
Comparative Analysis of rRNA Removal Methods for RNA-Seq Differential Expression in Halophilic Archaea


Abstract
:1. Introduction
2. Material and Methods
2.1. Media, Strains, and Growth Conditions
2.2. RNA-Seq Experimental Protocol
- Biotinylated probes with strepdavidin bead pull-down:
- Discontinued Ribo-Zero rRNA Removal Kit (Bacteria). Abbr: RZ;
- siTools HVO RiboPOOLTM with probes specific for HVO. Abbr: rP-HVO;
- siTools Pan-Archaea riboPOOLTM (probes included). Abbr: rP-PA.
- RNAse H and enzymatic depletion-based protocols with magnetic bead pull-down:
- Ribo-Zero Plus Kit (probes included). Abbr: RZ+;
- NEBNext Bacteria rRNA depletion Kit (New England Biolabs) with probes designed for bacteria (included in kit from NEB). Abbr: NEB-B;
- NEBNext Depletion Core Reagent Set with customized sequence-specific probes for HVO (Table S3). These probes were designed using the NEB web tool (https://depletion-design.neb.com/, accessed on 3 January 2020) and ordered from IDT technologies (idtdna.com, accessed on 3 January 2020). Abbr: NEB-HVO.
2.3. Data Analysis
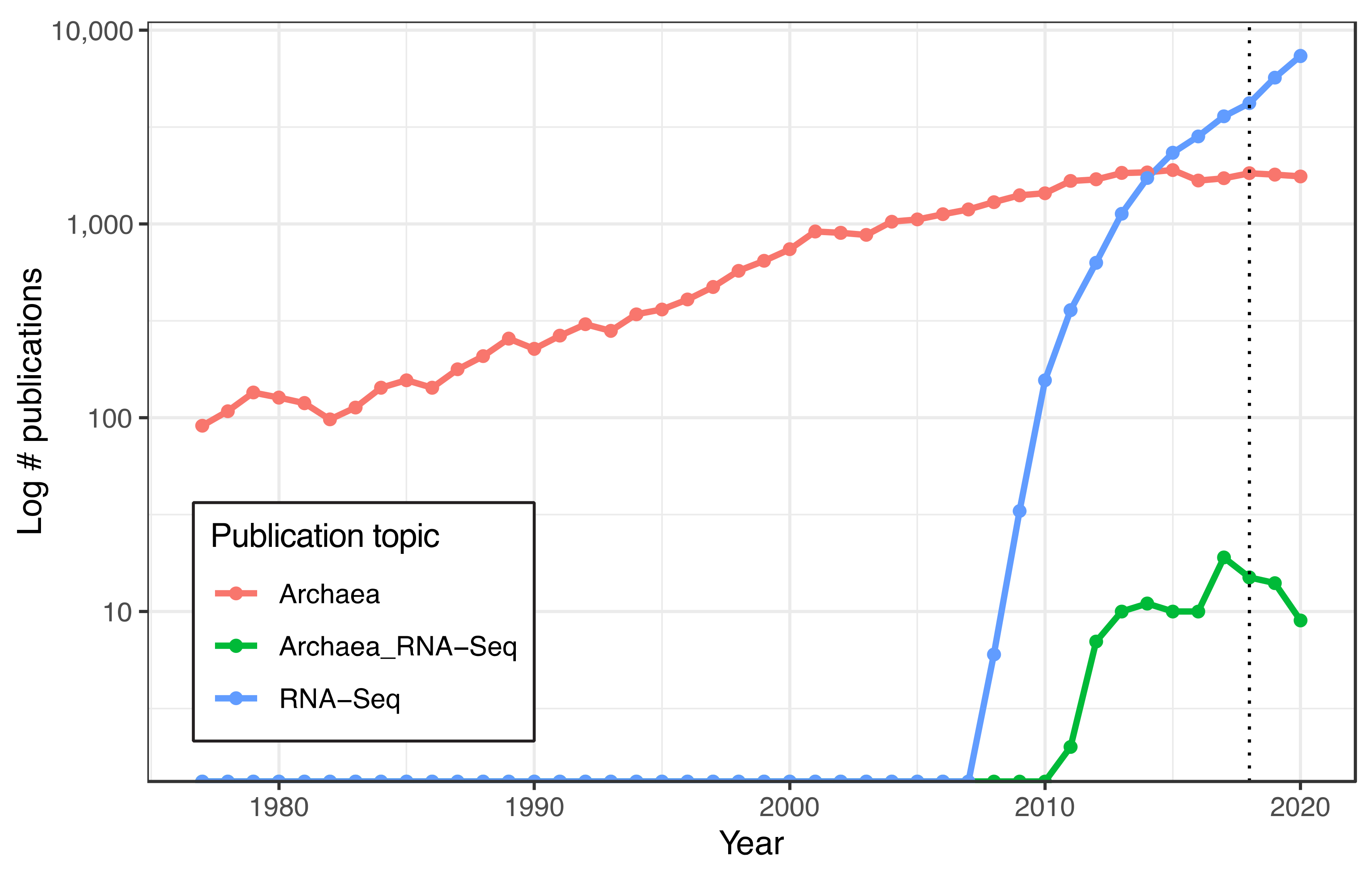
2.3.1. Publications on Archaeal RNA-Seq per Year
2.3.2. RNA-Seq Data Processing
2.3.3. Probe Specificity Analysis
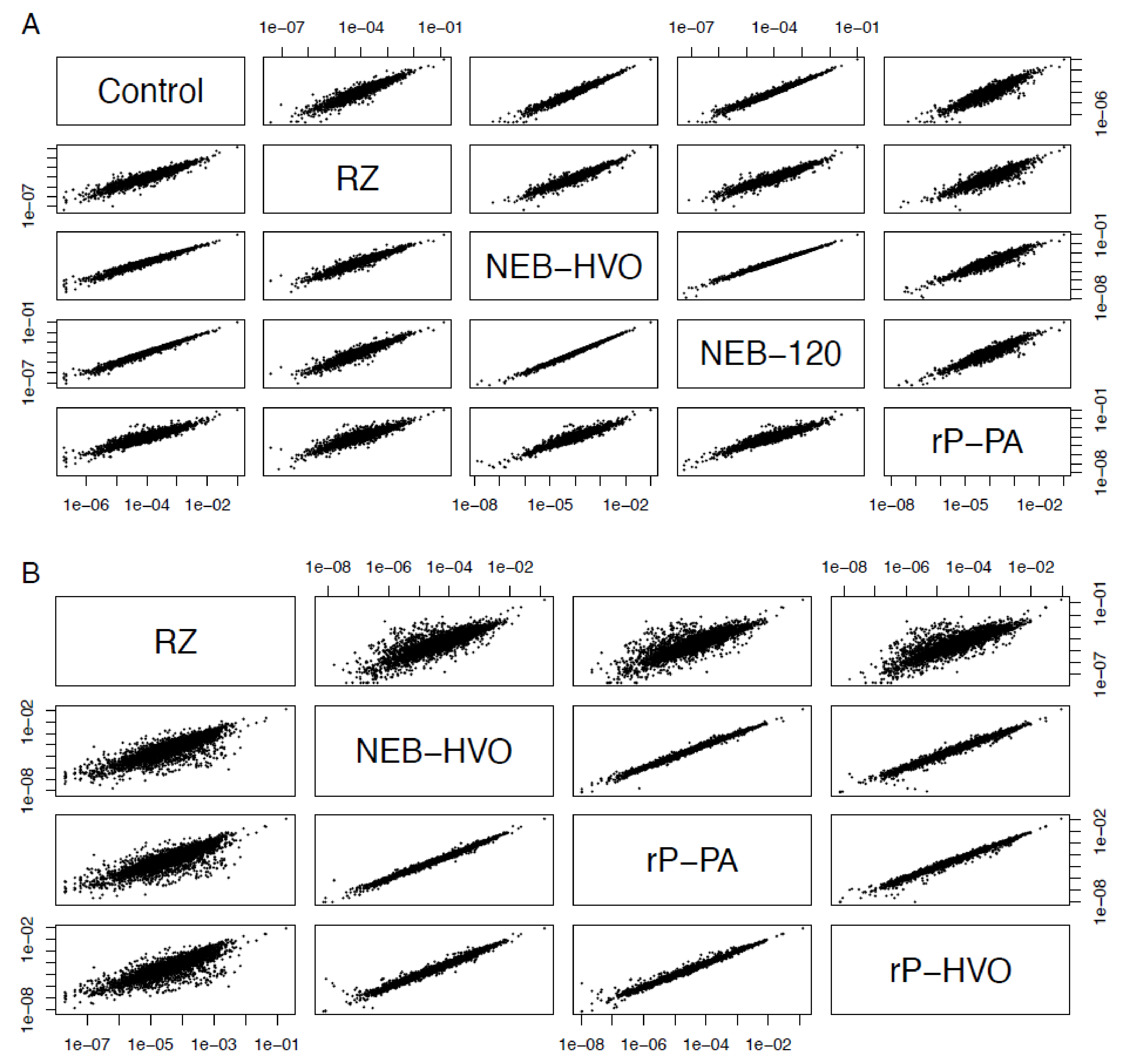
2.3.4. Count Correlations
2.3.5. Power Analysis
2.3.6. Probe Design for Other Species of Interest
3. Results
3.1. Discontinuation of the Illumina RiboZero Kit Is Associated with a Decline in Published Archaeal RNA-Seq Studies
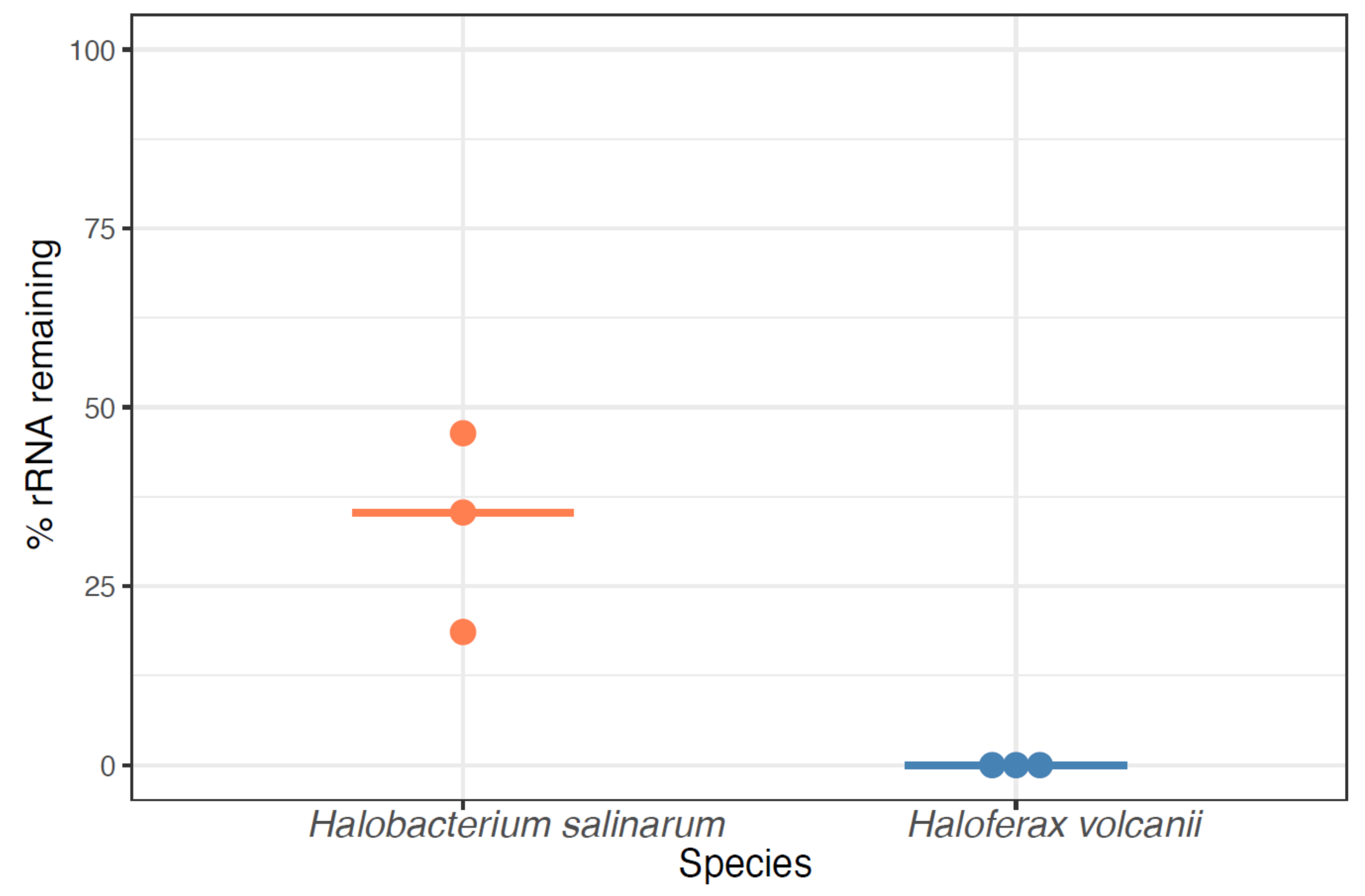
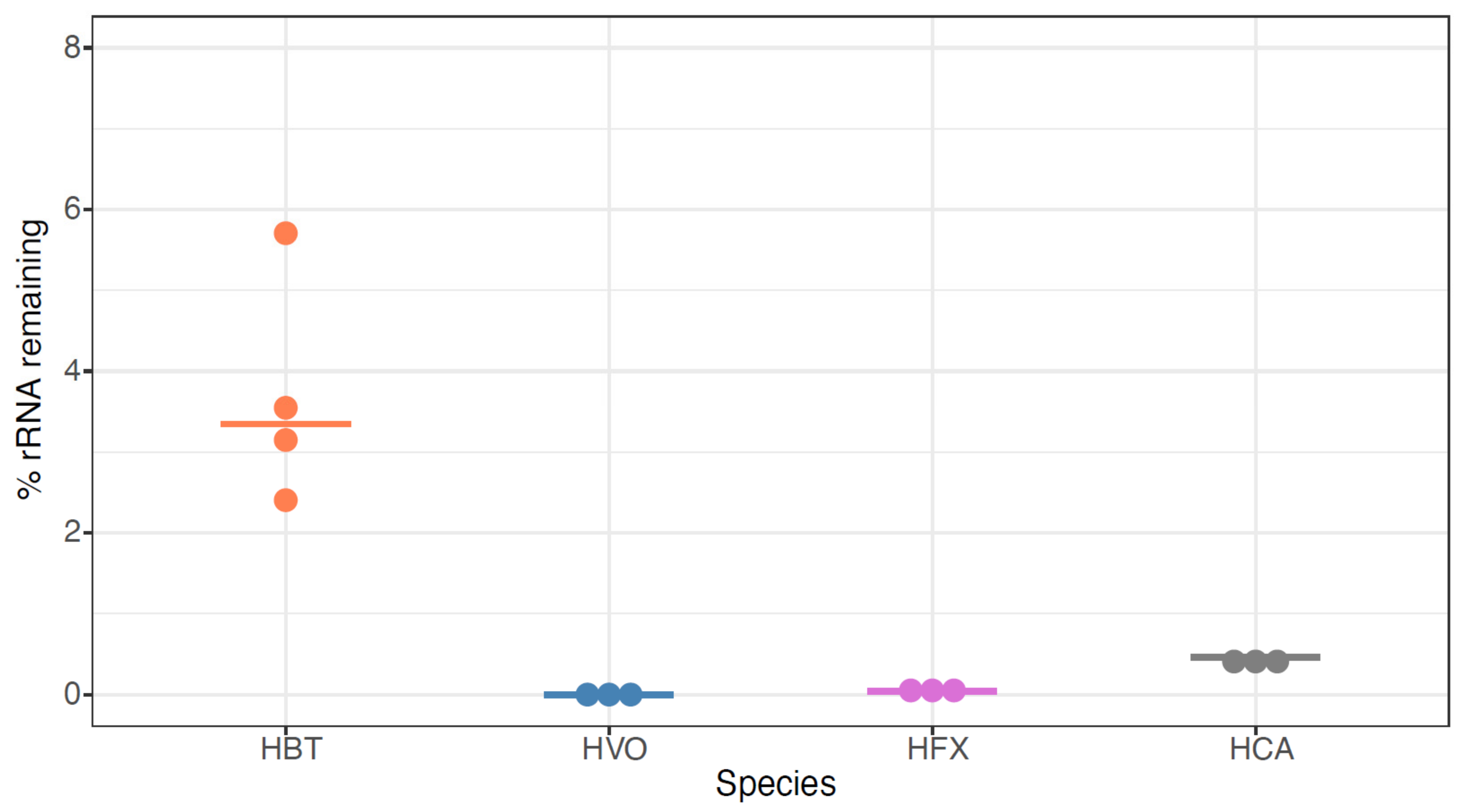
3.2. Testing New rRNA Depletion Strategies on Total RNA Samples from Halobacterium salinarum (HBT)
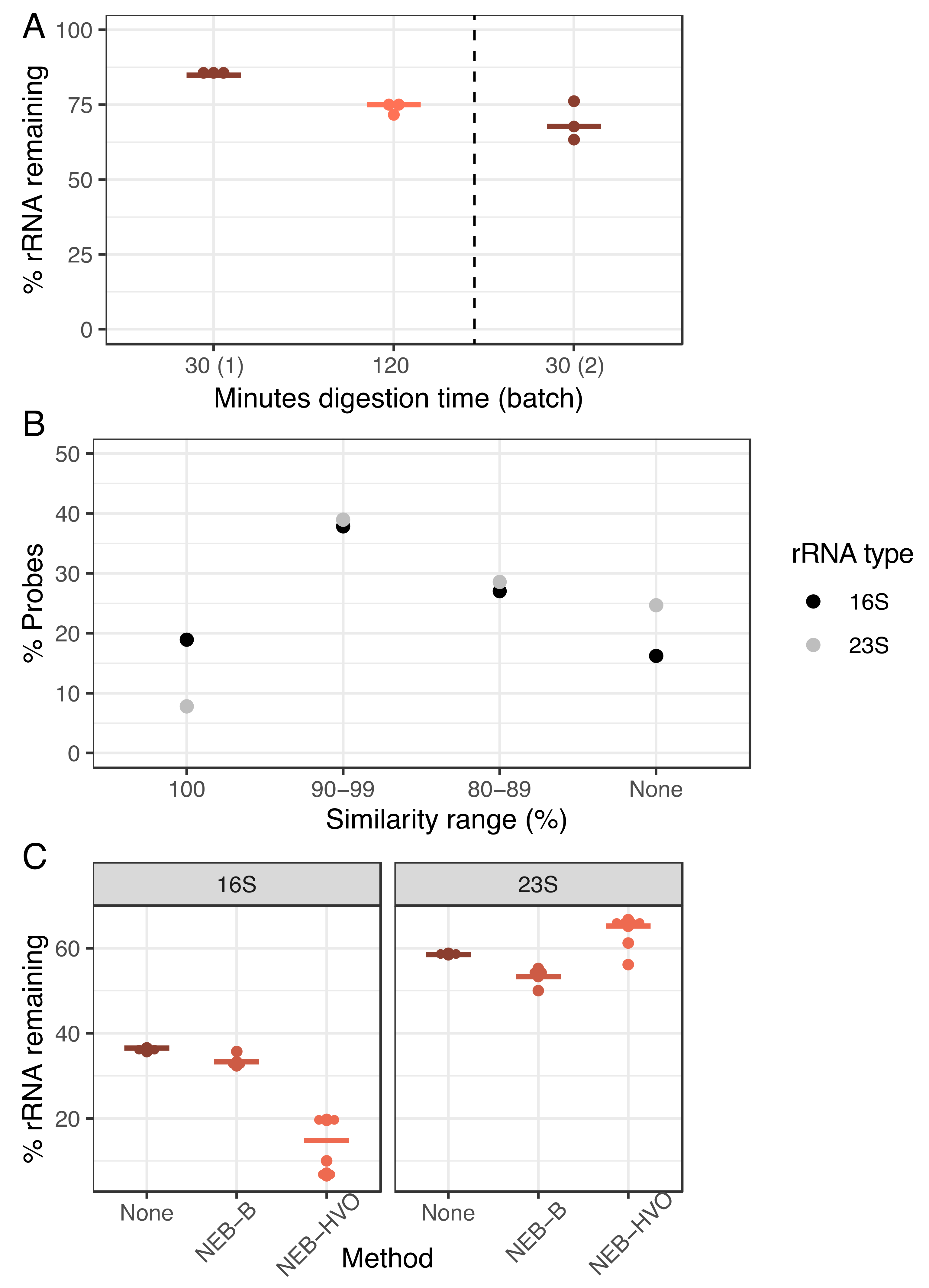
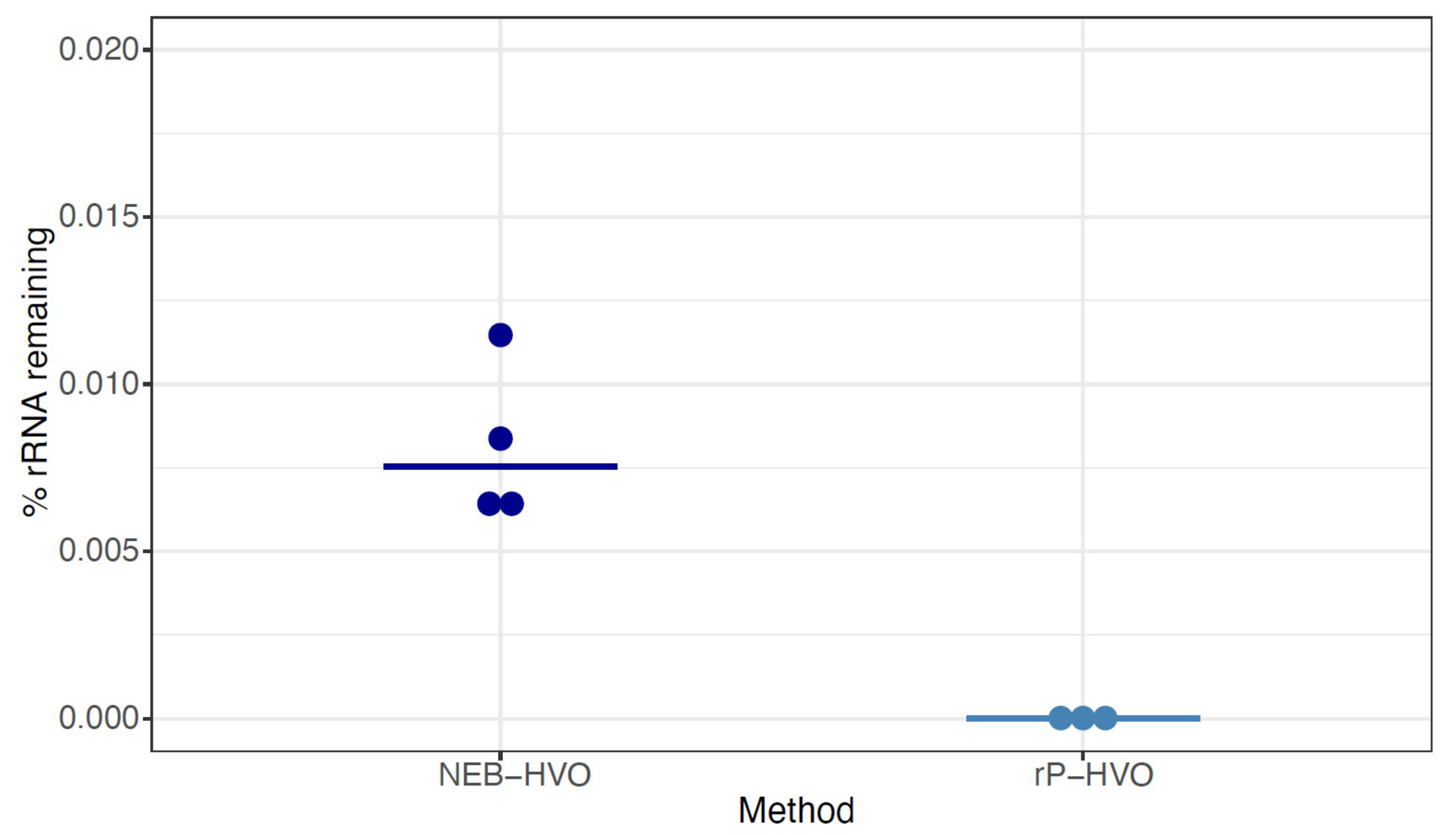
3.3. Species-Specific Probe Methods Efficiently Remove Haloferax volcanii (HVO) rRNA
3.4. siTools Panarchaea Kit Efficiently Removes rRNA from Diverse Halophilic Archaeal Species
3.5. Choice of Removal Method Does Not Affect Per-Gene Read Counts
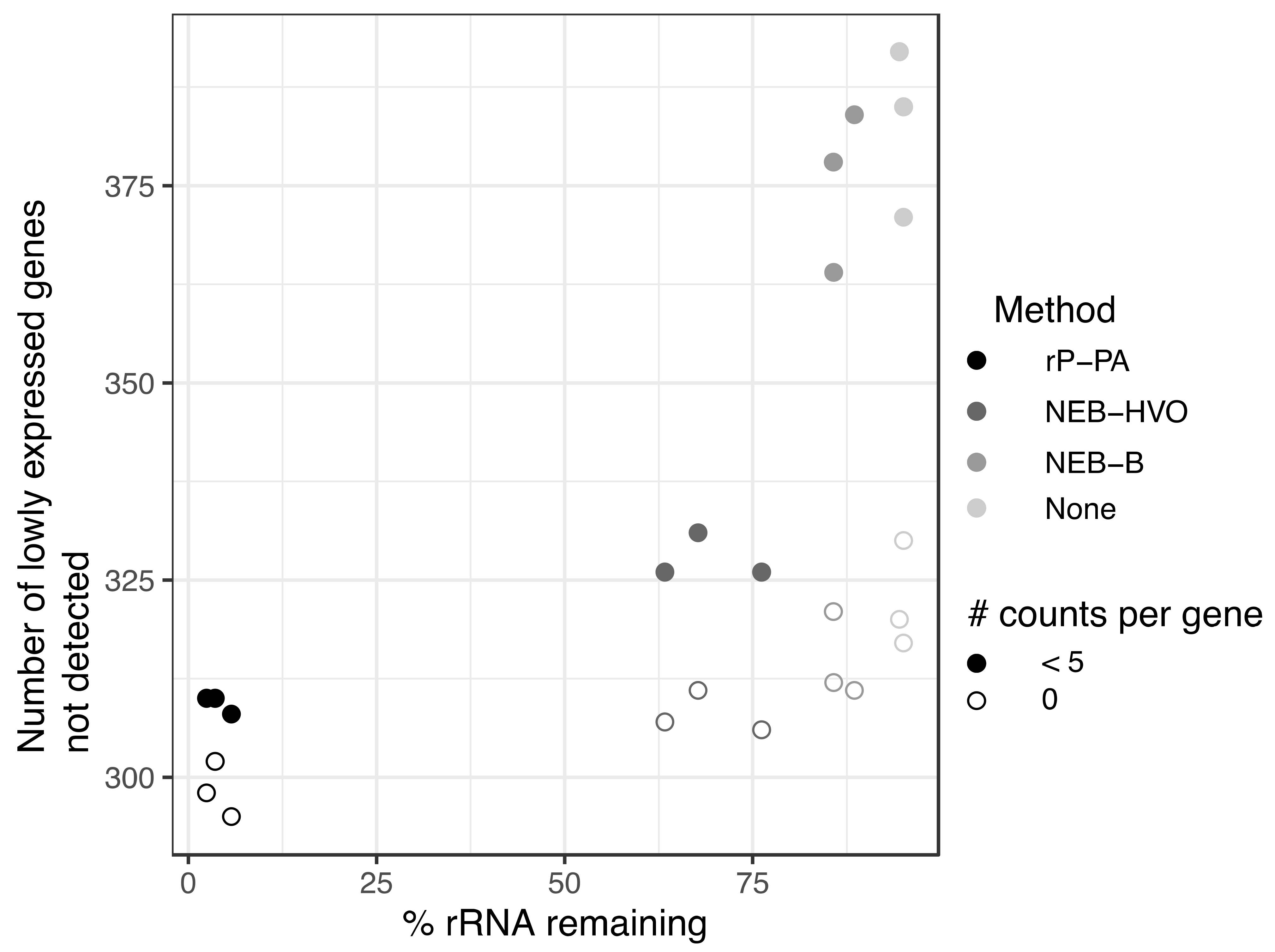
3.6. Utility of rRNA Removal Is Seen in Counts of Non-rRNA Genes
4. Discussion and Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Weber, A.P.M.; Weber, K.L.; Carr, K.; Wilkerson, C.; Ohlrogge, J.B. Sampling the Arabidopsis transcriptome with massively parallel pyrosequencing. Plant Physiol. 2007, 144, 32–42. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nagalakshmi, U.; Wang, Z.; Waern, K.; Shou, C.; Raha, D.; Gerstein, M.; Snyder, M. The transcriptional landscape of the yeast genome defined by RNA sequencing. Science 2008, 320, 1344–1349. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bainbridge, M.N.; Warren, R.L.; Hirst, M.; Romanuik, T.; Zeng, T.; Go, A.; Delaney, A.; Griffith, M.; Hickenbotham, M.; Magrini, V.; et al. Analysis of the prostate cancer cell line LNCaP transcriptome using a sequencing-by-synthesis approach. BMC Genom. 2006, 7, 246. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mortazavi, A.; Williams, B.A.; McCue, K.; Schaeffer, L.; Wold, B. Mapping and quantifying mammalian transcriptomes by RNA-Seq. Nat. Methods 2008, 5, 621–628. [Google Scholar] [CrossRef]
- Woese, C.R.; Fox, G.E. Phylogenetic structure of the prokaryotic domain: The primary kingdoms. Proc. Natl. Acad. Sci. USA 1977, 74, 5088–5090. [Google Scholar] [CrossRef] [Green Version]
- Zaremba-Niedzwiedzka, K.; Caceres, E.F.; Saw, J.H.; Backstrom, D.; Juzokaite, L.; Vancaester, E.; Seitz, K.W.; Anantharaman, K.; Starnawski, P.; Kjeldsen, K.U.; et al. Asgard archaea illuminate the origin of eukaryotic cellular complexity. Nature 2017, 541, 353–358. [Google Scholar] [CrossRef] [Green Version]
- Cai, M.; Liu, Y.; Zhou, Z.; Yang, Y.; Pan, J.; Gu, J.-D.; Li, M. Asgard archaea are diverse, ubiquitous, and transcriptionally active microbes. bioRxiv 2018, 374165. [Google Scholar] [CrossRef]
- Angel, R.; Claus, P.; Conrad, R. Methanogenic archaea are globally ubiquitous in aerated soils and become active under wet anoxic conditions. ISME J. 2012, 6, 847–862. [Google Scholar] [CrossRef] [Green Version]
- Browne, P.D.; Cadillo-Quiroz, H. Contribution of transcriptomics to systems-level understanding of methanogenic Archaea. Archaea 2013, 2013, 586369. [Google Scholar] [CrossRef] [Green Version]
- Hackley, R.K.; Schmid, A.K. Global Transcriptional Programs in Archaea Share Features with the Eukaryotic Environmental Stress Response. J. Mol. Biol. 2019, 431, 4147–4166. [Google Scholar] [CrossRef]
- Martinez-Pastor, M.; Tonner, P.D.; Darnell, C.L.; Schmid, A.K. Transcriptional Regulation in Archaea: From Individual Genes to Global Regulatory Networks. Annu. Rev. Genet. 2017, 51, 143–170. [Google Scholar] [CrossRef]
- Gelsinger, D.R.; DiRuggiero, J. Transcriptional Landscape and Regulatory Roles of Small Noncoding RNAs in the Oxidative Stress Response of the Haloarchaeon Haloferax Volcanii. J. Bacteriol. 2018, 200, e00779-17. [Google Scholar] [CrossRef] [Green Version]
- Cai, M.; Liu, Y.; Yin, X.; Zhou, Z.; Friedrich, M.W.; Richter-Heitmann, T.; Nimzyk, R.; Kulkarni, A.; Wang, X.; Li, W.; et al. Diverse Asgard archaea including the novel phylum Gerdarchaeota participate in organic matter degradation. Sci. China Life Sci. 2020, 63, 886–897. [Google Scholar] [CrossRef]
- Dulmage, K.A.; Darnell, C.L.; Vreugdenhil, A.; Schmid, A.K. Copy number variation is associated with gene expression change in archaea. Microb. Genom. 2018, 4, e000210. [Google Scholar] [CrossRef]
- Qi, L.; Yue, L.; Feng, D.; Qi, F.; Li, J.; Dong, X. Genome-wide mRNA processing in methanogenic archaea reveals post-transcriptional regulation of ribosomal protein synthesis. Nucleic Acids Res. 2017, 45, 7285–7298. [Google Scholar] [CrossRef] [Green Version]
- Zhou, X.; Stevens, M.J.A.; Neuenschwander, S.; Schwarm, A.; Kreuzer, M.; Bratus-Neuenschwander, A.; Zeitz, J.O. The transcriptome response of the ruminal methanogen Methanobrevibacter ruminantium strain M1 to the inhibitor lauric acid. BMC Res. Notes 2018, 11, 135. [Google Scholar] [CrossRef]
- Zhou, Z.; Liu, Y.; Lloyd, K.G.; Pan, J.; Yang, Y.; Gu, J.-D.; Li, M. Genomic and transcriptomic insights into the ecology and metabolism of benthic archaeal cosmopolitan, Thermoprofundales (MBG-D archaea). ISME J. 2019, 13, 885–901. [Google Scholar] [CrossRef] [Green Version]
- Tarazona, S.; García-Alcalde, F.; Dopazo, J.; Ferrer, A.; Conesa, A. Differential expression in RNA-seq: A matter of depth. Genome Res. 2011, 21, 2213–2223. [Google Scholar] [CrossRef] [Green Version]
- Baccarella, A.; Williams, C.R.; Parrish, J.Z.; Kim, C.C. Empirical assessment of the impact of sample number and read depth on RNA-Seq analysis workflow performance. BMC Bioinform. 2018, 19, 423. [Google Scholar] [CrossRef]
- Liu, Y.; Zhou, J.; White, K.P. RNA-seq differential expression studies: More sequence or more replication? Bioinformatics 2014, 30, 301–304. [Google Scholar] [CrossRef]
- Davis, C.A.; Hitz, B.C.; Sloan, C.A.; Chan, E.T.; Davidson, J.M.; Gabdank, I.; Hilton, J.A.; Jain, K.; Baymuradov, U.K.; Narayanan, A.K.; et al. The Encyclopedia of DNA elements (ENCODE): Data portal update. Nucleic Acids Res. 2018, 46, D794–D801. [Google Scholar] [CrossRef] [Green Version]
- Babski, J.; Haas, K.A.; Näther-Schindler, D.; Pfeiffer, F.; Förstner, K.U.; Hammelmann, M.; Hilker, R.; Becker, A.; Sharma, C.M.; Marchfelder, A.; et al. Genome-wide identification of transcriptional start sites in the haloarchaeon Haloferax volcanii based on differential RNA-Seq (dRNA-Seq). BMC Genom. 2016, 17, 629. [Google Scholar] [CrossRef] [Green Version]
- Allers, T.; Barak, S.; Liddell, S.; Wardell, K.; Mevarech, M. Improved strains and plasmid vectors for conditional overexpression of His-tagged proteins in Haloferax volcanii. Appl. Environ. Microbiol. 2010, 76, 1759–1769. [Google Scholar] [CrossRef] [Green Version]
- Peck, R.F.; DasSarma, S.; Krebs, M.P. Homologous gene knockout in the archaeon Halobacterium salinarum with ura3 as a counterselectable marker. Mol. Microbiol. 2000, 35, 667–676. [Google Scholar] [CrossRef]
- Liu, H.; Han, J.; Liu, X.; Zhou, J.; Xiang, H. Development of pyrF-based gene knockout systems for genome-wide manipulation of the archaea Haloferax mediterranei and Haloarcula hispanica. J. Genet. Genom. 2011, 38, 261–269. [Google Scholar] [CrossRef]
- Pfeiffer, F.; Broicher, A.; Gillich, T.; Klee, K.; Mejia, J.; Rampp, M.; Oesterhelt, D. Genome information management and integrated data analysis with HaloLex. Arch Microbiol. 2008, 190, 281–299. [Google Scholar] [CrossRef] [Green Version]
- Pfeiffer, F.; Dyall-Smith, M. Open Issues for Protein Function Assignment in Haloferax volcanii and Other Halophilic Archaea. Genes 2021, 12, 963. [Google Scholar] [CrossRef]
- Schmid, A.K.; Reiss, D.J.; Kaur, A.; Pan, M.; King, N.; Van, P.T.; Hohmann, L.; Martin, D.B.; Baliga, N.S. The anatomy of microbial cell state transitions in response to oxygen. Genom. Res. 2007, 17, 1399–1413. [Google Scholar] [CrossRef] [Green Version]
- Bonneau, R.; Baliga, N.S.; Deutsch, E.W.; Shannon, P.; Hood, L. Comprehensive de novo structure prediction in a systems-biology context for the archaea Halobacterium sp. NRC-1. Genom. Biol. 2004, 5, R52. [Google Scholar] [CrossRef] [Green Version]
- Culviner, P.H.; Guegler, C.K.; Laub, M.T. A Simple, Cost-Effective, and Robust Method for rRNA Depletion in RNA-Sequencing Studies. mBio 2020, 11, e00010-20. [Google Scholar] [CrossRef] [Green Version]
- Bitan-Banin, G.; Ortenberg, R.; Mevarech, M. Development of a gene knockout system for the halophilic archaeon Haloferax volcanii by use of the pyrE gene. J. Bacteriol. 2003, 185, 772–778. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Allers, T.; Ngo, H.-P.; Mevarech, M.; Lloyd, R.G. Development of additional selectable markers for the halophilic archaeon Haloferax volcanii based on the leuB and trpA genes. Appl. Environ. Microbiol. 2004, 70, 943–953. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schwarzer, S.; Rodriguez-Franco, M.; Oksanen, H.M.; Quax, T.E.F. Growth Phase Dependent Cell Shape of Haloarcula. Microorganisms 2021, 9, 231. [Google Scholar] [CrossRef] [PubMed]
- Sakrikar, S.; Schmid, A.K. An archaeal histone-like protein regulates gene expression in response to salt stress. Nucleic Acids Res. 2021, 49, 12732–12743. [Google Scholar] [CrossRef]
- Langmead, B.; Salzberg, S.L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 2012, 9, 357–359. [Google Scholar] [CrossRef] [Green Version]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R.; Genome Project Data Processing Subgroup. The Sequence Alignment/Map format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef] [Green Version]
- Anders, S.; Pyl, P.T.; Huber, W. HTSeq—a Python framework to work with high-throughput sequencing data. Bioinformatics 2015, 31, 166–169. [Google Scholar] [CrossRef]
- Busby, M.A.; Stewart, C.; Miller, C.A.; Grzeda, K.R.; Marth, G.T. Scotty: A web tool for designing RNA-Seq experiments to measure differential gene expression. Bioinformatics 2013, 29, 656–657. [Google Scholar] [CrossRef] [Green Version]
- Paya, G.; Bautista, V.; Camacho, M.; Castejon-Fernandez, N.; Alcaraz, L.A.; Bonete, M.J.; Esclapez, J. Small RNAs of Haloferax mediterranei: Identification and Potential Involvement in Nitrogen Metabolism. Genes 2018, 9, 83. [Google Scholar] [CrossRef] [Green Version]
- Harrington, C.A.; Fei, S.S.; Minnier, J.; Carbone, L.; Searles, R.; Davis, B.A.; Ogle, K.; Planck, S.R.; Rosenbaum, J.T.; Choi, D. RNA-Seq of human whole blood: Evaluation of globin RNA depletion on Ribo-Zero library method. Sci. Rep. 2020, 10, 6271. [Google Scholar] [CrossRef] [Green Version]
- Adiconis, X.; Borges-Rivera, D.; Satija, R.; DeLuca, D.S.; Busby, M.A.; Berlin, A.M.; Sivachenko, A.; Thompson, D.A.; Wysoker, A.; Fennell, T.; et al. Comparative analysis of RNA sequencing methods for degraded or low-input samples. Nat. Methods 2013, 10, 623–629. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pang, X.; Zhou, D.; Song, Y.; Pei, D.; Wang, J.; Guo, Z.; Yang, R. Bacterial mRNA Purification by Magnetic Capture-Hybridization Method. Microbiol. Immunol. 2004, 48, 91–96. [Google Scholar] [CrossRef] [PubMed]
- Su, C.; Sordillo, L.M. A simple method to enrich mRNA from total prokaryotic RNA. Mol. Biotechnol. 1998, 10, 83–85. [Google Scholar] [CrossRef] [PubMed]
- He, S.; Wurtzel, O.; Singh, K.; Froula, J.L.; Yilmaz, S.; Tringe, S.G.; Wang, Z.; Chen, F.; Lindquist, E.A.; Sorek, R.; et al. Validation of two ribosomal RNA removal methods for microbial metatranscriptomics. Nat. Methods 2010, 7, 807–812. [Google Scholar] [CrossRef] [Green Version]
- Giannoukos, G.; Ciulla, D.M.; Huang, K.; Haas, B.J.; Izard, J.; Levin, J.Z.; Livny, J.; Earl, A.M.; Gevers, D.; Ward, D.V.; et al. Efficient and robust RNA-seq process for cultured bacteria and complex community transcriptomes. Genom. Biol. 2012, 13, r23. [Google Scholar] [CrossRef] [Green Version]
- Farag, I.F.; Zhao, R.; Biddle, J.F. “Sifarchaeota,” a Novel Asgard Phylum from Costa Rican Sediment Capable of Polysaccharide Degradation and Anaerobic Methylotrophy. Appl. Environ. Microbiol. 2021, 87, e02584-20. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Name and Wild Type Strain | Species Abbreviation | Genotype Used Here | Reference Genome |
|---|---|---|---|
| Halobacterium salinarum NRC-1 [24] | HBT | Δura3 | GCF_000006805.1_ASM680v1 |
| Haloferax volcanii DS2 [31] | HVO | ΔpyrE | GCF_000025685.1_ASM2568v1 |
| Haloferax mediterranei ATCC33500 [25] | HFX | ΔpyrE | GCF_000306765.2_ASM30676v2 |
| Haloarcula hispainca DF60 [25] | HAH | ΔpyrF | GCF_000223905.1_ASM22390v1 |
| Name | Species Abbreviation | Ingredients (per L) | Supplement | pH |
|---|---|---|---|---|
| CM (rich media) | HBT | 250 g NaCl (Fisher Chemicals, Hampton, NH, USA); 20 g MgSO4.7H2O (Fisher Chemicals); C6H5Na3O7.2H2O (Fisher Chemicals); 2 g KCl (Fisher Chemicals); 10 g bacteriological peptone (Oxoid, Hampshire, UK) | 50 mL uracil (1 mg/mL) (Acros Organics, Geel, Belgium) | 6.8 |
| YPC 18% (rich media [32]) | HVO and HFX | 144 g NaCl (Fisher Chemicals);4.2 g KCl (Fisher Chemicals); 18 g MgCl2.6 H2O (Fisher Chemicals); 20 g MgSO4.7H2O (Fisher Chemicals); 12 mL 1 M TrisHCl (Fisher Chemicals) pH7.5; 5 g yeast extract (Fisher Chemicals); 1 g 10 g bacteriological peptone (Oxoid); 1 g Cas aminoacids (VWR, Radnor, PA, USA) | 50 mL uracil (1 mg/mL) (Acros Organics) | 7.5 |
| PR 18% (minimal media [this study]) | HVO | 170 g NaCl (Fisher Chemicals); 70 g MgCl2.6 H2O (Fisher Chemicals); 7 g KCl (Fisher Chemicals); 5 mL 1 M TrisHCl (Fisher Chemicals) pH7.5; 5 mL 1 M NH4Cl; 2 mL 0.25 M K2HPO4; 5 mL 1 M NaHCO3; 0.8 mL thiamine (1 mg/mL); 0.1 mL biotine (1 mg/mL); 0.5% glucose. | 50 mL uracil (1 mg/mL) (Acros Organics) | 7.2 |
| YPC 23% (rich media [33]) | HAH | 180 g NaCl (Fisher Chemicals); 4.2 g KCl (Fisher Chemicals); 18 g MgCl2.6 H2O (Fisher Chemicals); 20 g MgSO4.7H2O (Fisher Chemicals); 12 mL 1 M TrisHCl (Fisher Chemicals) pH7.5; 5 g yeast extract (Fisher Chemicals); 1 g 10 g bacteriological peptone (Oxoid); 1 g Cas aminoacids (VWR) | 50 mL uracil (1 mg/mL) (Acros Organics) | 7.5 |
| Species | Media | Doubling Time (h) | Days Until Stationary Phase |
|---|---|---|---|
| HBT | CM | 6 | 3 |
| HVO | YPC18% | 3 | 2.5 (36 h) |
| HVO | PR18% | 12 | 3 |
| HFX | YPC18% | 2.5 | 2 |
| HAH | YPC23% | 6 | 3 |
| Species | Forward Primer Sequence 5′-3′ | Reverse Primer Sequence 5′-3′ | Fragment Size |
|---|---|---|---|
| HBT | CGACATTCGGGTTGCGTTGTG | GGCGTTGTTCACGAAGCA | 1372 |
| HFX | CACATCAGCGAGGAGTTTGA | GACAGACGACGAGTTGGTCA | 162 |
| HVO | AGAAGTACAAGGGCGTCGAA | TTTTCGAACTCCTCGCTGAT | 171 |
| HAH | GCCGATTGCTCCGTCTACTA | ACTGCTCGGTGAGAAACGTC | 161 |
| Species | rRNA Type | Gene Identifier (s) | Alternate Gene Identifier (s) |
|---|---|---|---|
| HBT | 16S | VNG_RS09790 | VNG_r02 |
| 23S | VNG_RS09800 | VNG_r03 | |
| 5S | VHG_RS00395 | VNG_r04 | |
| HVO | 16S | HVO_RS13015, HVO_RS18920 | HVO_3038, HVO_3064 |
| 23S | HVO_RS13025, HVO_RS18910 | HVO_3040, HVO_3062 | |
| 5S | HVO_RS13030, VHO_RS18905 | HVO_3041, HVO_3061 | |
| HFX | 16S | HFX_RS14380, HFX_RS08900 | HFX_1820, HFX_2933 |
| 23S | HFX_RS14370, HFX_RS08910 | HFX_1822, HFX_2931 | |
| 5S | HFX_RS08915, HFX_RS14365 | HFX_2930, HFX_1823 | |
| HAH | 16S | HAH_RS08910, HAH_RS01110 | HAH_1834, HAH_0232 |
| 23S | HAH_RS08905, HAH_RS01120 | HAH_1833, HAH_0234 | |
| 5S | HAH_RS08900, HAH_RS01125 | HAH_1832, HAH_0235 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pastor, M.M.; Sakrikar, S.; Rodriguez, D.N.; Schmid, A.K. Comparative Analysis of rRNA Removal Methods for RNA-Seq Differential Expression in Halophilic Archaea. Biomolecules 2022, 12, 682. https://doi.org/10.3390/biom12050682
Pastor MM, Sakrikar S, Rodriguez DN, Schmid AK. Comparative Analysis of rRNA Removal Methods for RNA-Seq Differential Expression in Halophilic Archaea. Biomolecules. 2022; 12(5):682. https://doi.org/10.3390/biom12050682
Chicago/Turabian StylePastor, Mar Martinez, Saaz Sakrikar, Deyra N. Rodriguez, and Amy K. Schmid. 2022. "Comparative Analysis of rRNA Removal Methods for RNA-Seq Differential Expression in Halophilic Archaea" Biomolecules 12, no. 5: 682. https://doi.org/10.3390/biom12050682