
Alteration of Cellular Energy Metabolism through LPAR2-Axin2 Axis in Gastric Cancer
 , ,
, , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Cell Culture
2.3. LPA Treatment
2.4. Cell Proliferation Assay
2.5. ECIS Wound-Healing Assay (ECIS)
2.6. Luciferase Reporter Assay
2.7. Mitochondrial Bioenergetics
2.8. Data Analysis from the Clinical Cohort
2.9. Statistical Analysis
3. Results
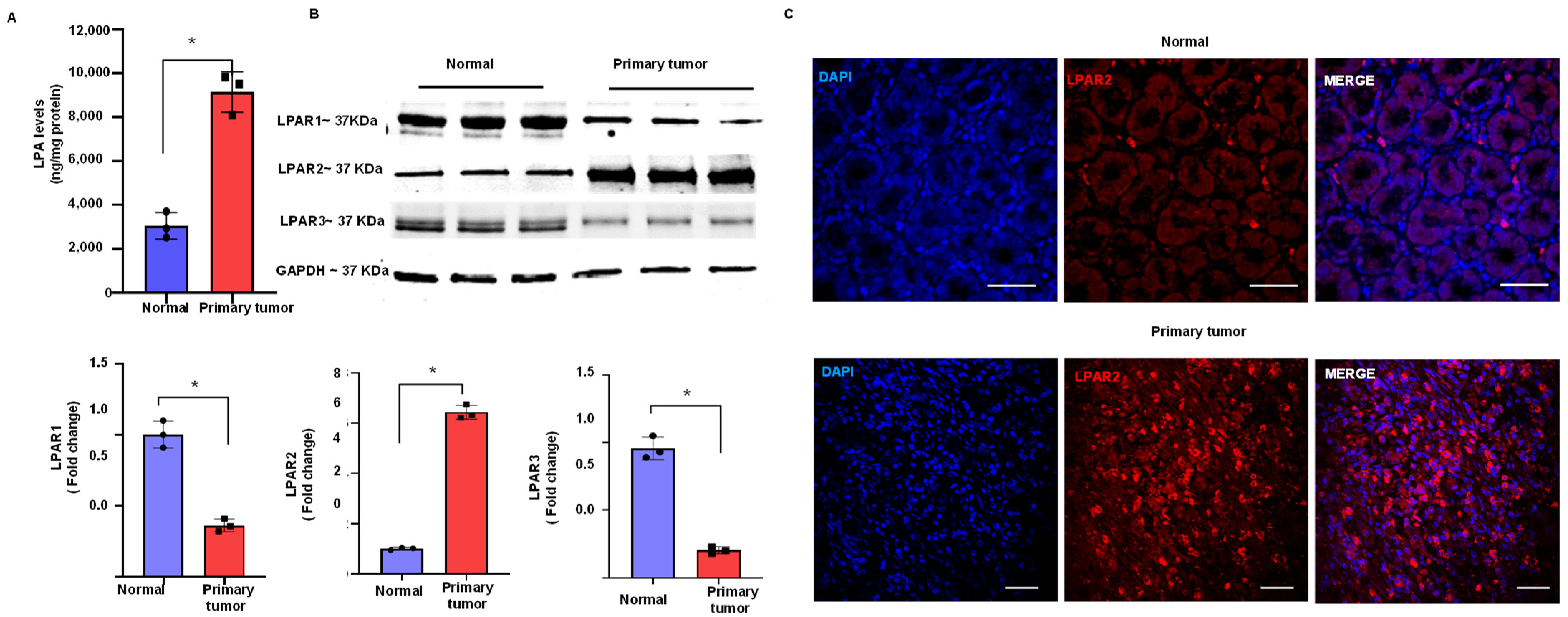
3.1. Upregulation of LPA and LPAR2 in Gastric Cancer
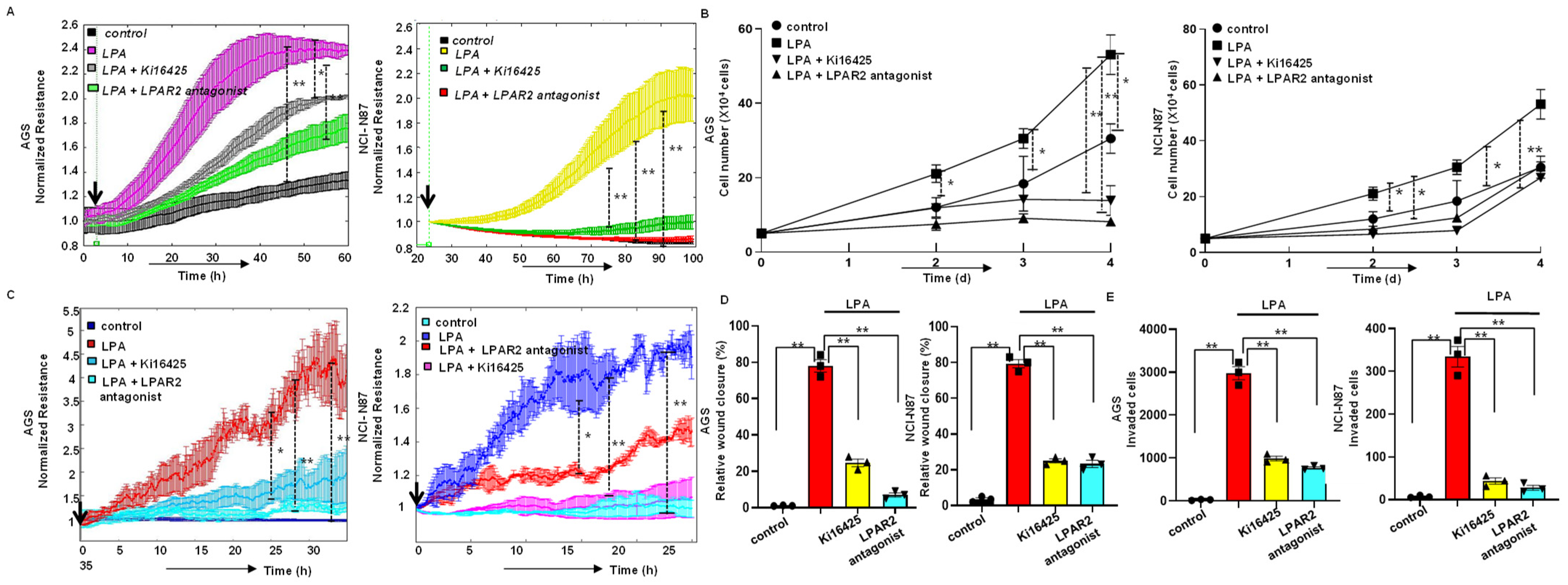
3.2. LPA Stimulates Proliferation of Gastric Cancer Cells through LPAR2
3.3. LPA-Induced Motility of Gastric Cancer Cells through LPAR2 Receptor
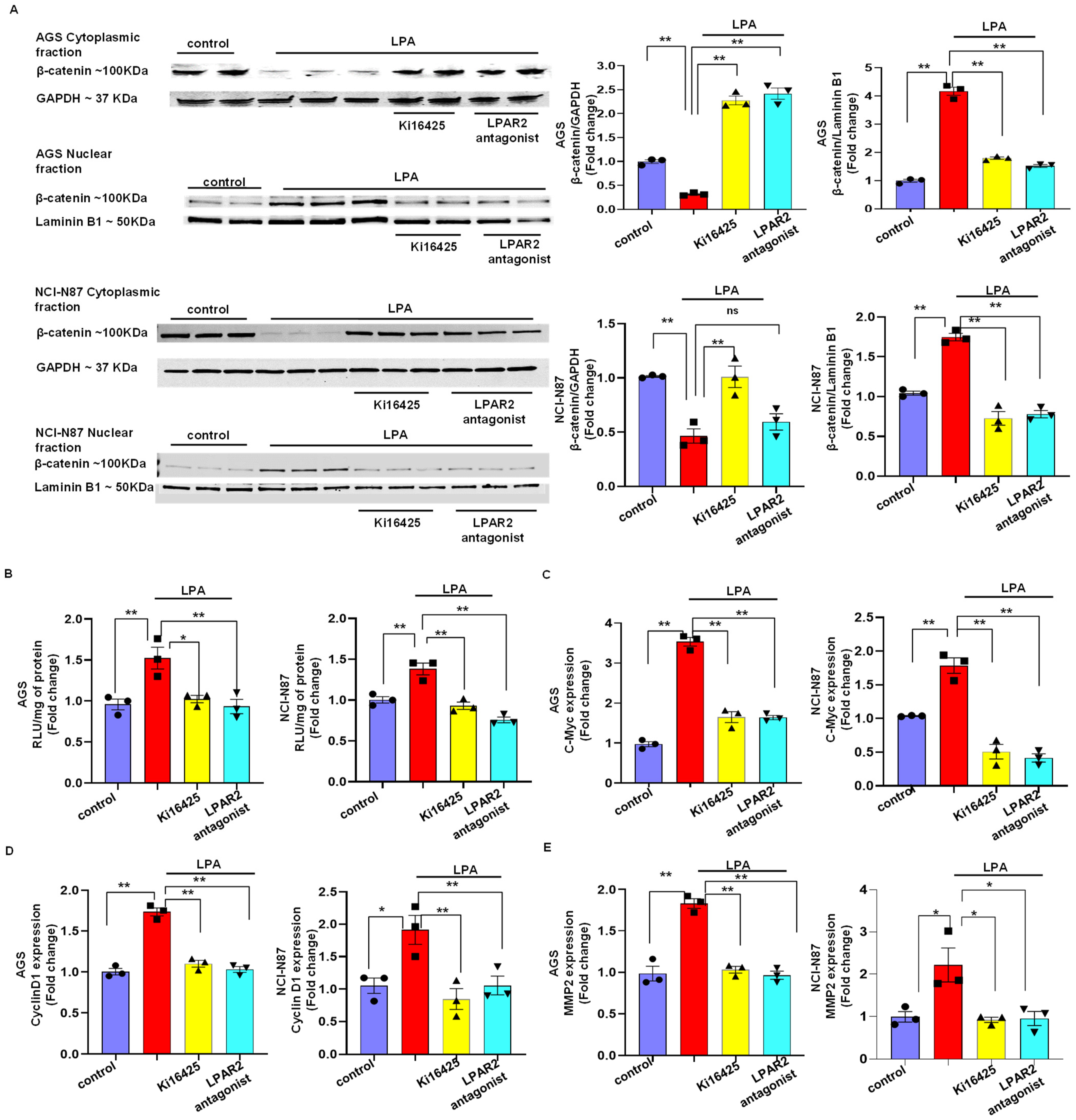
3.4. LPA Mediates Gastric Cancer through LPAR2 and the β-Catenin Signaling Pathway
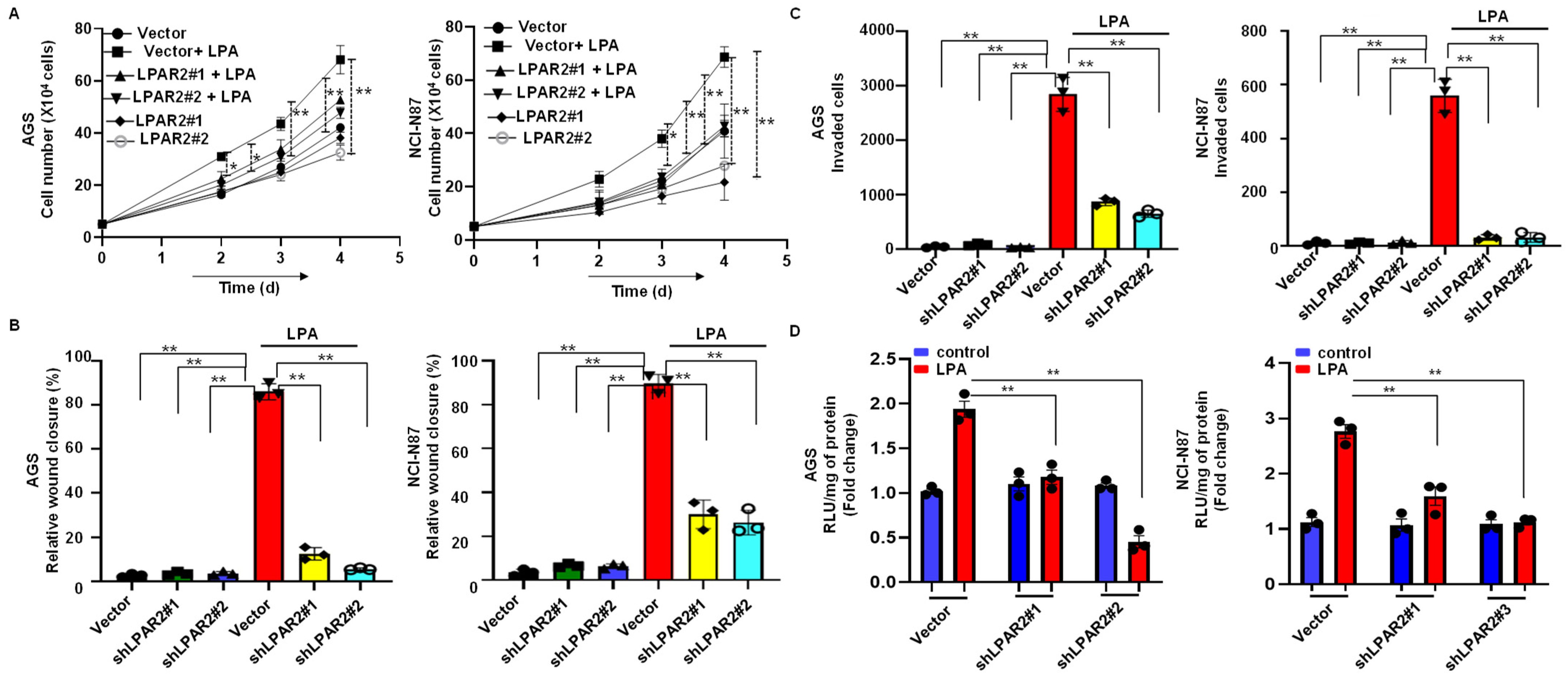
3.5. Silencing of LPAR2 by shRNA Inhibits LPA-Induced Progression of Gastric Cancer through the β-Catenin Signaling Pathway
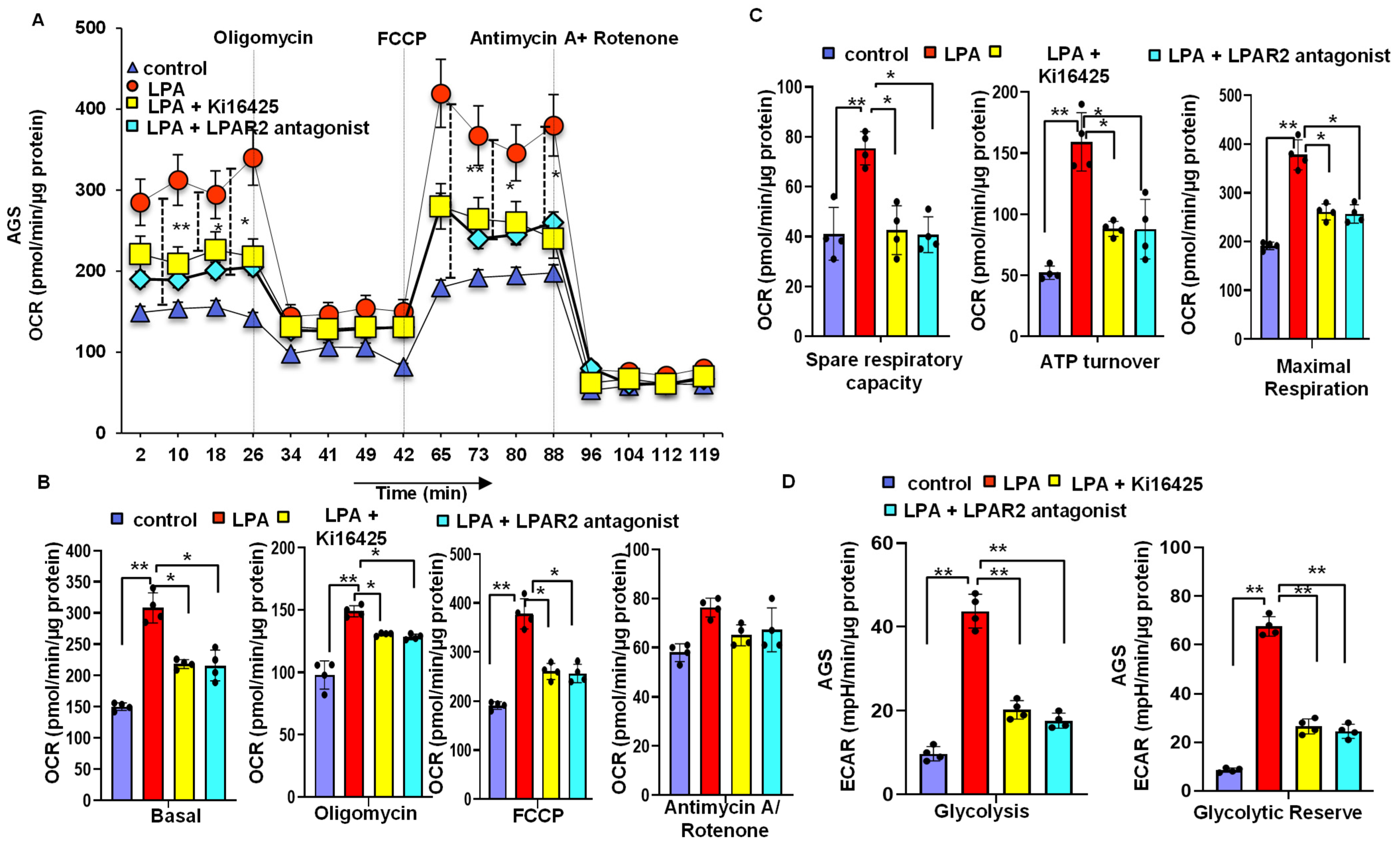
3.6. LPA Treatment Stimulates Cellular Energy Metabolism through the LPAR2 Antagonist
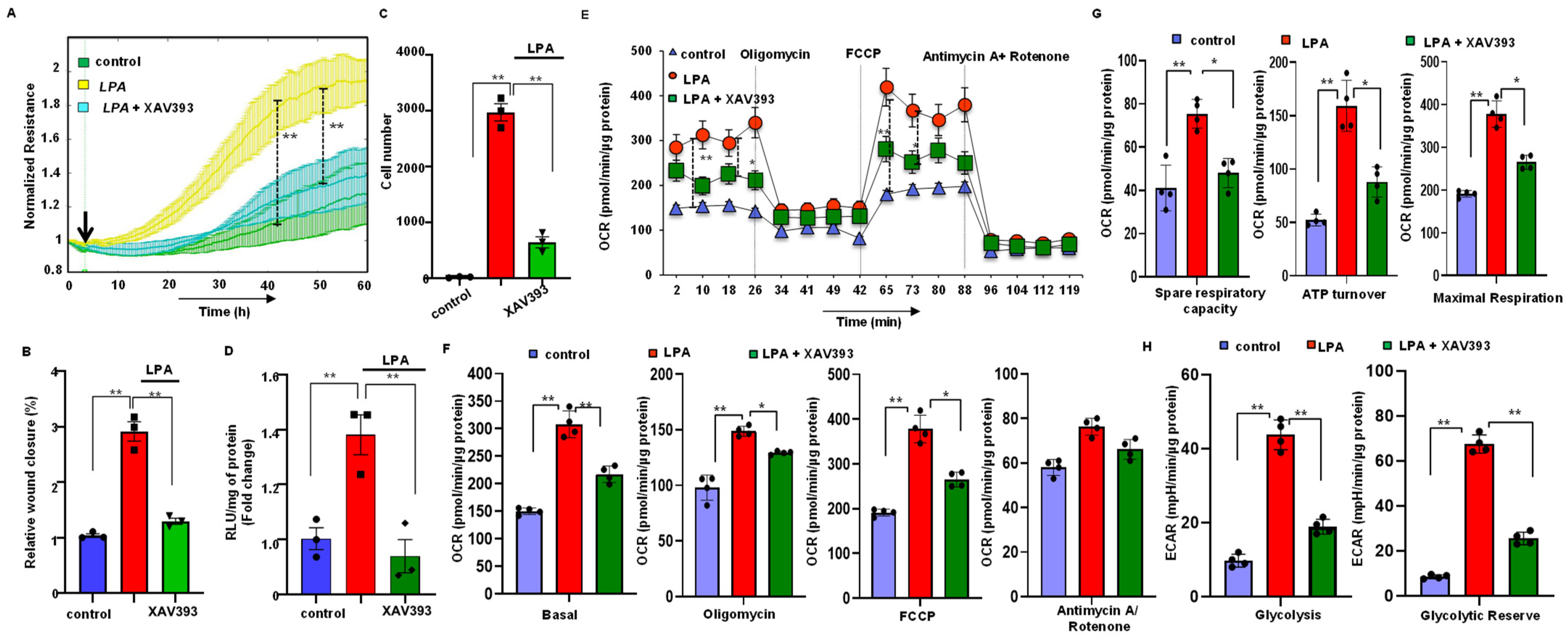
3.7. Stabilization of Axin2 Rescued LPA-Induced Cellular Energy Metabolism and Progression of Gastric Cancer Cells
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Rahman, R.; Asombang, A.W.; Ibdah, J.A. Characteristics of gastric cancer in Asia. World J. Gastroenterol. 2014, 20, 4483–4490. [Google Scholar] [CrossRef] [PubMed]
- Yang, W.; Raufi, A.; Klempner, S.J. Targeted therapy for gastric cancer: Molecular pathways and ongoing investigations. Biochim. Biophys. Acta 2014, 1846, 232–237. [Google Scholar] [CrossRef] [PubMed]
- Ara, H.; Takagishi, M.; Enomoto, A.; Asai, M.; Ushida, K.; Asai, N.; Shimoyama, Y.; Kaibuchi, K.; Kodera, Y.; Takahashi, M. Role for Daple in non-canonical Wnt signaling during gastric cancer invasion and metastasis. Cancer Sci. 2016, 107, 133–139. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Clements, W.M.; Wang, J.; Sarnaik, A.; Kim, O.J.; MacDonald, J.; Fenoglio-Preiser, C.; Groden, J.; Lowy, A.M. beta-Catenin mutation is a frequent cause of Wnt pathway activation in gastric cancer. Cancer Res. 2002, 62, 3503–3506. [Google Scholar] [PubMed]
- Ooi, C.H.; Ivanova, T.; Wu, J.; Lee, M.; Tan, I.B.; Tao, J.; Ward, L.; Koo, J.H.; Gopalakrishnan, V.; Zhu, Y.; et al. Oncogenic Pathway Combinations Predict Clinical Prognosis in Gastric Cancer. PLoS Genet. 2009, 5, e1000676. [Google Scholar] [CrossRef] [Green Version]
- Kikuchi, A. Tumor formation by genetic mutations in the components of the Wnt signaling pathway. Cancer Sci. 2003, 94, 225–229. [Google Scholar] [CrossRef]
- Polakis, P. The many ways of Wnt in cancer. Curr. Opin. Genet. Dev. 2007, 17, 45–51. [Google Scholar] [CrossRef]
- Chiurillo, M.A. Role of the Wnt/β-catenin pathway in gastric cancer: An in-depth literature review. World J. Exp. Med. 2015, 5, 84–102. [Google Scholar] [CrossRef]
- Abbruzzese, G.; Gorny, A.-K.; Kaufmann, L.T.; Cousin, H.; Kleino, I.; Steinbeisser, H.; Alfandari, D. The Wnt receptor Frizzled-4 modulates ADAM13 metalloprotease activity. J. Cell Sci. 2015, 128, 1139–1149. [Google Scholar] [CrossRef] [Green Version]
- Bhattarai, S.; Sharma, S.; Ara, H.; Subedi, U.; Sun, G.; Li, C.; Bhuiyan, M.S.; Kevil, C.; Armstrong, W.P.; Minvielle, M.T.; et al. Disrupted Blood-Brain Barrier and Mitochondrial Impairment by Autotaxin–Lysophosphatidic Acid Axis in Postischemic Stroke. J. Am. Heart Assoc. 2021, 10, e021511. [Google Scholar] [CrossRef]
- Deng, W.; Wang, D.-A.; Gosmanova, E.; Johnson, L.R.; Tigyi, G. LPA protects intestinal epithelial cells from apoptosis by inhibiting the mitochondrial pathway. Am. J. Physiol. Gastrointest. Liver Physiol. 2003, 284, G821–G829. [Google Scholar] [CrossRef] [PubMed]
- Sayas, C.L.; Ariaens, A.; Ponsioen, B.; Moolenaar, W.H. GSK-3 Is Activated by the Tyrosine Kinase Pyk2 during LPA1-mediated Neurite Retraction. Mol. Biol. Cell 2006, 17, 1834–1844. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Panchatcharam, M.; Miriyala, S.; Salous, A.; Wheeler, J.; Dong, A.; Mueller, P.; Sunkara, M.; Escalante-Alcalde, D.; Morris, A.J.; Smyth, S.S. Lipid phosphate phosphatase 3 negatively regulates smooth muscle cell phenotypic modulation to limit intimal hyperplasia. Arter. Thromb. Vasc. Biol. 2013, 33, 52–59. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gräler, M.H.; Goetzl, E.J. Lysophospholipids and their G protein-coupled receptors in inflammation and immunity. Biochim. Biophys. Acta 2002, 1582, 168–174. [Google Scholar] [CrossRef]
- Mills, G.B.; Moolenaar, W.H. The emerging role of lysophosphatidic acid in cancer. Nat. Rev. Cancer 2003, 3, 582–591. [Google Scholar] [CrossRef]
- Chandrashekar, D.S.; Karthikeyan, S.K.; Korla, P.K.; Patel, H.; Shovon, A.R.; Athar, M.; Netto, G.J.; Qin, Z.S.; Kumar, S.; Manne, U.; et al. UALCAN: An update to the integrated cancer data analysis platform. Neoplasia 2022, 25, 18–27. [Google Scholar] [CrossRef]
- Yang, D.; Yang, W.; Zhang, Q.; Hu, Y.; Bao, L.; Damirin, A. Migration of gastric cancer cells in response to lysophosphatidic acid is mediated by LPA receptor 2. Oncol. Lett. 2013, 5, 1048–1052. [Google Scholar] [CrossRef] [Green Version]
- Lin, Y.H.; Lin, Y.C.; Chen, C.C. Lysophosphatidic Acid Receptor Antagonists and Cancer: The Current Trends, Clinical Implications, and Trials. Cells 2021, 10, 1629. [Google Scholar] [CrossRef]
- Lin, S.; Lee, S.J.; Shim, H.; Chun, J.; Yun, C.C. The absence of LPA receptor 2 reduces the tumorigenesis by ApcMin mutation in the intestine. Am. J. Physiol. Gastrointest. Liver Physiol. 2010, 299, G1128–G1138. [Google Scholar] [CrossRef] [Green Version]
- Ara, H.; Bhattarai, S.; Sharma, S.; Subedi, U.; Yu, X.; Bhuiyan, M.S.; Miriyala, S.; Panchatcharam, M. LPAR2 receptor stimulates progression of gastric cancer through β-catenin signaling pathway. FASEB J. 2022, 36. [Google Scholar] [CrossRef]
- Giles, R.H.; van Es, J.H.; Clevers, H. Caught up in a Wnt storm: Wnt signaling in cancer. Biochim. Biophys. Acta 2003, 1653, 1–24. [Google Scholar] [CrossRef] [PubMed]
- Chandra, M.; Escalante-Alcalde, D.; Bhuiyan, M.S.; Orr, A.W.; Kevil, C.; Morris, A.J.; Nam, H.; Dominic, P.; McCarthy, K.J.; Miriyala, S.; et al. Cardiac-specific inactivation of LPP3 in mice leads to myocardial dysfunction and heart failure. Redox. Biol. 2018, 14, 261–271. [Google Scholar] [CrossRef] [PubMed]
- Sharma, S.; Bhattarai, S.; Ara, H.; Sun, G.; St Clair, D.K.; Bhuiyan, M.S.; Kevil, C.; Watts, M.N.; Dominic, P.; Shimizu, T.; et al. SOD2 deficiency in cardiomyocytes defines defective mitochondrial bioenergetics as a cause of lethal dilated cardiomyopathy. Redox. Biol. 2020, 37, 101740. [Google Scholar] [CrossRef] [PubMed]
- Cai, H.; Xu, Y. The role of LPA and YAP signaling in long-term migration of human ovarian cancer cells. Cell Commun. Signal. 2013, 11, 31. [Google Scholar] [CrossRef] [Green Version]
- Ren, Z.; Zhang, C.; Ma, L.; Zhang, X.; Shi, S.; Tang, D.; Xu, J.; Hu, Y.; Wang, B.; Zhang, F.; et al. Lysophosphatidic acid induces the migration and invasion of SGC-7901 gastric cancer cells through the LPA2 and Notch signaling pathways. Int. J. Mol. Med. 2019, 44, 67–78. [Google Scholar] [CrossRef] [Green Version]
- Stoddard, N.C.; Chun, J. Promising pharmacological directions in the world of lysophosphatidic Acid signaling. Biomol. Ther. 2015, 23, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Singla, A.; Kumar, A.; Priyamvada, S.; Tahniyath, M.; Saksena, S.; Gill, R.K.; Alrefai, W.A.; Dudeja, P.K. LPA stimulates intestinal DRA gene transcription via LPA2 receptor, PI3K/AKT, and c-Fos-dependent pathway. Am. J. Physiol. Gastrointest. Liver Physiol. 2012, 302, G618–G627. [Google Scholar] [CrossRef] [Green Version]
- Shin, K.-J.; Kim, Y.L.; Lee, S.; Kim, D.-k.; Ahn, C.; Chung, J.; Seong, J.Y.; Hwang, J.-I. Lysophosphatidic acid signaling through LPA receptor subtype 1 induces colony scattering of gastrointestinal cancer cells. J. Cancer Res. Clin. Oncol. 2008, 135, 45. [Google Scholar] [CrossRef]
- Hurlstone, A.; Clevers, H. T-cell factors: Turn-ons and turn-offs. EMBO J. 2002, 21, 2303–2311. [Google Scholar] [CrossRef] [Green Version]
- Cheon, S.S.; Nadesan, P.; Poon, R.; Alman, B.A. Growth factors regulate beta-catenin-mediated TCF-dependent transcriptional activation in fibroblasts during the proliferative phase of wound healing. Exp. Cell Res. 2004, 293, 267–274. [Google Scholar] [CrossRef]
- Sanchez-Tillo, E.; de Barrios, O.; Siles, L.; Cuatrecasas, M.; Castells, A.; Postigo, A. beta-catenin/TCF4 complex induces the epithelial-to-mesenchymal transition (EMT)-activator ZEB1 to regulate tumor invasiveness. Proc. Natl. Acad. Sci. USA 2011, 108, 19204–19209. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sekine, S.; Shibata, T.; Sakamoto, M.; Hirohashi, S. Target disruption of the mutant β-catenin gene in colon cancer cell line HCT116: Preservation of its malignant phenotype. Oncogene 2002, 21, 5906–5911. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chan, T.A.; Wang, Z.; Dang, L.H.; Vogelstein, B.; Kinzler, K.W. Targeted inactivation of CTNNB1 reveals unexpected effects of beta-catenin mutation. Proc. Natl. Acad. Sci. USA 2002, 99, 8265–8270. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fang, X.; Yu, S.; Tanyi, J.L.; Lu, Y.; Woodgett, J.R.; Mills, G.B. Convergence of multiple signaling cascades at glycogen synthase kinase 3: Edg receptor-mediated phosphorylation and inactivation by lysophosphatidic acid through a protein kinase C-dependent intracellular pathway. Mol. Cell. Biol. 2002, 22, 2099–2110. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beurel, E.; Grieco, S.F.; Jope, R.S. Glycogen synthase kinase-3 (GSK3): Regulation, actions, and diseases. Pharmacol. Ther. 2015, 148, 114–131. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Choi, S.H.; Estarás, C.; Moresco, J.J.; Yates, J.R., 3rd; Jones, K.A. α-Catenin interacts with APC to regulate β-catenin proteolysis and transcriptional repression of Wnt target genes. Genes Dev. 2013, 27, 2473–2488. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, M.; Zhong, W.W.; Srivastava, N.; Slavin, A.; Yang, J.; Hoey, T.; An, S. G protein-coupled lysophosphatidic acid receptors stimulate proliferation of colon cancer cells through the beta-catenin pathway. Proc. Natl. Acad. Sci. USA 2005, 102, 6027–6032. [Google Scholar] [CrossRef] [Green Version]
- Kessenbrock, K.; Plaks, V.; Werb, Z. Matrix metalloproteinases: Regulators of the tumor microenvironment. Cell 2010, 141, 52–67. [Google Scholar] [CrossRef] [Green Version]
- Griguer, C.E.; Oliva, C.R.; Gillespie, G.Y. Glucose Metabolism Heterogeneity in Human and Mouse Malignant Glioma Cell Lines. J. Neuro-Oncol. 2005, 74, 123–133. [Google Scholar] [CrossRef]
- Chen, J.L.-Y.; Lucas, J.E.; Schroeder, T.; Mori, S.; Wu, J.; Nevins, J.; Dewhirst, M.; West, M.; Chi, J.-T. The genomic analysis of lactic acidosis and acidosis response in human cancers. PLoS Genet. 2008, 4, e1000293. [Google Scholar] [CrossRef]
- Dong, C.; Yuan, T.; Wu, Y.; Wang, Y.; Fan, T.W.; Miriyala, S.; Lin, Y.; Yao, J.; Shi, J.; Kang, T.; et al. Loss of FBP1 by Snail-mediated repression provides metabolic advantages in basal-like breast cancer. Cancer Cell. 2013, 23, 316–331. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sreedhar, A.; Lefort, J.; Petruska, P.; Gu, X.; Shi, R.; Miriyala, S.; Panchatcharam, M.; Zhao, Y. UCP2 upregulation promotes PLCγ-1 signaling during skin cell transformation. Mol. Carcinog. 2017, 56, 2290–2300. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Miriyala, S.; Fang, F.; Bakthavatchalu, V.; Noel, T.; Schnell, D.M.; Wang, C.; St Clair, W.H.; St Clair, D.K. Manganese superoxide dismutase deficiency triggers mitochondrial uncoupling and the Warburg effect. Oncogene 2015, 34, 4229–4237. [Google Scholar] [CrossRef] [Green Version]
- Zu, X.L.; Guppy, M. Cancer metabolism: Facts, fantasy, and fiction. Biochem. Biophys. Res. Commun. 2004, 313, 459–465. [Google Scholar] [CrossRef] [PubMed]
- Scott, D.A.; Richardson, A.D.; Filipp, F.V.; Knutzen, C.A.; Chiang, G.G.; Ze’ev, A.R.; Osterman, A.L.; Smith, J.W. Comparative metabolic flux profiling of melanoma cell lines: Beyond the Warburg effect. J. Biol. Chem. 2011, 286, 42626–42634. [Google Scholar] [CrossRef] [Green Version]
- Hsu, P.P.; Sabatini, D.M. Cancer Cell Metabolism: Warburg and Beyond. Cell 2008, 134, 703–707. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moreno-Sánchez, R.; Rodríguez-Enríquez, S.; Marín-Hernández, A.; Saavedra, E. Energy metabolism in tumor cells. FEBS J. 2007, 274, 1393–1418. [Google Scholar] [CrossRef]
- Smolková, K.; Plecitá-Hlavatá, L.; Bellance, N.; Benard, G.; Rossignol, R.; Ježek, P. Waves of gene regulation suppress and then restore oxidative phosphorylation in cancer cells. Int. J. Biochem. Cell Biol. 2011, 43, 950–968. [Google Scholar] [CrossRef]
- Kroemer, G.; Pouyssegur, J. Tumor cell metabolism: Cancer’s Achilles’ heel. Cancer. Cell. 2008, 13, 472–482. [Google Scholar] [CrossRef]
- Dang, C.V.; Le, A.; Gao, P. MYC-induced cancer cell energy metabolism and therapeutic opportunities. Clin. Cancer Res. 2009, 15, 6479–6483. [Google Scholar] [CrossRef]
- Vaupel, P. Metabolic microenvironment of tumor cells: A key factor in malignant progression. Exp. Oncol. 2010, 32, 125–127. [Google Scholar] [PubMed]
- Shin, J.H.; Kim, H.W.; Rhyu, I.J.; Kee, S.H. Axin is expressed in mitochondria and suppresses mitochondrial ATP synthesis in HeLa cells. Exp. Cell. Res. 2016, 340, 12–21. [Google Scholar] [CrossRef] [PubMed]
- Park, S.Y.; Shin, J.H.; Kee, S.H. E-cadherin expression increases cell proliferation by regulating energy metabolism through nuclear factor-κB in AGS cells. Cancer Sci. 2017, 108, 1769–1777. [Google Scholar] [CrossRef] [PubMed]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ara, H.; Subedi, U.; Sharma, P.; Bhattarai, S.; Sharma, S.; Manikandan, S.; Yu, X.; Bhuiyan, M.S.; Sun, H.; Miriyala, S.; et al. Alteration of Cellular Energy Metabolism through LPAR2-Axin2 Axis in Gastric Cancer. Biomolecules 2022, 12, 1805. https://doi.org/10.3390/biom12121805
Ara H, Subedi U, Sharma P, Bhattarai S, Sharma S, Manikandan S, Yu X, Bhuiyan MS, Sun H, Miriyala S, et al. Alteration of Cellular Energy Metabolism through LPAR2-Axin2 Axis in Gastric Cancer. Biomolecules. 2022; 12(12):1805. https://doi.org/10.3390/biom12121805
Chicago/Turabian StyleAra, Hosne, Utsab Subedi, Papori Sharma, Susmita Bhattarai, Sudha Sharma, Shrivats Manikandan, Xiuping Yu, Md. Shenuarin Bhuiyan, Hong Sun, Sumitra Miriyala, and et al. 2022. "Alteration of Cellular Energy Metabolism through LPAR2-Axin2 Axis in Gastric Cancer" Biomolecules 12, no. 12: 1805. https://doi.org/10.3390/biom12121805