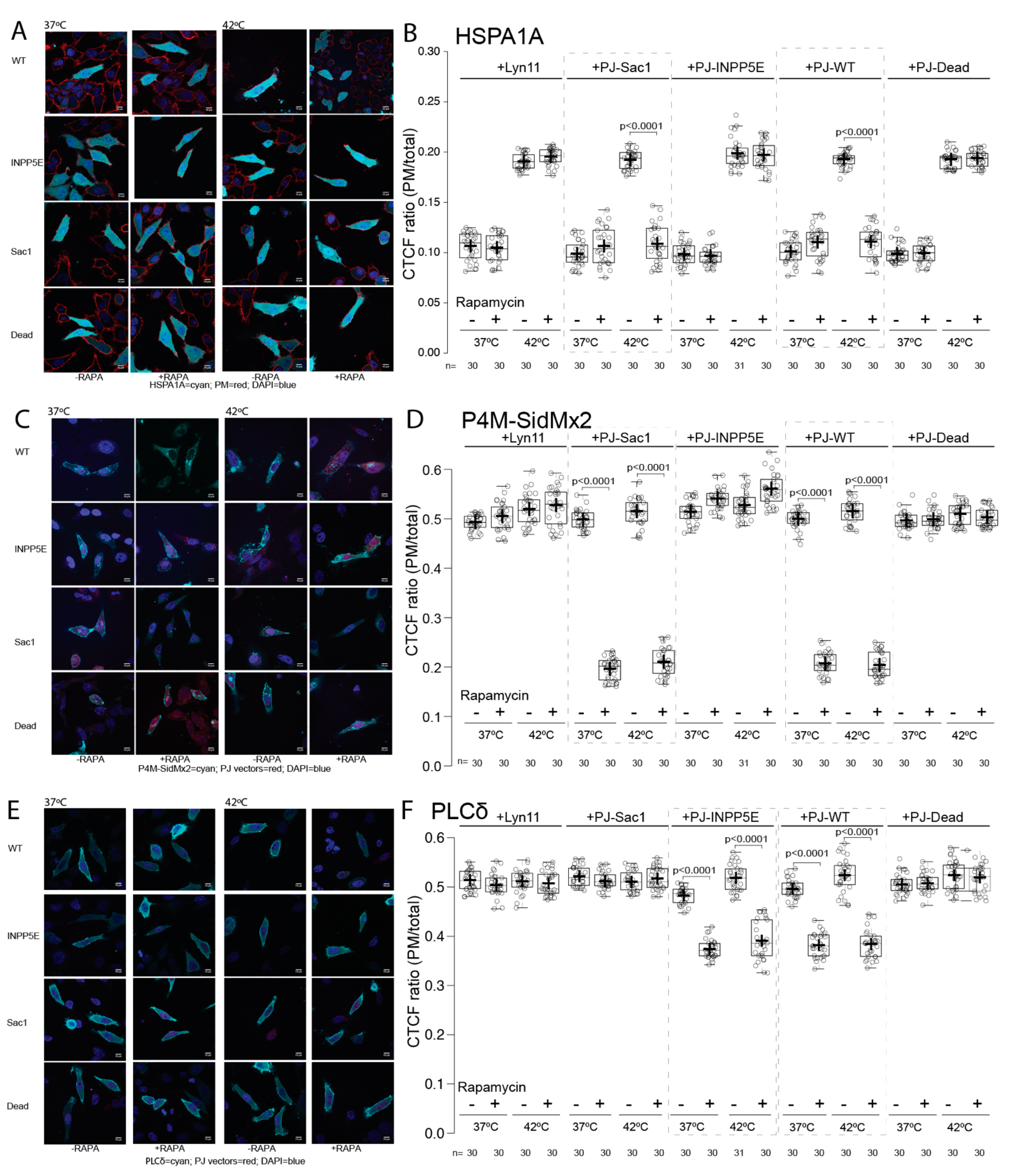
3.3. Phosphatase PIP Depletion Reduces HSPA1A’s PM Localization
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Lindquist, S.; Craig, E.A. The heat-shock proteins. Annu. Rev. Genet. 1988, 22, 631–677. [Google Scholar] [CrossRef] [PubMed]
- Kultz, D. Molecular and evolutionary basis of the cellular stress response. Annu. Rev. Physiol. 2005, 67, 225–257. [Google Scholar] [CrossRef] [PubMed]
- Albakova, Z.; Armeev, G.A.; Kanevskiy, L.M.; Kovalenko, E.I.; Sapozhnikov, A.M. HSP70 Multi-Functionality in Cancer. Cells 2020, 9, 587. [Google Scholar] [CrossRef] [Green Version]
- Elmallah, M.I.Y.; Cordonnier, M.; Vautrot, V.; Chanteloup, G.; Garrido, C.; Gobbo, J. Membrane-anchored heat-shock protein 70 (Hsp70) in cancer. Cancer Lett. 2020, 469, 134–141. [Google Scholar] [CrossRef]
- Vostakolaei, M.A.; Abdolalizadeh, J.; Hejazi, M.S.; Kordi, S.; Molavi, O. Hsp70 in Cancer: Partner or Traitor to Immune System. Iran J. Allergy Asthma Immunol. 2019, 18, 589–604. [Google Scholar] [CrossRef]
- Balogi, Z.; Multhoff, G.; Jensen, T.K.; Lloyd-Evans, E.; Yamashima, T.; Jaattela, M.; Harwood, J.L.; Vigh, L. Hsp70 interactions with membrane lipids regulate cellular functions in health and disease. Prog. Lipid Res. 2019, 74, 18–30. [Google Scholar] [CrossRef]
- Murakami, N.; Kuhnel, A.; Schmid, T.E.; Ilicic, K.; Stangl, S.; Braun, I.S.; Gehrmann, M.; Molls, M.; Itami, J.; Multhoff, G. Role of membrane Hsp70 in radiation sensitivity of tumor cells. Radiat. Oncol. 2015, 10, 149. [Google Scholar] [CrossRef] [Green Version]
- Gehrmann, M.; Liebisch, G.; Schmitz, G.; Anderson, R.; Steinem, C.; De Maio, A.; Pockley, G.; Multhoff, G. Tumor-specific Hsp70 plasma membrane localization is enabled by the glycosphingolipid Gb3. PLoS ONE 2008, 3, e1925. [Google Scholar] [CrossRef] [Green Version]
- Fellinger, H.; Stangl, S.; Hernandez Schnelzer, A.; Schwab, M.; Di Genio, T.; Pieper, M.; Werner, C.; Shevtsov, M.; Haller, B.; Multhoff, G. Time- and Dose-Dependent Effects of Ionizing Irradiation on the Membrane Expression of Hsp70 on Glioma Cells. Cells 2020, 9, 912. [Google Scholar] [CrossRef] [Green Version]
- Vega, V.L.; Rodriguez-Silva, M.; Frey, T.; Gehrmann, M.; Diaz, J.C.; Steinem, C.; Multhoff, G.; Arispe, N.; De Maio, A. Hsp70 translocates into the plasma membrane after stress and is released into the extracellular environment in a membrane-associated form that activates macrophages. J. Immunol. 2008, 180, 4299–4307. [Google Scholar] [CrossRef] [Green Version]
- Multhoff, G. Heat shock protein 70 (Hsp70): Membrane location, export and immunological relevance. Methods 2007, 43, 229–237. [Google Scholar] [CrossRef] [PubMed]
- Mambula, S.S.; Stevenson, M.A.; Ogawa, K.; Calderwood, S.K. Mechanisms for Hsp70 secretion: Crossing membranes without a leader. Methods 2007, 43, 168–175. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Maio, A. Extracellular Hsp70: Export and function. Curr. Protein Pept. Sci. 2014, 15, 225–231. [Google Scholar] [CrossRef] [PubMed]
- Tytell, M. Release of heat shock proteins (Hsps) and the effects of extracellular Hsps on neural cells and tissues. Int. J. Hyperthermia 2005, 21, 445–455. [Google Scholar] [CrossRef] [PubMed]
- Schilling, D.; Gehrmann, M.; Steinem, C.; De Maio, A.; Pockley, A.G.; Abend, M.; Molls, M.; Multhoff, G. Binding of heat shock protein 70 to extracellular phosphatidylserine promotes killing of normoxic and hypoxic tumor cells. FASEB J. 2009, 23, 2467–2477. [Google Scholar] [CrossRef] [Green Version]
- Schmitt, E.; Gehrmann, M.; Brunet, M.; Multhoff, G.; Garrido, C. Intracellular and extracellular functions of heat shock proteins: Repercussions in cancer therapy. J. Leukoc Biol. 2007, 81, 15–27. [Google Scholar] [CrossRef]
- Calderwood, S.K.; Mambula, S.S.; Gray, P.J., Jr. Extracellular heat shock proteins in cell signaling and immunity. Ann. N. Y. Acad. Sci. 2007, 1113, 28–39. [Google Scholar] [CrossRef]
- Breuninger, S.; Stangl, S.; Werner, C.; Sievert, W.; Lobinger, D.; Foulds, G.A.; Wagner, S.; Pickhard, A.; Piontek, G.; Kokowski, K.; et al. Membrane Hsp70-A Novel Target for the Isolation of Circulating Tumor Cells After Epithelial-to-Mesenchymal Transition. Front. Oncol. 2018, 8, 497. [Google Scholar] [CrossRef] [Green Version]
- Shevtsov, M.; Sato, H.; Multhoff, G.; Shibata, A. Novel Approaches to Improve the Efficacy of Immuno-Radiotherapy. Front. Oncol. 2019, 9, 156. [Google Scholar] [CrossRef] [Green Version]
- Shevtsov, M.; Multhoff, G.; Mikhaylova, E.; Shibata, A.; Guzhova, I.; Margulis, B. Combination of Anti-Cancer Drugs with Molecular Chaperone Inhibitors. Int. J. Mol. Sci. 2019, 20, 284. [Google Scholar] [CrossRef] [Green Version]
- Bilog, A.D.; Smulders, L.; Oliverio, R.; Labanieh, C.; Zapanta, J.; Stahelin, R.V.; Nikolaidis, N. Membrane Localization of HspA1A, a Stress Inducible 70-kDa Heat-Shock Protein, Depends on Its Interaction with Intracellular Phosphatidylserine. Biomolecules 2019, 9, 152. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McCallister, C.; Siracusa, M.C.; Shirazi, F.; Chalkia, D.; Nikolaidis, N. Functional diversification and specialization of cytosolic 70-kDa heat shock proteins. Sci. Rep. 2015, 5, 9363. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McCallister, C.; Kdeiss, B.; Nikolaidis, N. Biochemical characterization of the interaction between HspA1A and phospholipids. Cell Stress Chaperones 2015, 21, 41–53. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Petersen, N.H.; Kirkegaard, T.; Olsen, O.D.; Jaattela, M. Connecting Hsp70, sphingolipid metabolism and lysosomal stability. Cell Cycle 2010, 9, 2305–2309. [Google Scholar] [CrossRef] [Green Version]
- McCallister, C.; Kdeiss, B.; Oliverio, R.; Nikolaidis, N. Characterization of the binding between a 70-kDa heat shock protein, HspA1A, and phosphoinositides. Biochem. Biophys. Res. Commun. 2016, 472, 270–275. [Google Scholar] [CrossRef]
- McCallister, C.; Kdeiss, B.; Nikolaidis, N. HspA1A, a 70-kDa heat shock protein, differentially interacts with anionic lipids. Biochem. Biophys. Res. Commun. 2015, 467, 835–840. [Google Scholar] [CrossRef]
- Armijo, G.; Okerblom, J.; Cauvi, D.M.; Lopez, V.; Schlamadinger, D.E.; Kim, J.; Arispe, N.; De Maio, A. Interaction of heat shock protein 70 with membranes depends on the lipid environment. Cell Stress Chaperones 2014, 19, 877–886. [Google Scholar] [CrossRef] [Green Version]
- Arispe, N.; Doh, M.; Simakova, O.; Kurganov, B.; De Maio, A. Hsc70 and Hsp70 interact with phosphatidylserine on the surface of PC12 cells resulting in a decrease of viability. FASEB J. 2004, 18, 1636–1645. [Google Scholar] [CrossRef] [Green Version]
- Lamprecht, C.; Gehrmann, M.; Madl, J.; Romer, W.; Multhoff, G.; Ebner, A. Molecular AFM imaging of Hsp70-1A association with dipalmitoyl phosphatidylserine reveals membrane blebbing in the presence of cholesterol. Cell Stress Chaperones 2018, 23, 673–683. [Google Scholar] [CrossRef]
- Juhasz, K.; Thuenauer, R.; Spachinger, A.; Duda, E.; Horvath, I.; Vigh, L.; Sonnleitner, A.; Balogi, Z. Lysosomal rerouting of Hsp70 trafficking as a potential immune activating tool for targeting melanoma. Curr. Pharm. Des. 2013, 19, 430–440. [Google Scholar] [CrossRef] [Green Version]
- Shevtsov, M.; Huile, G.; Multhoff, G. Membrane heat shock protein 70: A theranostic target for cancer therapy. Philos Trans. R. Soc. Lond. B Biol. Sci. 2018, 373, 20160526. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hammond, G.R.; Fischer, M.J.; Anderson, K.E.; Holdich, J.; Koteci, A.; Balla, T.; Irvine, R.F. PI4P and PI(4,5)P2 are essential but independent lipid determinants of membrane identity. Science 2012, 337, 727–730. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hammond, G.R.; Machner, M.P.; Balla, T. A novel probe for phosphatidylinositol 4-phosphate reveals multiple pools beyond the Golgi. J. Cell Biol. 2014, 205, 113–126. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Johnson, K.A.; Taghon, G.J.; Scott, J.L.; Stahelin, R.V. The Ebola Virus matrix protein, VP40, requires phosphatidylinositol 4,5-bisphosphate (PI(4,5)P2) for extensive oligomerization at the plasma membrane and viral egress. Sci. Rep. 2016, 6, 19125. [Google Scholar] [CrossRef] [Green Version]
- Balla, T. Inositol-lipid binding motifs: Signal integrators through protein-lipid and protein-protein interactions. J. Cell Sci. 2005, 118, 2093–2104. [Google Scholar] [CrossRef] [Green Version]
- Stahelin, R.V. Lipid binding domains: More than simple lipid effectors. J. Lipid Res. 2009, 50 (Supplement), S299–S304. [Google Scholar] [CrossRef] [Green Version]
- Hess, K.; Oliverio, R.; Nguyen, P.; Le, D.; Ellis, J.; Kdeiss, B.; Ord, S.; Chalkia, D.; Nikolaidis, N. Concurrent action of purifying selection and gene conversion results in extreme conservation of the major stress-inducible Hsp70 genes in mammals. Sci. Rep. 2018, 8, 5082. [Google Scholar] [CrossRef]
- Yeung, T.; Gilbert, G.E.; Shi, J.; Silvius, J.; Kapus, A.; Grinstein, S. Membrane phosphatidylserine regulates surface charge and protein localization. Science 2008, 319, 210–213. [Google Scholar] [CrossRef]
- Stauffer, T.P.; Ahn, S.; Meyer, T. Receptor-induced transient reduction in plasma membrane PtdIns(4,5)P2 concentration monitored in living cells. Curr. Biol. 1998, 8, 343–346. [Google Scholar] [CrossRef] [Green Version]
- Lawe, D.C.; Patki, V.; Heller-Harrison, R.; Lambright, D.; Corvera, S. The FYVE domain of early endosome antigen 1 is required for both phosphatidylinositol 3-phosphate and Rab5 binding. Critical role of this dual interaction for endosomal localization. J. Biol. Chem. 2000, 275, 3699–3705. [Google Scholar] [CrossRef] [Green Version]
- Willars, G.B.; Nahorski, S.R.; Challiss, R.A. Differential regulation of muscarinic acetylcholine receptor-sensitive polyphosphoinositide pools and consequences for signaling in human neuroblastoma cells. J. Biol. Chem. 1998, 273, 5037–5046. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bojjireddy, N.; Botyanszki, J.; Hammond, G.; Creech, D.; Peterson, R.; Kemp, D.C.; Snead, M.; Brown, R.; Morrison, A.; Wilson, S.; et al. Pharmacological and genetic targeting of the PI4KA enzyme reveals its important role in maintaining plasma membrane phosphatidylinositol 4-phosphate and phosphatidylinositol 4,5-bisphosphate levels. J. Biol. Chem. 2014, 289, 6120–6132. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Collins, T.J. ImageJ for microscopy. Biotechniques 2007, 43, 25–30. [Google Scholar] [CrossRef] [PubMed]
- Burgess, A.; Vigneron, S.; Brioudes, E.; Labbe, J.C.; Lorca, T.; Castro, A. Loss of human Greatwall results in G2 arrest and multiple mitotic defects due to deregulation of the cyclin B-Cdc2/PP2A balance. Proc. Natl. Acad. Sci. USA 2010, 107, 12564–12569. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oliverio, R.; Nguyen, P.; Kdeiss, B.; Ord, S.; Daniels, A.J.; Nikolaidis, N. Functional characterization of natural variants found on the major stress inducible 70-kDa heat shock gene, HSPA1A, in humans. Biochem. Biophys. Res. Commun. 2018, 506, 799–804. [Google Scholar] [CrossRef]
- Spitzer, M.; Wildenhain, J.; Rappsilber, J.; Tyers, M. BoxPlotR: A web tool for generation of box plots. Nat. Methods 2014, 11, 121–122. [Google Scholar] [CrossRef]
- He, J.; Vora, M.; Haney, R.M.; Filonov, G.S.; Musselman, C.A.; Burd, C.G.; Kutateladze, A.G.; Verkhusha, V.V.; Stahelin, R.V.; Kutateladze, T.G. Membrane insertion of the FYVE domain is modulated by pH. Proteins 2009, 76, 852–860. [Google Scholar] [CrossRef] [Green Version]
- Sohn, M.; Korzeniowski, M.; Zewe, J.P.; Wills, R.C.; Hammond, G.R.V.; Humpolickova, J.; Vrzal, L.; Chalupska, D.; Veverka, V.; Fairn, G.D.; et al. PI(4,5)P2 controls plasma membrane PI4P and PS levels via ORP5/8 recruitment to ER-PM contact sites. J. Cell Biol. 2018, 217, 1797–1813. [Google Scholar] [CrossRef]
- Patki, V.; Virbasius, J.; Lane, W.S.; Toh, B.H.; Shpetner, H.S.; Corvera, S. Identification of an early endosomal protein regulated by phosphatidylinositol 3-kinase. Proc. Natl. Acad. Sci. USA 1997, 94, 7326–7330. [Google Scholar] [CrossRef] [Green Version]
- Kamentseva, R.; Kosheverova, V.; Kharchenko, M.; Zlobina, M.; Salova, A.; Belyaeva, T.; Nikolsky, N.; Kornilova, E. Functional cycle of EEA1-positive early endosome: Direct evidence for pre-existing compartment of degradative pathway. PLoS ONE 2020, 15, e0232532. [Google Scholar] [CrossRef]
- D’Hooghe, L.; Chalmers, A.D.; Heywood, S.; Whitley, P. Cell surface dynamics and cellular distribution of endogenous FcRn. PLoS ONE 2017, 12, e0182695. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bourseau-Guilmain, E.; Menard, J.A.; Lindqvist, E.; Indira Chandran, V.; Christianson, H.C.; Cerezo Magana, M.; Lidfeldt, J.; Marko-Varga, G.; Welinder, C.; Belting, M. Hypoxia regulates global membrane protein endocytosis through caveolin-1 in cancer cells. Nat. Commun. 2016, 7, 11371. [Google Scholar] [CrossRef]
- Maugeri, N.; Radhakrishnan, J.; Knight, J.C. Genetic determinants of HSP70 gene expression following heat shock. Hum. Mol. Genet. 2010, 19, 4939–4947. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Evdokimovskaya, Y.; Skarga, Y.; Vrublevskaya, V.; Morenkov, O. Secretion of the heat shock proteins HSP70 and HSC70 by baby hamster kidney (BHK-21) cells. Cell Biol. Int. 2010, 34, 985–990. [Google Scholar] [CrossRef] [PubMed]
- Mambula, S.S.; Calderwood, S.K. Heat shock protein 70 is secreted from tumor cells by a nonclassical pathway involving lysosomal endosomes. J. Immunol. 2006, 177, 7849–7857. [Google Scholar] [CrossRef] [Green Version]
- Gastpar, R.; Gehrmann, M.; Bausero, M.A.; Asea, A.; Gross, C.; Schroeder, J.A.; Multhoff, G. Heat shock protein 70 surface-positive tumor exosomes stimulate migratory and cytolytic activity of natural killer cells. Cancer Res. 2005, 65, 5238–5247. [Google Scholar] [CrossRef] [Green Version]
- Lancaster, G.I.; Febbraio, M.A. Exosome-dependent trafficking of HSP70: A novel secretory pathway for cellular stress proteins. J. Biol. Chem. 2005, 280, 23349–23355. [Google Scholar] [CrossRef] [Green Version]









Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Smulders, L.; Altman, R.; Briseno, C.; Saatchi, A.; Wallace, L.; AlSebaye, M.; Stahelin, R.V.; Nikolaidis, N. Phosphatidylinositol Monophosphates Regulate the Membrane Localization of HSPA1A, a Stress-Inducible 70-kDa Heat Shock Protein. Biomolecules 2022, 12, 856. https://doi.org/10.3390/biom12060856
Smulders L, Altman R, Briseno C, Saatchi A, Wallace L, AlSebaye M, Stahelin RV, Nikolaidis N. Phosphatidylinositol Monophosphates Regulate the Membrane Localization of HSPA1A, a Stress-Inducible 70-kDa Heat Shock Protein. Biomolecules. 2022; 12(6):856. https://doi.org/10.3390/biom12060856
Chicago/Turabian StyleSmulders, Larissa, Rachel Altman, Carolina Briseno, Alireza Saatchi, Leslie Wallace, Maha AlSebaye, Robert V. Stahelin, and Nikolas Nikolaidis. 2022. "Phosphatidylinositol Monophosphates Regulate the Membrane Localization of HSPA1A, a Stress-Inducible 70-kDa Heat Shock Protein" Biomolecules 12, no. 6: 856. https://doi.org/10.3390/biom12060856