
IntroSpect: Motif-Guided Immunopeptidome Database Building Tool to Improve the Sensitivity of HLA I Binding Peptide Identification by Mass Spectrometry

 ,
,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Generation of Cell Lines
2.2. Purification of HLA-I Peptides
2.3. LC-MS/MS Analysis of HLA-I Peptides
2.4. Sequencing and Analysis
2.5. Mass Spectrometry Database Search
2.6. Gibbs Clustering of HLA-I Peptides
2.7. PSSM Model Training and Filtering
2.8. Synthetic Peptide Validation
2.9. Peptide Pearson Correlation Coefficient (PCC) Calculation
2.10. Code Availability
3. Results
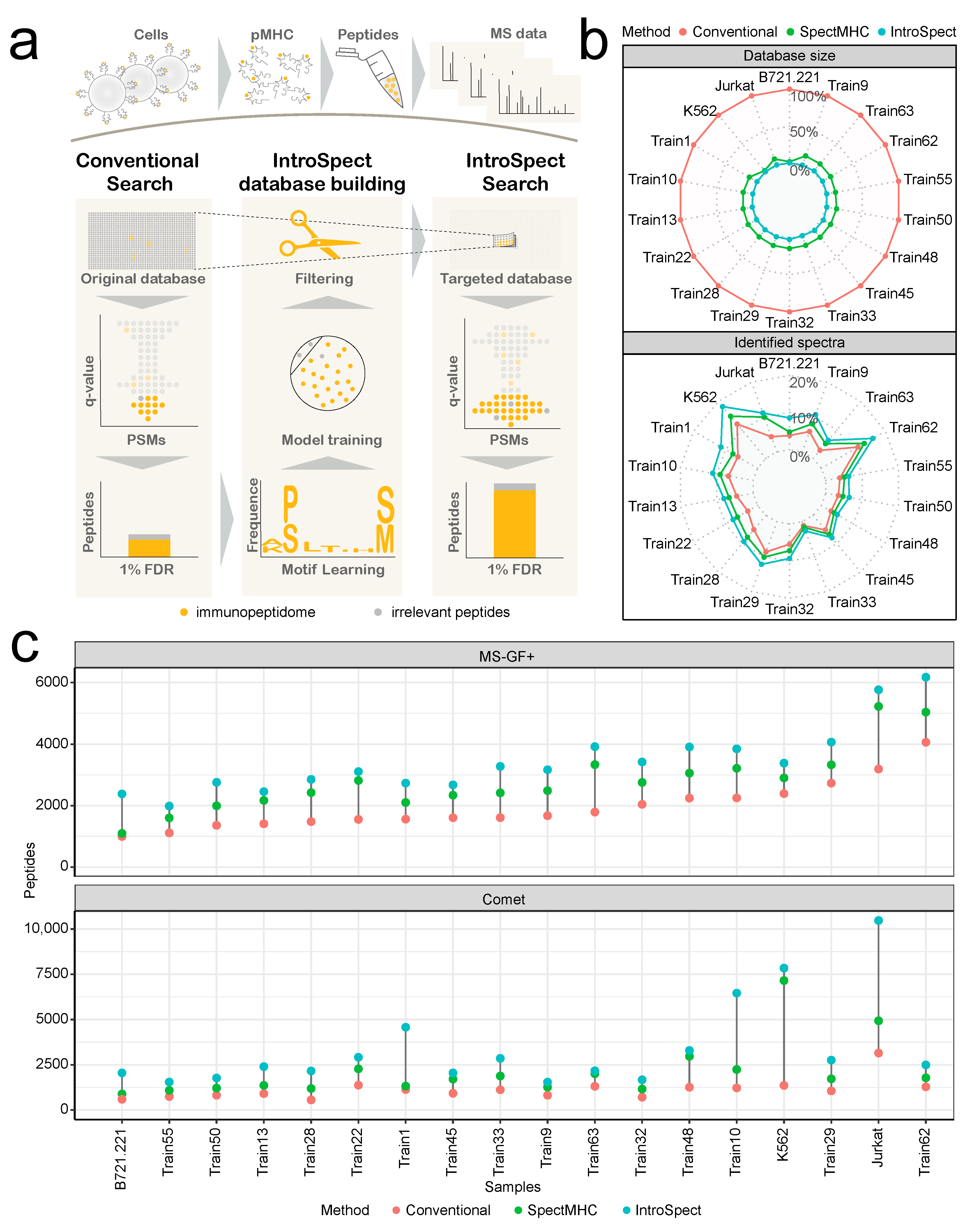
3.1. The Development of IntroSpect
3.2. IntroSpect Can Identify Substantially More Peptides
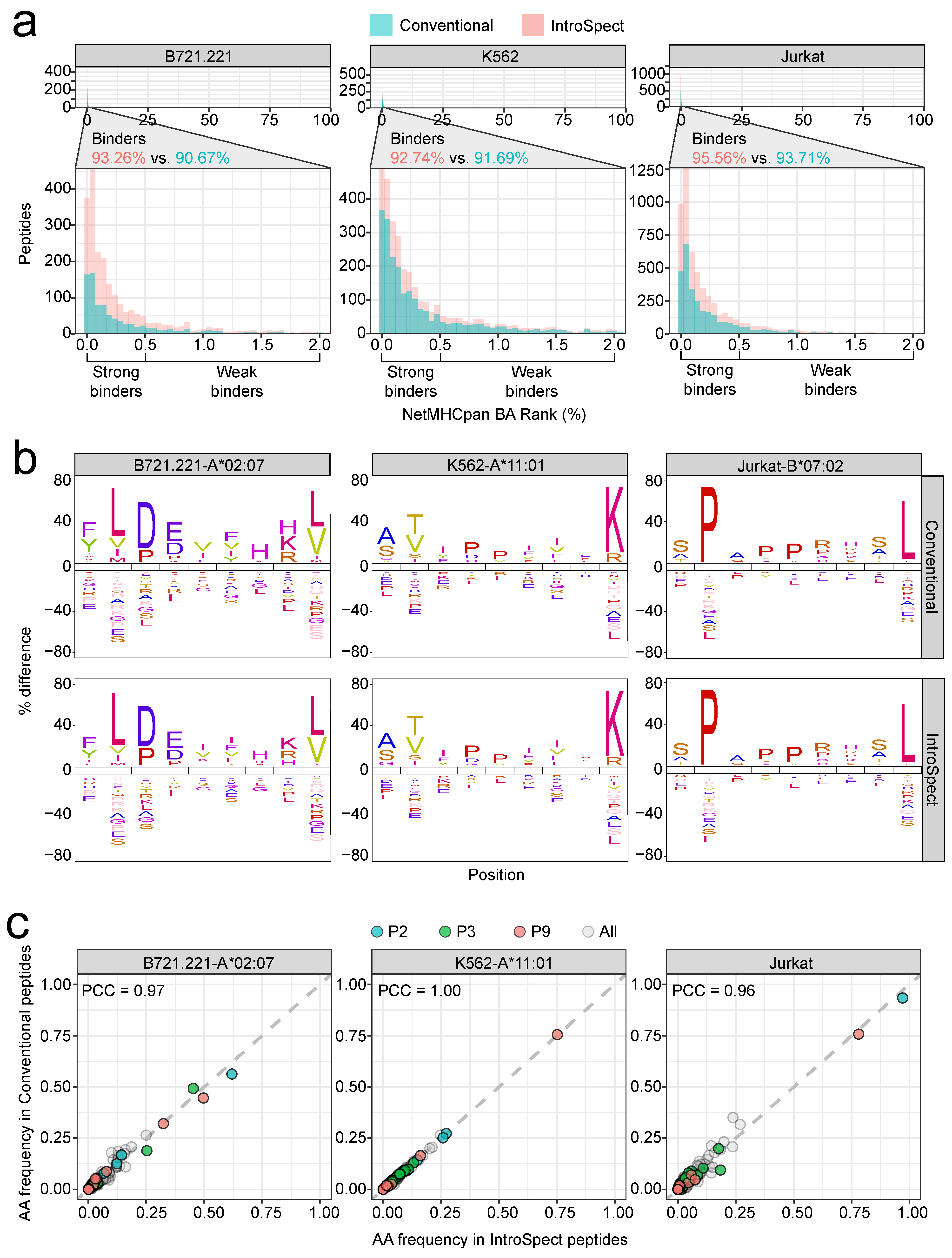
3.3. IntroSpect Achieved a Similar Accuracy as Conventional Search
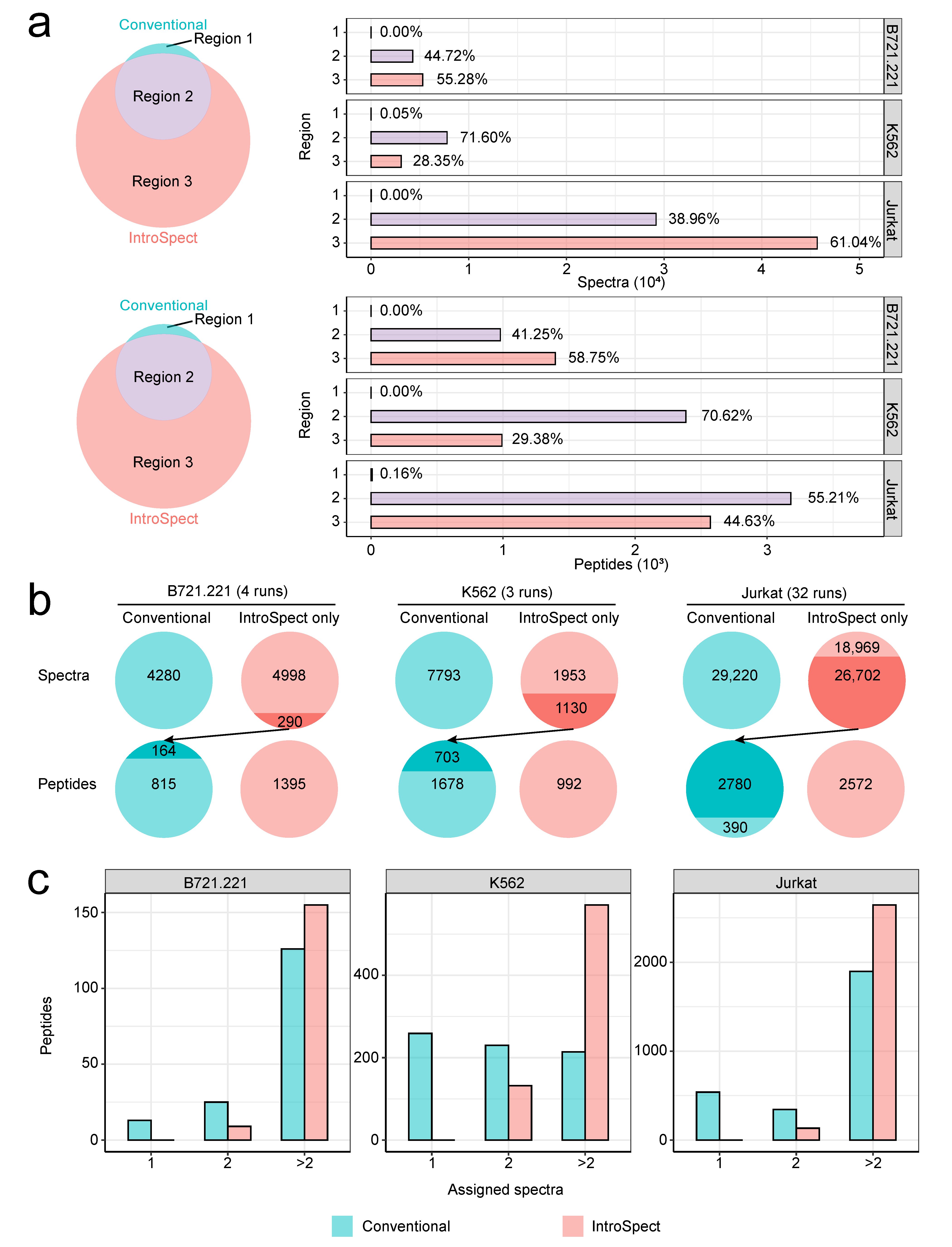
3.4. IntroSpect Inherits the Results of Conventional Database Search
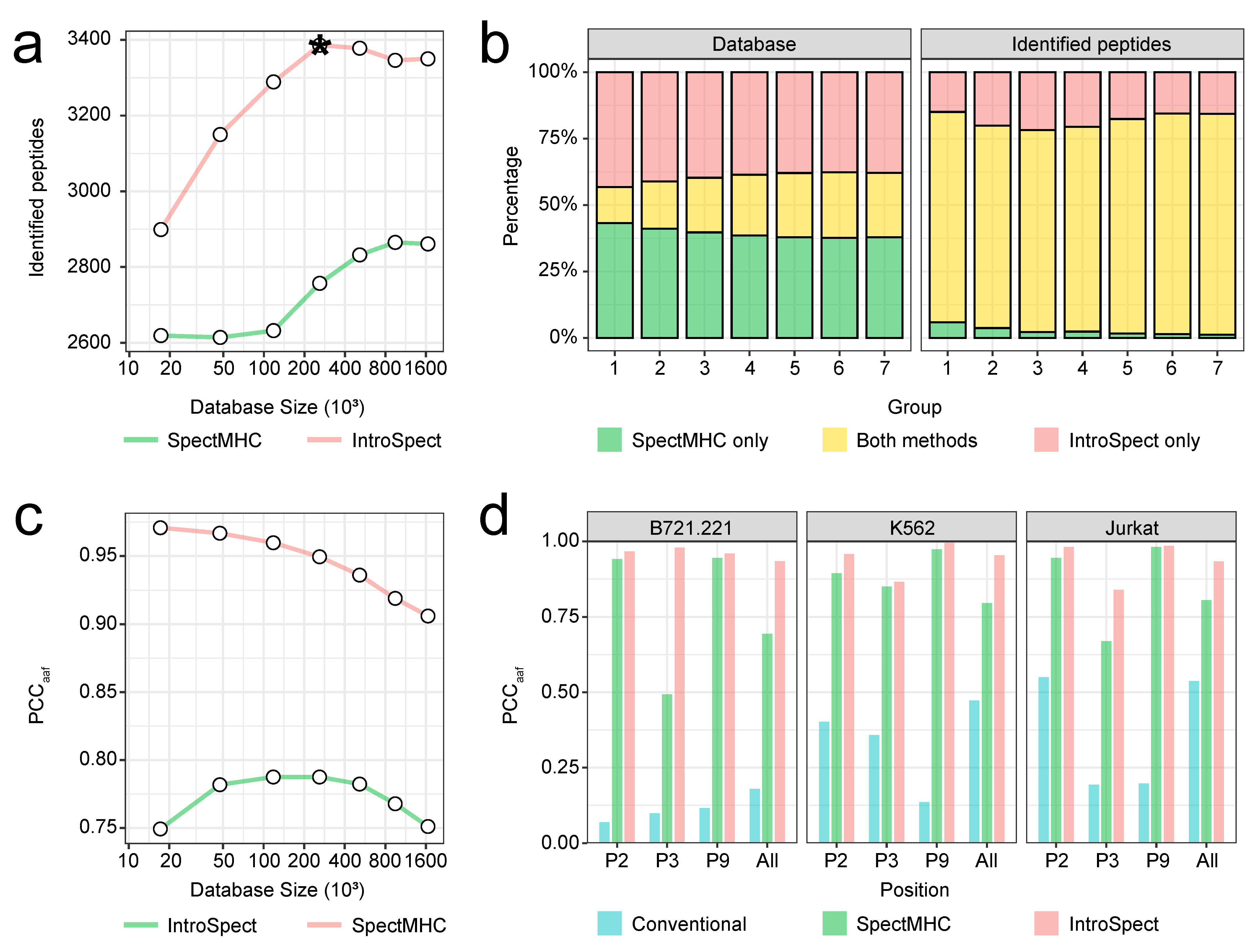
3.5. The Database Generated by IntroSpect Is Smaller and More Targeted Than SpectMHC
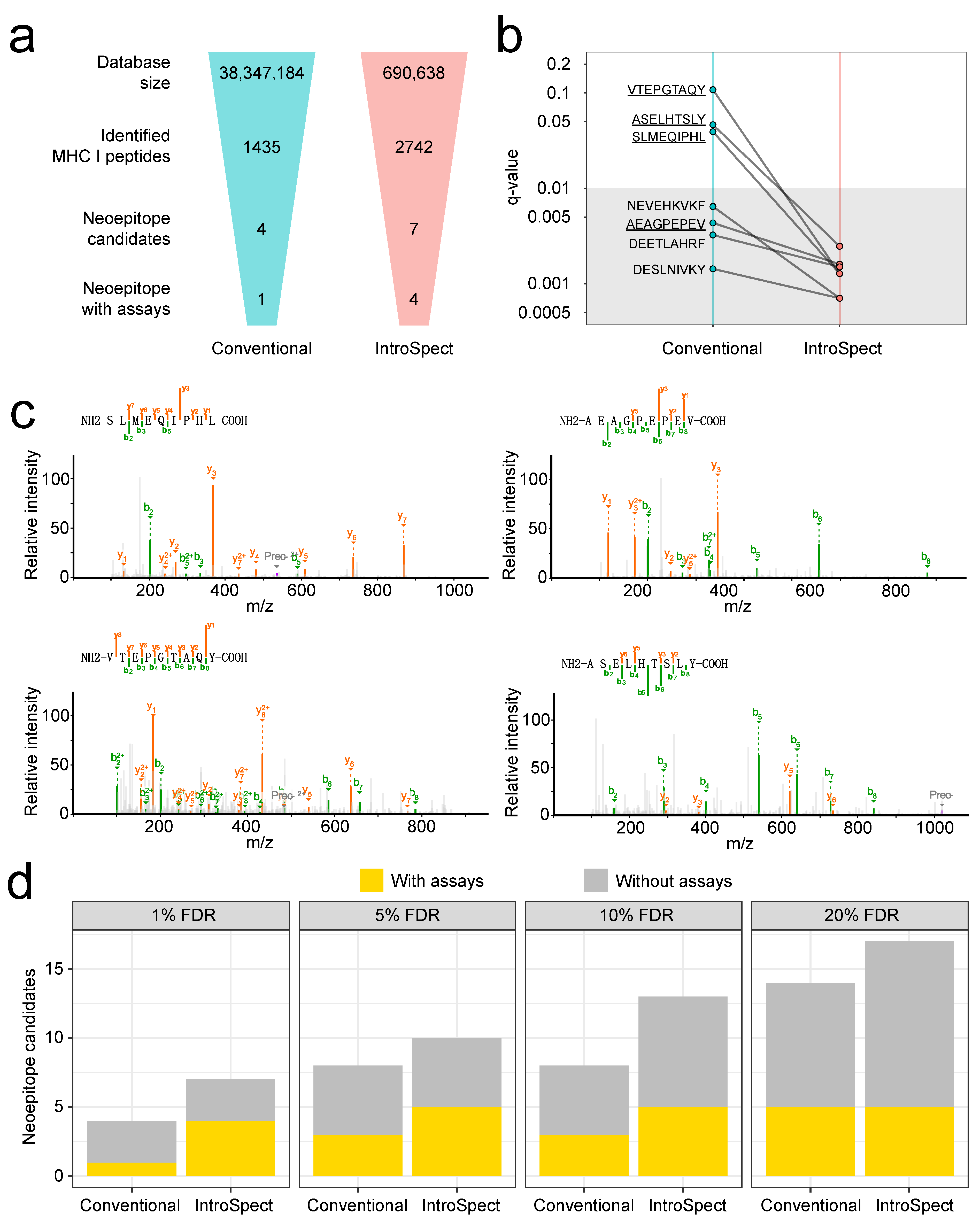
3.6. IntroSpect Identified More Neoepitopes Than Conventional Method
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Neefjes, J.; Jongsma, M.L.M.; Paul, P.; Bakke, O. Towards a systems understanding of MHC class I and MHC class II antigen presentation. Nat. Rev. Immunol. 2011, 11, 823–836. [Google Scholar] [CrossRef] [PubMed]
- Vizcaíno, J.A.; Kubiniok, P.; Kovalchik, K.A.; Ma, Q.; Duquette, J.D.; Mongrain, I.; Deutsch, E.W.; Peters, B.; Sette, A.; Sirois, I.; et al. The Human Immunopeptidome Project, A roadmap to predict and treat immune diseases. Mol. Cell. Proteom. 2020, 19, 31–49. [Google Scholar] [CrossRef] [PubMed]
- Bassani-Sternberg, M.; Bräunlein, E.; Klar, R.; Engleitner, T.; Sinitcyn, P.; Audehm, S.; Straub, M.; Weber, J.; Slota-Huspenina, J.; Specht, K.; et al. Direct identification of clinically relevant neoepitopes presented on native human melanoma tissue by mass spectrometry. Nat. Commun. 2016, 7, 13404. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hu, Z.; Ott, P.A.; Wu, C.J. Towards personalized, tumour-specific, therapeutic vaccines for cancer. Nat. Rev. Immunol. 2018, 18, 168. [Google Scholar] [CrossRef]
- Fritsche, J.; Rakitsch, B.; Hoffgaard, F.; Römer, M.; Schuster, H.; Kowalewski, D.J.; Priemer, M.; Stos-Zweifel, V.; Hörzer, H.; Satelli, A.; et al. Translating immunopeptidomics to immunotherapy-decision-making for patient and personalized target selection. Proteomics 2018, 18, 1700284. [Google Scholar] [CrossRef] [Green Version]
- Bassani-Sternberg, M.; Coukos, G. Mass spectrometry-based antigen discovery for cancer immunotherapy. Curr. Opin. Immunol. 2016, 41, 9–17. [Google Scholar] [CrossRef]
- Di Marco, M.; Peper, J.K.; Rammensee, H.G. Identification of immunogenic epitopes by MS/MS. Cancer J. 2017, 23, 102–107. [Google Scholar] [CrossRef]
- Kalaora, S.; Barnea, E.; Merhavi-Shoham, E.; Qutob, N.; Teer, J.K.; Shimony, N.; Schachter, J.; Rosenberg, S.A.; Besser, M.J.; Admon, A.; et al. Use of HLA peptidomics and whole exome sequencing to identify human immunogenic neo-antigens. Oncotarget 2016, 7, 5110. [Google Scholar] [CrossRef] [Green Version]
- Purcell, A.W.; Ramarathinam, S.H.; Ternette, N. Mass spectrometry–based identification of MHC-bound peptides for immunopeptidomics. Nat. Protoc. 2019, 14, 1687. [Google Scholar] [CrossRef]
- Kote, S.; Pirog, A.; Bedran, G.; Alfaro, J.; Dapic, I. Mass spectrometry-based identification of MHC-associated peptides. Cancers 2020, 12, 535. [Google Scholar] [CrossRef] [Green Version]
- Kim, S.; Pevzner, P.A. MS-GF+ makes progress towards a universal database search tool for proteomics. Nat. Commun. 2014, 5, 5277. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eng, J.K.; Jahan, T.A.; Hoopmann, M.R. Comet, An open-source MS/MS sequence database search tool. Proteomics 2013, 13, 22–24. [Google Scholar] [CrossRef] [PubMed]
- Fenyö, D.; Beavis, R.C. A method for assessing the statistical significance of mass spectrometry-based protein identifications using general scoring schemes. Anal. Chem. 2003, 75, 768–774. [Google Scholar] [CrossRef] [PubMed]
- Tyanova, S.; Temu, T.; Cox, J. The MaxQuant computational platform for mass spectrometry-based shotgun proteomics. Nat. Protoc. 2016, 11, 2301. [Google Scholar] [CrossRef] [PubMed]
- Noble, W.S. Mass spectrometrists should search only for peptides they care about. Nat. Methods 2015, 12, 605–608. [Google Scholar] [CrossRef] [Green Version]
- Frewen, W.E.; Merrihew, G.E.; Wu, C.C.; Noble, W.S.; MacCoss, M.J. Analysis of peptide MS/MS spectra from large-scale proteomics experiments using spectrum libraries. Anal. Chem. 2006, 78, 5678–5684. [Google Scholar] [CrossRef]
- Faridi, P.; Purcell, A.W.; Croft, N.P. In immunopeptidomics we need a sniper instead of a shotgun. Proteomics 2018, 18, 1700464. [Google Scholar] [CrossRef] [Green Version]
- Murphy, J.P.; Konda, P.; Kowalewski, D.J.; Schuster, H.; Clements, D.; Kim, Y.; Cohen, A.M.; Sharif, T.; Nielsen, M.; Stevanovic, S.; et al. MHC-I ligand discovery using targeted database searches of mass spectrometry data, Implications for T-cell immunotherapies. J. Proteome Res. 2017, 16, 1806–1816. [Google Scholar] [CrossRef] [Green Version]
- Li, S.; Tang, H.; Ye, Y. A meta-proteogenomic approach to peptide identification incorporating assembly uncertainty and genomic variation. Mol. Cell. Proteom. 2019, 18, S183–S192. [Google Scholar] [CrossRef] [Green Version]
- Ivanov, M.V.; Levitsky, L.I.; Gorshkov, M.V. Adaptation of decoy fusion strategy for existing multi-stage search workflows. J. Am. Soc. Mass Spectrom. 2016, 27, 1579–1582. [Google Scholar] [CrossRef]
- Andreatta, M.; Nicastri, A.; Peng, X.; Hancock, G.; Dorrell, L.; Ternette, N.; Nielsen, M. MS-Rescue, A Computational Pipeline to Increase the Quality and Yield of Immunopeptidomics Experiments. Proteomics 2019, 19, 1800357. [Google Scholar] [CrossRef] [PubMed]
- Li, K.; Jain, A.; Malovannaya, A.; Wen, B.; Zhang, B. DeepRescore, Leveraging Deep Learning to Improve Peptide Identification in Immunopeptidomics. Proteomics 2020, 20, 1900334. [Google Scholar] [CrossRef] [PubMed]
- Knudsen, G.M.; Chalkley, R.J. The effect of using an inappropriate protein database for proteomic data analysis. PLoS ONE 2011, 6, e20873. [Google Scholar]
- Blakeley, P.; Overton, I.M.; Hubbard, S.J. Addressing statistical biases in nucleotide-derived protein databases for proteogenomic search strategies. J. Proteome Res. 2012, 11, 5221–5234. [Google Scholar] [CrossRef]
- Li, H.; Joh, Y.S.; Kim, H.; Paek, E.; Lee, S.W.; Hwang, K.B. Evaluating the effect of database inflation in proteogenomic search on sensitive and reliable peptide identification. BMC Genom. 2016, 17, 1031. [Google Scholar] [CrossRef] [Green Version]
- Krug, K.; Carpy, A.; Behrends, G.; Matic, K.; Soares, N.C.; Macek, B. Deep coverage of the Escherichia coli proteome enables the assessment of false discovery rates in simple proteogenomic experiments. Mol. Cell. Proteom. 2013, 12, 3420–3430. [Google Scholar] [CrossRef] [Green Version]
- Sinitcyn, P.; Rudolph, J.D.; Cox, J. Computational methods for understanding mass spectrometry–based shotgun proteomics data. Annu. Rev. Biomed. Data Sci. 2018, 1, 207–234. [Google Scholar] [CrossRef]
- Nesvizhskii, A.I. Proteogenomics, Concepts, applications and computational strategies. Nat. Methods 2014, 11, 1114. [Google Scholar] [CrossRef]
- Bichmann, L.; Nelde, A.; Ghosh, M.; Heumos, L.; Mohr, C.; Peltzer, A.; Kuchenbecker, L.; Sachsenberg, T.; Walz, J.S.; Stevanović, S. MHCquant, Automated and reproducible data analysis for immunopeptidomics. J. Proteome Res. 2019, 18, 3876–3884. [Google Scholar] [CrossRef]
- Elias, J.E.; Gygi, S.P. Target-decoy search strategy for increased confidence in large-scale protein identifications by mass spectrometry. Nat. Methods 2007, 4, 207–214. [Google Scholar] [CrossRef]
- Nesvizhskii, A.I. A survey of computational methods and error rate estimation procedures for peptide and protein identification in shotgun proteomics. J. Proteom. 2010, 73, 2092–2123. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reche, P.A.; Keskin, D.B.; Hussey, R.E.; Ancuta, P.; Gabuzda, D.; Reinherz, E.L. Elicitation from virus-naive individuals of cytotoxic T lymphocytes directed against conserved HIV-1 epitopes. Med. Immunol. 2006, 5, 1. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bassani-Sternberg, M.; Pletscher-Frankild, S.; Jensen, L.J.; Mann, M. Mass spectrometry of human leukocyte antigen class I peptidomes reveals strong effects of protein abundance and turnover on antigen presentation. Mol. Cell. Proteom. 2015, 14, 658–673. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, Y.; Chen, Y.; Shi, C.; Huang, Z.; Zhang, Y.; Li, S.; Li, Y.; Ye, J.; Yu, C.; Li, Z.; et al. SOAPnuke: A MapReduce acceleration-supported software for integrated quality control and preprocessing of high-throughput sequencing data. Gigascience 2018, 7, gix120. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, H. Minimap2, Pairwise alignment for nucleotide sequences. Bioinformatics 2018, 34, 3094–3100. [Google Scholar] [CrossRef] [PubMed]
- McKenna, A.; Hanna, M.; Banks, E.; Sivachenko, A.; Cibulskis, K.; Kernytsky, A.; Garimella, K.; Altshuler, D.; Gabriel, S.; Daly, M.; et al. The Genome Analysis Toolkit, A MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010, 20, 1297–1303. [Google Scholar] [CrossRef] [Green Version]
- Kim, D.; Langmead, B.; Salzberg, S.L. HISAT, A fast spliced aligner with low memory requirements. Nat. Methods 2015, 12, 357–360. [Google Scholar] [CrossRef] [Green Version]
- Li, B.; Dewey, C.N. RSEM, Accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinform. 2011, 12, 323. [Google Scholar] [CrossRef] [Green Version]
- Abelin, J.G.; Keskin, D.B.; Sarkizova, S.; Hartigan, C.R.; Zhang, W.; Sidney, J.; Stevens, J.; Lane, W.; Zhang, G.L.; Eisenhaure, T.M.; et al. Mass spectrometry profiling of HLA-associated peptidomes in mono-allelic cells enables more accurate epitope prediction. Immunity 2017, 46, 315–326. [Google Scholar] [CrossRef] [Green Version]
- Bulik-Sullivan, B.; Busby, J.; Palmer, C.D.; Davis, M.J.; Murphy, T.; Clark, A.; Busby, M.; Duke, F.; Yang, A.; Young, L.; et al. Deep learning using tumor HLA peptide mass spectrometry datasets improves neoantigen identification. Nat. Biotechnol. 2019, 37, 55–63. [Google Scholar] [CrossRef]
- Comai, L.; Katz, J.E.; Mallick, P. Data Conversion with ProteoWizard msConvert. Methods Mol. Biol. 2017, 1550, 339. [Google Scholar]
- Apweiler, R.; Bairoch, A.; Wu, C.H.; Barker, W.C.; Boeckmann, B.; Ferro, S.; Gasteiger, E.; Huang, H.; Lopez, R.; Magrane, M.; et al. UniProt, The Universal Protein knowledgebase. Nucleic Acids Res. 2004, 32, 115–119. [Google Scholar] [CrossRef] [PubMed]
- MacCoss, M.J.; Noble, W.S.; Käll, L. Fast and accurate protein false discovery rates on large-scale proteomics data sets with percolator 3.0. J. Am. Soc. Mass Spectrom. 2016, 27, 1719–1727. [Google Scholar]
- Andreatta, M.; Alvarez, B.; Nielsen, M. GibbsCluster, Unsupervised clustering and alignment of peptide sequences. Nucleic Acids Res. 2017, 45, W458–W463. [Google Scholar] [CrossRef] [Green Version]
- Liu, G.; Li, D.; Li, Z.; Qiu, S.; Li, W.; Chao, C.C.; Yang, N.; Li, H.; Cheng, Z.; Song, X.; et al. PSSMHCpan, A novel PSSM-based software for predicting class I peptide-HLA binding affinity. Giga Sci. 2017, 6, gix017. [Google Scholar] [CrossRef] [Green Version]
- Altschul, S.F.; Gertz, E.M.; Agarwala, R.; Schäffer, A.A.; Yu, Y.K. PSI-BLAST pseudo counts and the minimum description length principle. Nucleic Acids Res. 2009, 37, 815–824. [Google Scholar] [CrossRef] [Green Version]
- Li, K.; Vaudel, M.; Zhang, B.; Ren, Y.; Wen, B. PDV: An integrative proteomics data viewer. Bioinformatics 2019, 35, 1249–1251. [Google Scholar] [CrossRef]
- Gfeller, D.; Guillaume, P.; Michaux, J.; Pak, H.S.; Daniel, R.T.; Racle, J.; Coukos, G.; Bassani-Sternberg, J. The length distribution and multiple specificity of naturally presented HLA-I ligands. J. Immunol. 2018, 201, 3705–3716. [Google Scholar] [CrossRef] [Green Version]
- Trolle, T.; McMurtrey, C.P.; Sidney, J.; Bardet, W.; Osborn, S.C.; Kaever, T.; Sette, A.; Hildebrand, W.H.; Nielsen, M.; Peters, B. The length distribution of class I–restricted T cell epitopes is determined by both peptide supply and MHC allele–specific binding preference. J. Immunol. 2016, 196, 1480–1487. [Google Scholar] [CrossRef] [Green Version]
- Newey, A.; Griffiths, B.; Michaux, J.; Pak, H.S.; Stevenson, B.J.; Woolston, A.; Semiannikova, M.; Spain, G.; Barber, L.J.; Matthews, N.; et al. Immunopeptidomics of colorectal cancer organoids reveals a sparse HLA class I neoantigen landscape and no increase in neoantigens with interferon or MEK-inhibitor treatment. J. Immunother. Cancer 2019, 7, 309. [Google Scholar] [CrossRef]
- Jurtz, V.; Paul, S.; Andreatta, M.; Marcatili, P.; Peters, B.; Nielsen, M. NetMHCpan-4.0, Improved peptide–MHC class I interaction predictions integrating eluted ligand and peptide binding affinity data. J. Immunol. 2017, 199, 3360–3368. [Google Scholar] [CrossRef] [PubMed]
- Maddelein, D.; Colaert, N.; Buchanan, I.; Hulstaert, N.; Gevaert, K.; Martens, L. The iceLogo web server and SOAP service for determining protein consensus sequences. Nucleic Acids Res. 2015, 43, W543–W546. [Google Scholar] [CrossRef] [PubMed]
- Colaert, N.; Helsens, K.; Martens, L.; Vandekerckhove, J.; Gevaert, K. Improved visualization of protein consensus sequences by iceLogo. Nat. Methods 2009, 6, 786–787. [Google Scholar] [CrossRef] [PubMed]
- Vita, R.; Mahajan, S.; Overton, J.A.; Dhanda, S.K.; Martini, S.; Cantrell, J.R.; Wheeler, D.K.; Sette, A.; Peters, B. The immune epitope database (IEDB), 2018 update. Nucleic Acids Res. 2019, 47, D339–D343. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chujoh, Y.; Sobao, Y.; Miwa, K.; Kaneko, Y.; Takiguchi, M. The role of anchor residues in the binding of peptides to HLA-A* 1101 molecules. Tissue Antigens 1998, 52, 501–509. [Google Scholar] [CrossRef] [PubMed]
- Giam, K.; Ayala-Perez, R.; Illing, P.T.; Schittenhelm, R.B.; Croft, N.P.; Purcell, A.W.; Dudek, N.L. A comprehensive analysis of peptides presented by HLA-A1. Tissue Antigens 2015, 85, 492–496. [Google Scholar] [CrossRef]
- Toh, H.; Savoie, C.J.; Kamikawaji, N.; Muta, S.; Sasazuki, T.; Kuhara, S. Changes at the floor of the peptide-binding groove induce a strong preference for Proline at position 3 of the bound peptide, Molecular dynamics simulations of HLA-A* 0217. Biopolym. Orig. Res. Biomol. 2000, 54, 318–327. [Google Scholar] [CrossRef]
- Rognan, D.; Scapozza, L.; Folkers, G.; Daser, A. Rational design of nonnatural peptides as high-affinity ligands for the HLA-B* 2705 human leukocyte antigen. Proc. Natl. Acad. Sci. USA 1995, 92, 753–757. [Google Scholar] [CrossRef] [Green Version]
- Laumont, C.M.; Vincent, K.; Hesnard, L.; Audemard, É.; Bonneil, É.; Laverdure, J.P.; Gendron, P.; Courcelles, M.; Hardy, M.-P.; Perreault, C.; et al. Noncoding regions are the main source of targetable tumor-specific antigens. Sci. Transl. Med. 2018, 10, eaau5516. [Google Scholar] [CrossRef] [Green Version]
- Chen, R.; Fauteux, F.; Foote, S.; Stupak, J.; Tremblay, T.L.; Gurnani, K.; Fulton, K.M.; Weeratna, R.D.; Twine, S.M.; Li, J. Chemical Derivatization Strategy for Extending the Identification of MHC Class I Immunopeptides. Anal Chem. 2018, 90, 11409–11416. [Google Scholar] [CrossRef]
- Koumantou, D.; Barnea, E.; Martin-Esteban, A.; Maben, Z.; Papakyriakou, A.; Mpakali, A.; Kokkala, P.; Pratsinis, H.; Georgiadis, D.; Stern, L.J.; et al. Editing the immunopeptidome of melanoma cells using a potent inhibitor of endoplasmic reticulum aminopeptidase 1 (ERAP1). Cancer Immunol. Immunother. 2019, 68, 1245–1261. [Google Scholar] [CrossRef] [PubMed]
- Steinbach, A.; Winter, J.; Reuschenbach, M.; Blatnik, R.; Klevenz, A.; Bertrand, M.; Hoppe, S.; von Knebel Doeberitz, M.; Grabowska, A.K.; Riemer, A.B. ERAP1 overexpression in HPV-induced malignancies, A possible novel immune evasion mechanism. Oncoimmunology 2017, 6, e1336594. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Becker, J.P.; Helm, D.; Rettel, M.; Stein, F.; Hernandez-Sanchez, A.; Urban, K.; Gebert, J.; Kloor, M.; Neu-Yilik, G.; von Knebel Doeberitz, M.; et al. NMD inhibition by 5-azacytidine augments presentation of immunogenic frameshift-derived neoepitopes. Iscience 2021, 24, 102389. [Google Scholar] [CrossRef] [PubMed]
- Tran, N.H.; Qiao, R.; Xin, L.; Chen, X.; Liu, C.; Zhang, X.; Shan, B.; Ghodsi, A.; Li, M. Deep learning enables de novo peptide sequencing from data-independent-acquistion mass spectrometry. Nat. Methods 2019, 16, 63–66. [Google Scholar] [CrossRef]
- Tran, N.H.; Zhang, X.; Xin, L.; Shan, B.; Li, M. De novo peptide sequencing by deep learning. Proc. Natl. Acad. Sci. USA 2017, 114, 8247–8252. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tran, N.H.; Rahman, M.Z.; He, L.; Xin, L.; Shan, B.; Li, M. Complete De Novo Assembly of Monoclonal Antibody Sequences. Sci. Rep. 2016, 6, 31730. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, S.; DeCourcy, A.; Tang, H. Constrained De Novo Sequencing of neo-Epitope Peptides Using Tandem Mass Spectrometry. In Proceedings of the International Conference on Research in Computational Molecular Biology, 21–24 April 2018; Springer: Cham, Switzerland, 2018; pp. 138–153. [Google Scholar]
- Faridi, P.; Li, C.; Ramarathinam, S.H.; Vivian, J.P.; Illing, P.T.; Mifsud, N.A.; Ayala, R.; Song, J.; Gearing, L.J.; Hertzog, P.J.; et al. A subset of HLA-I peptides are not genomically templated, Evidence for cis-and trans-spliced peptide ligands. Sci. Immunol. 2018, 3, eaar3947. [Google Scholar] [CrossRef] [Green Version]
- Chong, C.; Müller, M.; Pak, H.S.; Harnett, D.; Huber, F.; Grun, D.; Leleu, M.; Auger, A.; Arnaud, M.; Stevenson, B.J.; et al. Integrated proteogenomic deep sequencing and analytics accurately identify non-canonical peptides in tumor immunopeptidomes. Nat. Commun. 2020, 11, 1293. [Google Scholar] [CrossRef] [Green Version]
- Guo, X.; Chen, F.; Gao, F.; Li, L.; Liu, K.; You, L.; Hua, C.; Yang, F.; Liu, W.; Peng, C.; et al. CNSA, A data repository for archiving omics data. Database 2020, 2020, baaa055. [Google Scholar] [CrossRef]
- Chen, F.Z.; You, L.J.; Yang, F.; Wang, L.N.; Guo, X.Q.; Gao, F.; Hua, C.; Tan, C.; Fang, L.; Shan, R.Q.; et al. CNGBdb, China National GeneBank DataBase. Yi Chuan Hered. 2020, 42, 799–809. [Google Scholar]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Dataset | Source | Spectra | HLA Alleles |
|---|---|---|---|
| K562 | Inhouse | 64,572 | transduced to express only A*11:01 |
| B721.221 | Public 39 | 111,662 | transduced to express only A*02:07 |
| Jurkat | Public 21 | 670,119 | A*03:01, B*07:02, C*04:01, A*03:01, B*35:03, C*07:02 |
| Train1 | Public 40 | 84,453 | A*11:01, B*27:02, C*03:03, A*11:01, B*55:01, C*05:01 |
| Train9 | Public 40 | 88,437 | A*11:01, B*51:01, C*01:02, A*68:01, B*56:01, C*07:02 |
| Train10 | Public 40 | 170,101 | A*29:02, B*44:03, C*04:01, A*29:02, B*35:01, C*16:01 |
| Train13 | Public 40 | 128,712 | A*01:01, B*08:01, C*07:01, A*03:01, B*35:01, C*04:01 |
| Train22 | Public 40 | 273,039 | A*31:01, B*08:01, C*12:03, A*03:01, B*38:01, C*07:01 |
| Train28 | Public 40 | 192,712 | A*03:01, B*35:03, C*03:03, A*03:01, B*51:01, C*04:01 |
| Train29 | Public 40 | 175,619 | A*03:02, B*44:03, C*03:03, A*26:01, B*35:02, C*16:01 |
| Train32 | Public 40 | 123,863 | A*29:02, B*44:03, C*07:02, A*03:01, B*07:02, C*16:01 |
| Train33 | Public 40 | 463,383 | A*02:03, B*15:02, C*08:01, A*68:01, B*15:13, C*08:01 |
| Train45 | Public 40 | 178,449 | A*31:01, B*44:02, C*05:01, A*01:01, B*67:01, C*12:03 |
| Train48 | Public 40 | 468,069 | A*24:02, B*18:01, C*07:02, A*25:01, B*07:02, C*12:03 |
| Train50 | Public 40 | 142,681 | A*33:03, B*44:03, C*07:06, A*68:01, B*35:01, C*04:01 |
| Train55 | Public 40 | 281,891 | A*01:01, B*08:01, C*07:01, A*24:02, B*08:01, C*07:01 |
| Train62 | Public 40 | 168,243 | A*02:01, B*44:02, C*05:01, A*68:01, B*44:02, C*07:04 |
| Train63 | Public 68 | 329,221 | A*31:01, B*44:02, C*05:01, A*02:01, B*27:05, C*02:02 |
| Software | Source | Identified | Selected for Synthesis | Confirmed Positive | Precision (%) |
|---|---|---|---|---|---|
| MS-GF+ | Both conventional and IntroSpect | 2385 | 91 | 89 | 97.80 |
| IntroSpect only | 993 | 27 | 26 | 96.30 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, L.; Liu, G.; Hou, G.; Xiang, H.; Zhang, X.; Huang, Y.; Zhang, X.; Li, B.; Lee, L.J. IntroSpect: Motif-Guided Immunopeptidome Database Building Tool to Improve the Sensitivity of HLA I Binding Peptide Identification by Mass Spectrometry. Biomolecules 2022, 12, 579. https://doi.org/10.3390/biom12040579
Zhang L, Liu G, Hou G, Xiang H, Zhang X, Huang Y, Zhang X, Li B, Lee LJ. IntroSpect: Motif-Guided Immunopeptidome Database Building Tool to Improve the Sensitivity of HLA I Binding Peptide Identification by Mass Spectrometry. Biomolecules. 2022; 12(4):579. https://doi.org/10.3390/biom12040579
Chicago/Turabian StyleZhang, Le, Geng Liu, Guixue Hou, Haitao Xiang, Xi Zhang, Ying Huang, Xiuqing Zhang, Bo Li, and Leo J. Lee. 2022. "IntroSpect: Motif-Guided Immunopeptidome Database Building Tool to Improve the Sensitivity of HLA I Binding Peptide Identification by Mass Spectrometry" Biomolecules 12, no. 4: 579. https://doi.org/10.3390/biom12040579