
One-Carbon and Polyamine Metabolism as Cancer Therapy Targets

{kind=link}
Abstract
:1. Introduction
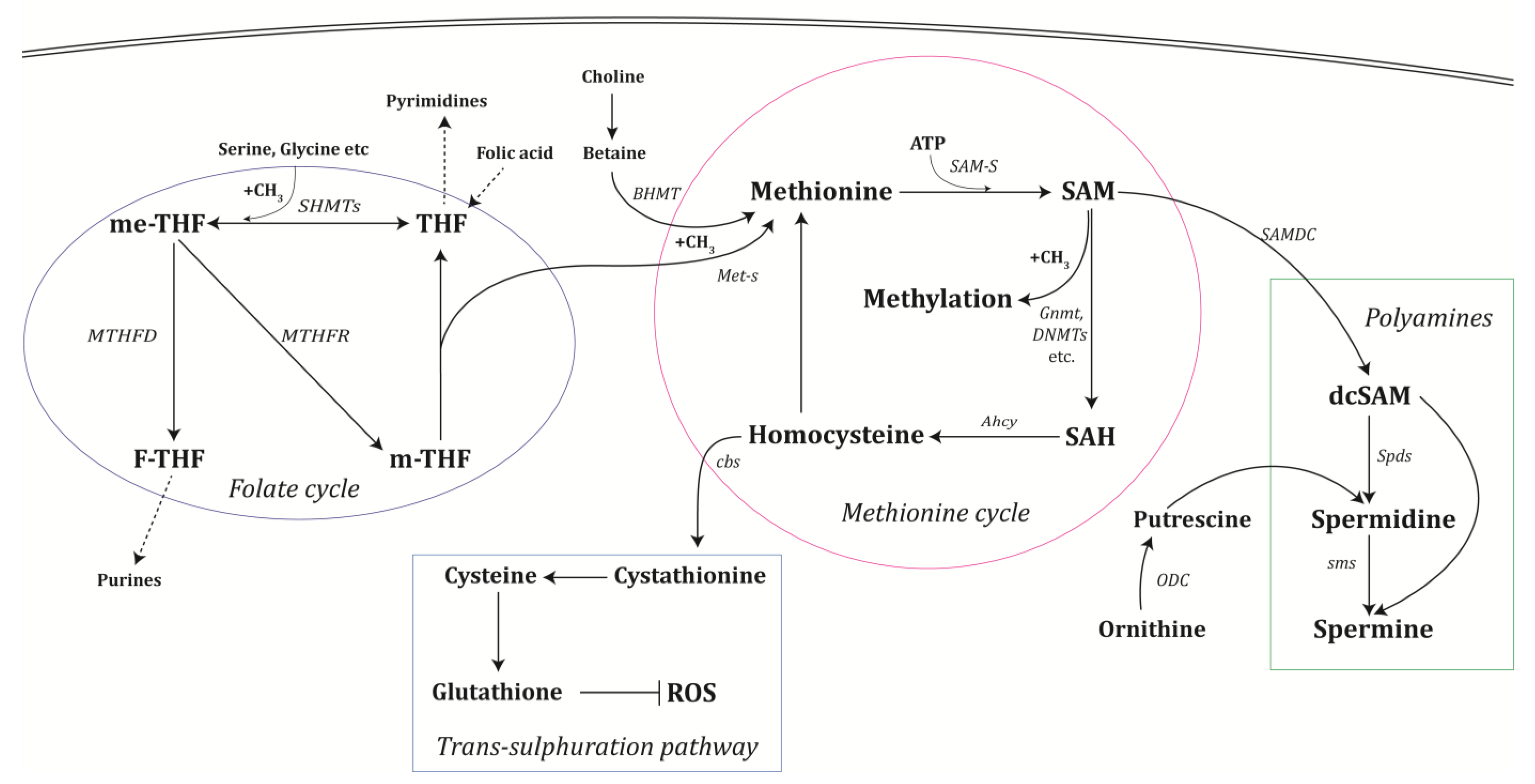
2. One-Carbon and Polyamine Metabolism
2.1. The Methionine Cycle
2.2. The Folate Cycle
2.3. Polyamine Synthesis
3. The Implications of One-Carbon and Polyamine Metabolism for Cancer
3.1. Folate Metabolism and Cancer
3.2. Serine Metabolism in Cancer
3.3. SAM-S Metabolism in Cancer
3.4. Methionine Dependency in Cancer
3.5. Homocysteine Metabolism and Cancer
3.6. The Role of One-Carbon Metabolism in Nucleotide Synthesis in Cancer
3.7. Polyamine Metabolism in Cancer
4. Mechanisms Relating One-Carbon and Polyamine Metabolism to Cancer
4.1. The Function of One-Carbon Metabolism in Methylation Reactions
4.2. Oxidative Stress and One-Carbon Metabolism in Cancer
4.3. The Linkage of Autophagy to the One-Carbon and Polyamine Metabolism in Cancer
5. Metabolic Cancer Therapy
5.1. Metabolic Therapy Targeting One-Carbon and Folate Metabolism
5.2. Therapy Targeting Polyamine Metabolism
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Locasale, J.W. Serine, glycine and one-carbon units: Cancer metabolism in full circle. Nat. Rev. Cancer 2013, 13, 572–583. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ducker, G.S.; Rabinowitz, J.D. One-Carbon Metabolism in Health and Disease. Cell Metab. 2017, 25, 27–42. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Clare, C.E.; Brassington, A.H.; Kwong, W.Y.; Sinclair, K.D. One-Carbon Metabolism: Linking Nutritional Biochemistry to Epigenetic Programming of Long-Term Development. Annu. Rev. Anim. Biosci. 2019, 7, 263–287. [Google Scholar] [CrossRef] [PubMed]
- Padmanabhan, N.; Watson, E.D. Lessons from the one-carbon metabolism: Passing it along to the next generation. Reprod. Biomed. Online 2013, 27, 637–643. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cavuoto, P.; Fenech, M.F. A review of methionine dependency and the role of methionine restriction in cancer growth control and life-span extension. Cancer Treat. Rev. 2012, 38, 726–736. [Google Scholar] [CrossRef] [PubMed]
- Sanderson, S.M.; Gao, X.; Dai, Z.; Locasale, J.W. Methionine metabolism in health and cancer: A nexus of diet and precision medicine. Nat. Rev. Cancer 2019, 19, 625–637. [Google Scholar] [CrossRef] [PubMed]
- Brosnan, J.T.; da Silva, R.P.; Brosnan, M.E. The metabolic burden of creatine synthesis. Amino Acids 2011, 40, 1325–1331. [Google Scholar] [CrossRef]
- Mato, J.M.; Corrales, F.J.; Lu, S.C.; Avila, M.A. S-Adenosylmethionine: A control switch that regulates liver function. FASEB J. 2002, 16, 15–26. [Google Scholar] [CrossRef] [Green Version]
- Zuhra, K.; Augsburger, F.; Majtan, T.; Szabo, C. Cystathionine-beta-Synthase: Molecular Regulation and Pharmacological Inhibition. Biomolecules 2020, 10, 697. [Google Scholar] [CrossRef]
- Cyr, A.R.; Domann, F.E. The redox basis of epigenetic modifications: From mechanisms to functional consequences. Antioxid. Redox. Signal 2011, 15, 551–589. [Google Scholar] [CrossRef]
- Nijhout, H.F.; Reed, M.C.; Anderson, D.F.; Mattingly, J.C.; James, S.J.; Ulrich, C.M. Long-range allosteric interactions between the folate and methionine cycles stabilize DNA methylation reaction rate. Epigenetics 2006, 1, 81–87. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stipanuk, M.H.; Ueki, I. Dealing with methionine/homocysteine sulfur: Cysteine metabolism to taurine and inorganic sulfur. J. Inherit. Metab. Dis. 2011, 34, 17–32. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kok, D.E.; Steegenga, W.T.; Smid, E.J.; Zoetendal, E.G.; Ulrich, C.M.; Kampman, E. Bacterial folate biosynthesis and colorectal cancer risk: More than just a gut feeling. Crit. Rev. Food Sci. Nutr. 2020, 60, 244–256. [Google Scholar] [CrossRef]
- Brosnan, M.E.; MacMillan, L.; Stevens, J.R.; Brosnan, J.T. Division of labour: How does folate metabolism partition between one-carbon metabolism and amino acid oxidation? Biochem. J. 2015, 472, 135–146. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tibbetts, A.S.; Appling, D.R. Compartmentalization of Mammalian folate-mediated one-carbon metabolism. Annu. Rev. Nutr. 2010, 30, 57–81. [Google Scholar] [CrossRef]
- Pegg, A.E. Mammalian polyamine metabolism and function. IUBMB Life 2009, 61, 880–894. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Igarashi, K.; Kashiwagi, K. Polyamines: Mysterious modulators of cellular functions. Biochem. Biophys. Res. Commun. 2000, 271, 559–564. [Google Scholar] [CrossRef] [PubMed]
- Dever, T.E.; Ivanov, I.P. Roles of polyamines in translation. J. Biol. Chem. 2018, 293, 18719–18729. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hesterberg, R.S.; Cleveland, J.L.; Epling-Burnette, P.K. Role of Polyamines in Immune Cell Functions. Med. Sci. 2018, 6, 22. [Google Scholar] [CrossRef] [Green Version]
- Pegg, A.E. Functions of Polyamines in Mammals. J. Biol. Chem. 2016, 291, 14904–14912. [Google Scholar] [CrossRef]
- Kus, K.; Kij, A.; Zakrzewska, A.; Jasztal, A.; Stojak, M.; Walczak, M.; Chlopicki, S. Alterations in arginine and energy metabolism, structural and signalling lipids in metastatic breast cancer in mice detected in plasma by targeted metabolomics and lipidomics. Breast Cancer Res. 2018, 20, 148. [Google Scholar] [CrossRef] [PubMed]
- Raimondi, M.V.; Randazzo, O.; La Franca, M.; Barone, G.; Vignoni, E.; Rossi, D.; Collina, S. DHFR Inhibitors: Reading the Past for Discovering Novel Anticancer Agents. Molecules 2019, 24, 1140. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wojtuszkiewicz, A.; Peters, G.J.; van Woerden, N.L.; Dubbelman, B.; Escherich, G.; Schmiegelow, K.; Sonneveld, E.; Pieters, R.; van de Ven, P.M.; Jansen, G.; et al. Methotrexate resistance in relation to treatment outcome in childhood acute lymphoblastic leukemia. J. Hematol. Oncol. 2015, 8, 61. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nilsson, R.; Jain, M.; Madhusudhan, N.; Sheppard, N.G.; Strittmatter, L.; Kampf, C.; Huang, J.; Asplund, A.; Mootha, V.K. Metabolic enzyme expression highlights a key role for MTHFD2 and the mitochondrial folate pathway in cancer. Nat. Commun. 2014, 5, 3128. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, G.Y.; Haverty, P.M.; Li, L.; Kljavin, N.M.; Bourgon, R.; Lee, J.; Stern, H.; Modrusan, Z.; Seshagiri, S.; Zhang, Z.; et al. Comparative oncogenomics identifies PSMB4 and SHMT2 as potential cancer driver genes. Cancer Res. 2014, 74, 3114–3126. [Google Scholar] [CrossRef] [Green Version]
- Min, D.J.; Vural, S.; Krushkal, J. Association of transcriptional levels of folate-mediated one-carbon metabolism-related genes in cancer cell lines with drug treatment response. Cancer Genet. 2019, 237, 19–38. [Google Scholar] [CrossRef]
- Ose, J.; Botma, A.; Balavarca, Y.; Buck, K.; Scherer, D.; Habermann, N.; Beyerle, J.; Pfutze, K.; Seibold, P.; Kap, E.J.; et al. Pathway analysis of genetic variants in folate-mediated one-carbon metabolism-related genes and survival in a prospectively followed cohort of colorectal cancer patients. Cancer Med. 2018, 7, 2797–2807. [Google Scholar] [CrossRef]
- Fan, J.; Ye, J.; Kamphorst, J.J.; Shlomi, T.; Thompson, C.B.; Rabinowitz, J.D. Quantitative flux analysis reveals folate-dependent NADPH production. Nature 2014, 510, 298–302. [Google Scholar] [CrossRef] [Green Version]
- Herbig, K.; Chiang, E.P.; Lee, L.R.; Hills, J.; Shane, B.; Stover, P.J. Cytoplasmic serine hydroxymethyltransferase mediates competition between folate-dependent deoxyribonucleotide and S-adenosylmethionine biosyntheses. J. Biol. Chem. 2002, 277, 38381–38389. [Google Scholar] [CrossRef] [Green Version]
- Paone, A.; Marani, M.; Fiascarelli, A.; Rinaldo, S.; Giardina, G.; Contestabile, R.; Paiardini, A.; Cutruzzola, F. SHMT1 knockdown induces apoptosis in lung cancer cells by causing uracil misincorporation. Cell Death Dis. 2014, 5, e1525. [Google Scholar] [CrossRef]
- Li, H.; Fu, X.; Yao, F.; Tian, T.; Wang, C.; Yang, A. MTHFD1L-Mediated Redox Homeostasis Promotes Tumor Progression in Tongue Squamous Cell Carcinoma. Front. Oncol. 2019, 9, 1278. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nikkanen, J.; Forsstrom, S.; Euro, L.; Paetau, I.; Kohnz, R.A.; Wang, L.; Chilov, D.; Viinamaki, J.; Roivainen, A.; Marjamaki, P.; et al. Mitochondrial DNA Replication Defects Disturb Cellular dNTP Pools and Remodel One-Carbon Metabolism. Cell Metab. 2016, 23, 635–648. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sullivan, M.R.; Mattaini, K.R.; Dennstedt, E.A.; Nguyen, A.A.; Sivanand, S.; Reilly, M.F.; Meeth, K.; Muir, A.; Darnell, A.M.; Bosenberg, M.W.; et al. Increased Serine Synthesis Provides an Advantage for Tumors Arising in Tissues Where Serine Levels Are Limiting. Cell Metab. 2019, 29, 1410–1421.e1414. [Google Scholar] [CrossRef] [PubMed]
- Maddocks, O.D.K.; Athineos, D.; Cheung, E.C.; Lee, P.; Zhang, T.; van den Broek, N.J.F.; Mackay, G.M.; Labuschagne, C.F.; Gay, D.; Kruiswijk, F.; et al. Modulating the therapeutic response of tumours to dietary serine and glycine starvation. Nature 2017, 544, 372–376. [Google Scholar] [CrossRef] [Green Version]
- Montrose, D.C.; Saha, S.; Foronda, M.; McNally, E.M.; Chen, J.; Zhou, X.K.; Ha, T.; Krumsiek, J.; Buyukozkan, M.; Verma, A.; et al. Exogenous and Endogenous Sources of Serine Contribute to Colon Cancer Metabolism, Growth, and Resistance to 5-Fluorouracil. Cancer Res. 2021, 81, 2275–2288. [Google Scholar] [CrossRef] [PubMed]
- Kalhan, S.C.; Hanson, R.W. Resurgence of serine: An often neglected but indispensable amino Acid. J. Biol. Chem. 2012, 287, 19786–19791. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ducker, G.S.; Chen, L.; Morscher, R.J.; Ghergurovich, J.M.; Esposito, M.; Teng, X.; Kang, Y.; Rabinowitz, J.D. Reversal of Cytosolic One-Carbon Flux Compensates for Loss of the Mitochondrial Folate Pathway. Cell Metab. 2016, 23, 1140–1153. [Google Scholar] [CrossRef] [Green Version]
- Kalhan, S.C.; Uppal, S.O.; Moorman, J.L.; Bennett, C.; Gruca, L.L.; Parimi, P.S.; Dasarathy, S.; Serre, D.; Hanson, R.W. Metabolic and genomic response to dietary isocaloric protein restriction in the rat. J. Biol. Chem. 2011, 286, 5266–5277. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pasternack, L.B.; Laude, D.A., Jr.; Appling, D.R. 13C NMR detection of folate-mediated serine and glycine synthesis in vivo in Saccharomyces cerevisiae. Biochemistry 1992, 31, 8713–8719. [Google Scholar] [CrossRef]
- Gregory, J.F., 3rd; Cuskelly, G.J.; Shane, B.; Toth, J.P.; Baumgartner, T.G.; Stacpoole, P.W. Primed, constant infusion with [2H3]serine allows in vivo kinetic measurement of serine turnover, homocysteine remethylation, and transsulfuration processes in human one-carbon metabolism. Am. J. Clin. Nutr. 2000, 72, 1535–1541. [Google Scholar] [CrossRef]
- Maddocks, O.D.; Berkers, C.R.; Mason, S.M.; Zheng, L.; Blyth, K.; Gottlieb, E.; Vousden, K.H. Serine starvation induces stress and p53-dependent metabolic remodelling in cancer cells. Nature 2013, 493, 542–546. [Google Scholar] [CrossRef] [PubMed]
- DeNicola, G.M.; Chen, P.H.; Mullarky, E.; Sudderth, J.A.; Hu, Z.; Wu, D.; Tang, H.; Xie, Y.; Asara, J.M.; Huffman, K.E.; et al. NRF2 regulates serine biosynthesis in non-small cell lung cancer. Nat. Genet. 2015, 47, 1475–1481. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, S.C. S-Adenosylmethionine. Int. J. Biochem. Cell Biol. 2000, 32, 391–395. [Google Scholar] [CrossRef] [PubMed]
- Lu, S.C.; Mato, J.M. S-adenosylmethionine in liver health, injury, and cancer. Physiol. Rev. 2012, 92, 1515–1542. [Google Scholar] [CrossRef] [Green Version]
- Struck, A.W.; Thompson, M.L.; Wong, L.S.; Micklefield, J. S-adenosyl-methionine-dependent methyltransferases: Highly versatile enzymes in biocatalysis, biosynthesis and other biotechnological applications. Chembiochem 2012, 13, 2642–2655. [Google Scholar] [CrossRef]
- Mentch, S.J.; Mehrmohamadi, M.; Huang, L.; Liu, X.; Gupta, D.; Mattocks, D.; Gomez Padilla, P.; Ables, G.; Bamman, M.M.; Thalacker-Mercer, A.E.; et al. Histone Methylation Dynamics and Gene Regulation Occur through the Sensing of One-Carbon Metabolism. Cell Metab. 2015, 22, 861–873. [Google Scholar] [CrossRef] [Green Version]
- Smith, A.D.; Warren, M.J.; Refsum, H. Vitamin B12. Adv. Food Nutr. Res. 2018, 83, 215–279. [Google Scholar] [CrossRef]
- Belardo, A.; Gevi, F.; Zolla, L. The concomitant lower concentrations of vitamins B6, B9 and B12 may cause methylation deficiency in autistic children. J. Nutr. Biochem. 2019, 70, 38–46. [Google Scholar] [CrossRef]
- Parkhitko, A.A.; Jouandin, P.; Mohr, S.E.; Perrimon, N. Methionine metabolism and methyltransferases in the regulation of aging and lifespan extension across species. Aging Cell 2019, 18, e13034. [Google Scholar] [CrossRef] [Green Version]
- Orentreich, N.; Matias, J.R.; DeFelice, A.; Zimmerman, J.A. Low methionine ingestion by rats extends life span. J. Nutr. 1993, 123, 269–274. [Google Scholar] [CrossRef]
- Lee, B.C.; Kaya, A.; Ma, S.; Kim, G.; Gerashchenko, M.V.; Yim, S.H.; Hu, Z.; Harshman, L.G.; Gladyshev, V.N. Methionine restriction extends lifespan of Drosophila melanogaster under conditions of low amino-acid status. Nat. Commun. 2014, 5, 3592. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luka, Z.; Mudd, S.H.; Wagner, C. Glycine N-methyltransferase and regulation of S-adenosylmethionine levels. J. Biol. Chem. 2009, 284, 22507–22511. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bale, S.; Ealick, S.E. Structural biology of S-adenosylmethionine decarboxylase. Amino Acids 2010, 38, 451–460. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Simile, M.M.; Saviozzi, M.; De Miglio, M.R.; Muroni, M.R.; Nufris, A.; Pascale, R.M.; Malvaldi, G.; Feo, F. Persistent chemopreventive effect of S-adenosyl-L-methionine on the development of liver putative preneoplastic lesions induced by thiobenzamide in diethylnitrosamine-initiated rats. Carcinogenesis 1996, 17, 1533–1537. [Google Scholar] [CrossRef] [PubMed]
- Pascale, R.M.; Simile, M.M.; De Miglio, M.R.; Nufris, A.; Daino, L.; Seddaiu, M.A.; Rao, P.M.; Rajalakshmi, S.; Sarma, D.S.; Feo, F. Chemoprevention by S-adenosyl-L-methionine of rat liver carcinogenesis initiated by 1,2-dimethylhydrazine and promoted by orotic acid. Carcinogenesis 1995, 16, 427–430. [Google Scholar] [CrossRef]
- Garcea, R.; Pascale, R.; Daino, L.; Frassetto, S.; Cozzolino, P.; Ruggiu, M.E.; Vannini, M.G.; Gaspa, L.; Feo, F. Variations of ornithine decarboxylase activity and S-adenosyl-L-methionine and 5’-methylthioadenosine contents during the development of diethylnitrosamine-induced liver hyperplastic nodules and hepatocellular carcinoma. Carcinogenesis 1987, 8, 653–658. [Google Scholar] [CrossRef]
- Garcea, R.; Daino, L.; Pascale, R.; Simile, M.M.; Puddu, M.; Ruggiu, M.E.; Seddaiu, M.A.; Satta, G.; Sequenza, M.J.; Feo, F. Protooncogene methylation and expression in regenerating liver and preneoplastic liver nodules induced in the rat by diethylnitrosamine: Effect of variations of S-adenosylmethionine:S-adenosylhomocysteine ratio. Carcinogenesis 1989, 10, 1183–1192. [Google Scholar] [CrossRef]
- Garcea, R.; Daino, L.; Pascale, R.; Simile, M.M.; Puddu, M.; Frassetto, S.; Cozzolino, P.; Seddaiu, M.A.; Gaspa, L.; Feo, F. Inhibition of promotion and persistent nodule growth by S-adenosyl-L-methionine in rat liver carcinogenesis: Role of remodeling and apoptosis. Cancer Res. 1989, 49, 1850–1856. [Google Scholar]
- Feo, F.; Garcea, R.; Daino, L.; Pascale, R.; Pirisi, L.; Frassetto, S.; Ruggiu, M.E. Early stimulation of polyamine biosynthesis during promotion by phenobarbital of diethylnitrosamine-induced rat liver carcinogenesis. The effects of variations of the S-adenosyl-L-methionine cellular pool. Carcinogenesis 1985, 6, 1713–1720. [Google Scholar] [CrossRef]
- Feo, F.; Garcea, R.; Pascale, R.; Pirisi, L.; Daino, L.; Donaera, A. The variations of S-adenosyl-L-methionine content modulate hepatocyte growth during phenobarbital promotion of diethylnitrosamine-induced rat liver carcinogenesis. Toxicol. Pathol. 1987, 15, 109–114. [Google Scholar] [CrossRef] [Green Version]
- Pascale, R.M.; Marras, V.; Simile, M.M.; Daino, L.; Pinna, G.; Bennati, S.; Carta, M.; Seddaiu, M.A.; Massarelli, G.; Feo, F. Chemoprevention of rat liver carcinogenesis by S-adenosyl-L-methionine: A long-term study. Cancer Res. 1992, 52, 4979–4986. [Google Scholar] [PubMed]
- Mahmood, N.; Cheishvili, D.; Arakelian, A.; Tanvir, I.; Khan, H.A.; Pepin, A.S.; Szyf, M.; Rabbani, S.A. Methyl donor S-adenosylmethionine (SAM) supplementation attenuates breast cancer growth, invasion, and metastasis in vivo; therapeutic and chemopreventive applications. Oncotarget 2018, 9, 5169–5183. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shukeir, N.; Stefanska, B.; Parashar, S.; Chik, F.; Arakelian, A.; Szyf, M.; Rabbani, S.A. Pharmacological methyl group donors block skeletal metastasis in vitro and in vivo. Br. J. Pharmacol. 2015, 172, 2769–2781. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parashar, S.; Cheishvili, D.; Arakelian, A.; Hussain, Z.; Tanvir, I.; Khan, H.A.; Szyf, M.; Rabbani, S.A. S-adenosylmethionine blocks osteosarcoma cells proliferation and invasion in vitro and tumor metastasis in vivo: Therapeutic and diagnostic clinical applications. Cancer Med. 2015, 4, 732–744. [Google Scholar] [CrossRef]
- Wang, Y.; Sun, Z.; Szyf, M. S-adenosyl-methionine (SAM) alters the transcriptome and methylome and specifically blocks growth and invasiveness of liver cancer cells. Oncotarget 2017, 8, 111866–111881. [Google Scholar] [CrossRef] [Green Version]
- Borrego, S.L.; Fahrmann, J.; Datta, R.; Stringari, C.; Grapov, D.; Zeller, M.; Chen, Y.; Wang, P.; Baldi, P.; Gratton, E.; et al. Metabolic changes associated with methionine stress sensitivity in MDA-MB-468 breast cancer cells. Cancer Metab. 2016, 4, 9. [Google Scholar] [CrossRef] [Green Version]
- Stern, P.H.; Wallace, C.D.; Hoffman, R.M. Altered methionine metabolism occurs in all members of a set of diverse human tumor cell lines. J. Cell Physiol. 1984, 119, 29–34. [Google Scholar] [CrossRef]
- Mecham, J.O.; Rowitch, D.; Wallace, C.D.; Stern, P.H.; Hoffman, R.M. The metabolic defect of methionine dependence occurs frequently in human tumor cell lines. Biochem. Biophys. Res. Commun. 1983, 117, 429–434. [Google Scholar] [CrossRef]
- Lapa, C.; Garcia-Velloso, M.J.; Luckerath, K.; Samnick, S.; Schreder, M.; Otero, P.R.; Schmid, J.S.; Herrmann, K.; Knop, S.; Buck, A.K.; et al. (11)C-Methionine-PET in Multiple Myeloma: A Combined Study from Two Different Institutions. Theranostics 2017, 7, 2956–2964. [Google Scholar] [CrossRef]
- McDonald, L.; Bray, C.; Field, C.; Love, F.; Davies, B. Homocystinuria, Thrombosis, and the Blood-Platelets. Lancet 1964, 1, 745–746. [Google Scholar] [CrossRef]
- Giovannucci, E.; Rimm, E.B.; Ascherio, A.; Stampfer, M.J.; Colditz, G.A.; Willett, W.C. Alcohol, low-methionine–low-folate diets, and risk of colon cancer in men. J. Natl. Cancer Inst. 1995, 87, 265–273. [Google Scholar] [CrossRef] [PubMed]
- Ma, J.; Stampfer, M.J.; Christensen, B.; Giovannucci, E.; Hunter, D.J.; Chen, J.; Willett, W.C.; Selhub, J.; Hennekens, C.H.; Gravel, R.; et al. A polymorphism of the methionine synthase gene: Association with plasma folate, vitamin B12, homocyst(e)ine, and colorectal cancer risk. Cancer Epidemiol. Biomarkers. Prev. 1999, 8, 825–829. [Google Scholar] [PubMed]
- Bravata, V. Controversial roles of methylenetetrahydrofolate reductase polymorphisms and folate in breast cancer disease. Int. J. Food. Sci. Nutr. 2015, 66, 43–49. [Google Scholar] [CrossRef] [PubMed]
- Kato, I.; Dnistrian, A.M.; Schwartz, M.; Toniolo, P.; Koenig, K.; Shore, R.E.; Akhmedkhanov, A.; Zeleniuch-Jacquotte, A.; Riboli, E. Serum folate, homocysteine and colorectal cancer risk in women: A nested case-control study. Br. J. Cancer 1999, 79, 1917–1922. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- de Jong, M.M.; Nolte, I.M.; te Meerman, G.J.; van der Graaf, W.T.; de Vries, E.G.; Sijmons, R.H.; Hofstra, R.M.; Kleibeuker, J.H. Low-penetrance genes and their involvement in colorectal cancer susceptibility. Cancer Epidemiol. Biomarkers. Prev. 2002, 11, 1332–1352. [Google Scholar]
- Matsuo, K.; Hamajima, N.; Hirai, T.; Kato, T.; Inoue, M.; Takezaki, T.; Tajima, K. Methionine Synthase Reductase Gene A66G Polymorphism is Associated with Risk of Colorectal Cancer. Asian Pac. J. Cancer Prev. 2002, 3, 353–359. [Google Scholar]
- Robien, K.; Ulrich, C.M. 5,10-Methylenetetrahydrofolate reductase polymorphisms and leukemia risk: A HuGE minireview. Am. J. Epidemiol. 2003, 157, 571–582. [Google Scholar] [CrossRef] [Green Version]
- Krajinovic, M.; Lamothe, S.; Labuda, D.; Lemieux-Blanchard, E.; Theoret, Y.; Moghrabi, A.; Sinnett, D. Role of MTHFR genetic polymorphisms in the susceptibility to childhood acute lymphoblastic leukemia. Blood 2004, 103, 252–257. [Google Scholar] [CrossRef] [Green Version]
- Singal, R.; Ferdinand, L.; Das, P.M.; Reis, I.M.; Schlesselman, J.J. Polymorphisms in the methylenetetrahydrofolate reductase gene and prostate cancer risk. Int. J. Oncol. 2004, 25, 1465–1471. [Google Scholar] [CrossRef]
- Matsuo, K.; Ito, H.; Wakai, K.; Hirose, K.; Saito, T.; Suzuki, T.; Kato, T.; Hirai, T.; Kanemitsu, Y.; Hamajima, H.; et al. One-carbon metabolism related gene polymorphisms interact with alcohol drinking to influence the risk of colorectal cancer in Japan. Carcinogenesis 2005, 26, 2164–2171. [Google Scholar] [CrossRef] [Green Version]
- Mancardi, D.; Penna, C.; Merlino, A.; Del Soldato, P.; Wink, D.A.; Pagliaro, P. Physiological and pharmacological features of the novel gasotransmitter: Hydrogen sulfide. Biochim. Biophys. Acta 2009, 1787, 864–872. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Safarinejad, M.R.; Shafiei, N.; Safarinejad, S. Relationship between three polymorphisms of methylenetetrahydrofolate reductase (MTHFR C677T, A1298C, and G1793A) gene and risk of prostate cancer: A case-control study. Prostate 2010, 70, 1645–1657. [Google Scholar] [CrossRef] [PubMed]
- Fang, D.H.; Ji, Q.; Fan, C.H.; An, Q.; Li, J. Methionine synthase reductase A66G polymorphism and leukemia risk: Evidence from published studies. Leuk. Lymphoma 2014, 55, 1910–1914. [Google Scholar] [CrossRef] [PubMed]
- Wang, P.; Li, S.; Wang, M.; He, J.; Xi, S. Association of MTRR A66G polymorphism with cancer susceptibility: Evidence from 85 studies. J. Cancer 2017, 8, 266–277. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qin, Y.T.; Zhang, Y.; Wu, F.; Su, Y.; Lu, G.N.; Wang, R.S. Association between MTHFR polymorphisms and acute myeloid leukemia risk: A meta-analysis. PLoS ONE 2014, 9, e88823. [Google Scholar] [CrossRef] [PubMed]
- Haghighi, M.M.; Mohebbi, S.R.; Najjar Sadeghi, R.; Vahedi, M.; Ghiasi, S.; Zali, M.R. Association between the 1793G> A MTHFR polymorphism and sporadic colorectal cancer in Iran. Asian Pac. J. Cancer Prev. 2008, 9, 659–662. [Google Scholar] [PubMed]
- Paynter, R.A.; Hankinson, S.E.; Hunter, D.J.; De Vivo, I. No association between MTHFR 677 C->T or 1298 A->C polymorphisms and endometrial cancer risk. Cancer Epidemiol. Biomarkers Prev. 2004, 13, 1088–1089. [Google Scholar] [CrossRef]
- Shujuan, Y.; Jianxing, Z.; Xin-Yue, C. Methylenetetrahydrofolate reductase genetic polymorphisms and esophageal squamous cell carcinoma susceptibility: A meta-analysis of case-control studies. Pak. J. Med. Sci. 2013, 29, 693–698. [Google Scholar]
- He, L.; Shen, Y. MTHFR C677T polymorphism and breast, ovarian cancer risk: A meta-analysis of 19,260 patients and 26,364 controls. Oncol. Targets Ther. 2017, 10, 227–238. [Google Scholar] [CrossRef] [Green Version]
- Tao, M.H.; Shields, P.G.; Nie, J.; Marian, C.; Ambrosone, C.B.; McCann, S.E.; Platek, M.; Krishnan, S.S.; Xie, B.; Edge, S.B.; et al. DNA promoter methylation in breast tumors: No association with genetic polymorphisms in MTHFR and MTR. Cancer Epidemiol. Biomarkers Prev. 2009, 18, 998–1002. [Google Scholar] [CrossRef] [Green Version]
- Skibola, C.F.; Smith, M.T.; Kane, E.; Roman, E.; Rollinson, S.; Cartwright, R.A.; Morgan, G. Polymorphisms in the methylenetetrahydrofolate reductase gene are associated with susceptibility to acute leukemia in adults. Proc. Natl. Acad. Sci. USA 1999, 96, 12810–12815. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, C.F.; Haven, T.R.; Wu, T.L.; Tsao, K.C.; Wu, J.T. Serum total homocysteine increases with the rapid proliferation rate of tumor cells and decline upon cell death: A potential new tumor marker. Clin. Chim. Acta 2002, 321, 55–62. [Google Scholar] [CrossRef] [PubMed]
- Akoglu, B.; Milovic, V.; Caspary, W.F.; Faust, D. Hyperproliferation of homocysteine-treated colon cancer cells is reversed by folate and 5-methyltetrahydrofolate. Eur. J. Nutr. 2004, 43, 93–99. [Google Scholar] [CrossRef] [PubMed]
- He, Q.; Yang, Z.; Sun, Y.; Qu, Z.; Jia, X.; Li, J.; Lin, Y.; Luo, Y. The Impact of Homocysteine on the Risk of Hormone-Related Cancers: A Mendelian Randomization Study. Front. Nutr. 2021, 8, 645371. [Google Scholar] [CrossRef] [PubMed]
- Zeng, J.D.; Wu, W.K.K.; Wang, H.Y.; Li, X.X. Serine and one-carbon metabolism, a bridge that links mTOR signaling and DNA methylation in cancer. Pharmacol. Res. 2019, 149, 104352. [Google Scholar] [CrossRef] [PubMed]
- Sanborn, T.A.; Kowle, R.L.; Sallach, H.J. Regulation of enzymes of serine and one-carbon metabolism by testosterone in rat prostate, liver, and kidney. Endocrinology 1975, 97, 1000–1007. [Google Scholar] [CrossRef]
- Webb, M.; Nickerson, W.J. Differential reversal of inhibitory effects of folic acid analogues on growth, division, and deoxyribonucleic acid synthesis of microorganisms. J. Bacteriol. 1956, 71, 140–148. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Labuschagne, C.F.; van den Broek, N.J.; Mackay, G.M.; Vousden, K.H.; Maddocks, O.D. Serine, but not glycine, supports one-carbon metabolism and proliferation of cancer cells. Cell Rep. 2014, 7, 1248–1258. [Google Scholar] [CrossRef] [Green Version]
- Maddocks, O.D.; Labuschagne, C.F.; Adams, P.D.; Vousden, K.H. Serine Metabolism Supports the Methionine Cycle and DNA/RNA Methylation through De Novo ATP Synthesis in Cancer Cells. Mol. Cell 2016, 61, 210–221. [Google Scholar] [CrossRef] [Green Version]
- Ducker, G.S.; Ghergurovich, J.M.; Mainolfi, N.; Suri, V.; Jeong, S.K.; Hsin-Jung Li, S.; Friedman, A.; Manfredi, M.G.; Gitai, Z.; Kim, H.; et al. Human SHMT inhibitors reveal defective glycine import as a targetable metabolic vulnerability of diffuse large B-cell lymphoma. Proc. Natl. Acad. Sci. USA 2017, 114, 11404–11409. [Google Scholar] [CrossRef] [Green Version]
- Asai, A.; Koseki, J.; Konno, M.; Nishimura, T.; Gotoh, N.; Satoh, T.; Doki, Y.; Mori, M.; Ishii, H. Drug discovery of anticancer drugs targeting methylenetetrahydrofolate dehydrogenase 2. Heliyon 2018, 4, e01021. [Google Scholar] [CrossRef] [Green Version]
- Corbin, J.M.; Ruiz-Echevarria, M.J. One-Carbon Metabolism in Prostate Cancer: The Role of Androgen Signaling. Int. J. Mol. Sci. 2016, 17, 1208. [Google Scholar] [CrossRef] [Green Version]
- Tsoi, T.H.; Chan, C.F.; Chan, W.L.; Chiu, K.F.; Wong, W.T.; Ng, C.F.; Wong, K.L. Urinary Polyamines: A Pilot Study on Their Roles as Prostate Cancer Detection Biomarkers. PLoS ONE 2016, 11, e0162217. [Google Scholar] [CrossRef] [Green Version]
- Zabala-Letona, A.; Arruabarrena-Aristorena, A.; Martin-Martin, N.; Fernandez-Ruiz, S.; Sutherland, J.D.; Clasquin, M.; Tomas-Cortazar, J.; Jimenez, J.; Torres, I.; Quang, P.; et al. mTORC1-dependent AMD1 regulation sustains polyamine metabolism in prostate cancer. Nature 2017, 547, 109–113, Erratum in Nature 2018, 554, 554. [Google Scholar] [CrossRef]
- Gomes, A.P.; Schild, T.; Blenis, J. Adding Polyamine Metabolism to the mTORC1 Toolkit in Cell Growth and Cancer. Dev. Cell 2017, 42, 112–114. [Google Scholar] [CrossRef] [Green Version]
- Kaminski, L.; Torrino, S.; Dufies, M.; Djabari, Z.; Haider, R.; Roustan, F.R.; Jaune, E.; Laurent, K.; Nottet, N.; Michiels, J.F.; et al. PGC1alpha Inhibits Polyamine Synthesis to Suppress Prostate Cancer Aggressiveness. Cancer Res. 2019, 79, 3268–3280. [Google Scholar] [CrossRef] [Green Version]
- Bello-Fernandez, C.; Packham, G.; Cleveland, J.L. The ornithine decarboxylase gene is a transcriptional target of c-Myc. Proc. Natl. Acad. Sci. USA 1993, 90, 7804–7808. [Google Scholar] [CrossRef] [Green Version]
- Bai, G.; Kasper, S.; Matusik, R.J.; Rennie, P.S.; Moshier, J.A.; Krongrad, A. Androgen regulation of the human ornithine decarboxylase promoter in prostate cancer cells. J. Androl. 1998, 19, 127–135. [Google Scholar]
- Shukla-Dave, A.; Castillo-Martin, M.; Chen, M.; Lobo, J.; Gladoun, N.; Collazo-Lorduy, A.; Khan, F.M.; Ponomarev, V.; Yi, Z.; Zhang, W.; et al. Ornithine Decarboxylase Is Sufficient for Prostate Tumorigenesis via Androgen Receptor Signaling. Am. J. Pathol. 2016, 186, 3131–3145. [Google Scholar] [CrossRef] [Green Version]
- Lee, Y.R.; Lee, J.W.; Hong, J.; Chung, B.C. Simultaneous Determination of Polyamines and Steroids in Human Serum from Breast Cancer Patients Using Liquid Chromatography-Tandem Mass Spectrometry. Molecules 2021, 26, 1153. [Google Scholar] [CrossRef]
- Zhu, D.; Zhao, Z.; Cui, G.; Chang, S.; Hu, L.; See, Y.X.; Liang Lim, M.G.; Guo, D.; Chen, X.; Poudel, B.; et al. Single-Cell Transcriptome Analysis Reveals Estrogen Signaling Coordinately Augments One-Carbon, Polyamine, and Purine Synthesis in Breast Cancer. Cell Rep. 2019, 27, 2798. [Google Scholar] [CrossRef] [Green Version]
- Thomas, T.J.; Thomas, T.; John, S.; Hsu, H.C.; Yang, P.; Keinanen, T.A.; Hyvonen, M.T. Tamoxifen metabolite endoxifen interferes with the polyamine pathway in breast cancer. Amino Acids 2016, 48, 2293–2302. [Google Scholar] [CrossRef]
- Celik, V.K.; Kapancik, S.; Kacan, T.; Kacan, S.B.; Kapancik, S.; Kilicgun, H. Serum levels of polyamine synthesis enzymes increase in diabetic patients with breast cancer. Endocr. Connect. 2017, 6, 574–579. [Google Scholar] [CrossRef] [Green Version]
- Cervelli, M.; Pietropaoli, S.; Signore, F.; Amendola, R.; Mariottini, P. Polyamines metabolism and breast cancer: State of the art and perspectives. Breast Cancer Res. Treat. 2014, 148, 233–248. [Google Scholar] [CrossRef]
- Singh, R.; Avliyakulov, N.K.; Braga, M.; Haykinson, M.J.; Martinez, L.; Singh, V.; Parveen, M.; Chaudhuri, G.; Pervin, S. Proteomic identification of mitochondrial targets of arginase in human breast cancer. PLoS ONE 2013, 8, e79242. [Google Scholar] [CrossRef]
- Avtandilyan, N.; Javrushyan, H.; Petrosyan, G.; Trchounian, A. The Involvement of Arginase and Nitric Oxide Synthase in Breast Cancer Development: Arginase and NO Synthase as Therapeutic Targets in Cancer. BioMed Res. Int. 2018, 2018, 8696923. [Google Scholar] [CrossRef] [Green Version]
- Clemente, G.S.; Van Waarde, A.; Antunes, I.F.; Dömling, A.; Elsinga, P.H. Arginase as a Potential Biomarker of Disease Progression: A Molecular Imaging Perspective. Int. J. Mol. Sci. 2020, 21, 5291. [Google Scholar] [CrossRef]
- Arruabarrena-Aristorena, A.; Zabala-Letona, A.; Carracedo, A. Oil for the cancer engine: The cross-talk between oncogenic signaling and polyamine metabolism. Sci. Adv. 2018, 4, eaar2606. [Google Scholar] [CrossRef] [Green Version]
- Fedorova, N.E.; Chernoryzh, Y.Y.; Vinogradskaya, G.R.; Emelianova, S.S.; Zavalyshina, L.E.; Yurlov, K.I.; Zakirova, N.F.; Verbenko, V.N.; Kochetkov, S.N.; Kushch, A.A.; et al. Inhibitor of polyamine catabolism MDL72.527 restores the sensitivity to doxorubicin of monocytic leukemia Thp-1 cells infected with human cytomegalovirus. Biochimie 2019, 158, 82–89. [Google Scholar] [CrossRef]
- Asai, Y.; Itoi, T.; Sugimoto, M.; Sofuni, A.; Tsuchiya, T.; Tanaka, R.; Tonozuka, R.; Honjo, M.; Mukai, S.; Fujita, M.; et al. Elevated Polyamines in Saliva of Pancreatic Cancer. Cancers 2018, 10, 43. [Google Scholar] [CrossRef] [Green Version]
- Loser, C.; Folsch, U.R.; Paprotny, C.; Creutzfeldt, W. Polyamine concentrations in pancreatic tissue, serum, and urine of patients with pancreatic cancer. Pancreas 1990, 5, 119–127. [Google Scholar] [CrossRef] [PubMed]
- Mendez, R.; Kesh, K.; Arora, N.; Di Martino, L.; McAllister, F.; Merchant, N.; Banerjee, S.; Banerjee, S. Microbial dysbiosis and polyamine metabolism as predictive markers for early detection of pancreatic cancer. Carcinogenesis 2020, 41, 561–570. [Google Scholar] [CrossRef] [PubMed]
- Tomita, A.; Mori, M.; Hiwatari, K.; Yamaguchi, E.; Itoi, T.; Sunamura, M.; Soga, T.; Tomita, M.; Sugimoto, M. Effect of storage conditions on salivary polyamines quantified via liquid chromatography-mass spectrometry. Sci. Rep. 2018, 8, 12075. [Google Scholar] [CrossRef] [PubMed]
- Phanstiel, O.t. An overview of polyamine metabolism in pancreatic ductal adenocarcinoma. Int. J. Cancer 2018, 142, 1968–1976. [Google Scholar] [CrossRef] [Green Version]
- Gysin, S.; Rickert, P.; Kastury, K.; McMahon, M. Analysis of genomic DNA alterations and mRNA expression patterns in a panel of human pancreatic cancer cell lines. Genes Chromosomes Cancer 2005, 44, 37–51. [Google Scholar] [CrossRef]
- Gitto, S.B.; Pandey, V.; Oyer, J.L.; Copik, A.J.; Hogan, F.C.; Phanstiel, O.t.; Altomare, D.A. Difluoromethylornithine Combined with a Polyamine Transport Inhibitor Is Effective against Gemcitabine Resistant Pancreatic Cancer. Mol. Pharm. 2018, 15, 369–376. [Google Scholar] [CrossRef]
- Mohammed, A.; Janakiram, N.B.; Madka, V.; Ritchie, R.L.; Brewer, M.; Biddick, L.; Patlolla, J.M.; Sadeghi, M.; Lightfoot, S.; Steele, V.E.; et al. Eflornithine (DFMO) prevents progression of pancreatic cancer by modulating ornithine decarboxylase signaling. Cancer Prev. Res. 2014, 7, 1198–1209. [Google Scholar] [CrossRef] [Green Version]
- Black, O., Jr.; Chang, B.K. Ornithine decarboxylase enzyme activity in human and hamster pancreatic tumor cell lines. Cancer Lett. 1982, 17, 87–93. [Google Scholar] [CrossRef]
- Subhi, A.L.; Tang, B.; Balsara, B.R.; Altomare, D.A.; Testa, J.R.; Cooper, H.S.; Hoffman, J.P.; Meropol, N.J.; Kruger, W.D. Loss of methylthioadenosine phosphorylase and elevated ornithine decarboxylase is common in pancreatic cancer. Clin. Cancer Res. 2004, 10, 7290–7296. [Google Scholar] [CrossRef] [Green Version]
- Massaro, C.; Thomas, J.; Phanstiel Iv, O. Investigation of Polyamine Metabolism and Homeostasis in Pancreatic Cancers. Med. Sci. 2017, 5, 32. [Google Scholar] [CrossRef] [Green Version]
- Forshell, T.P.; Rimpi, S.; Nilsson, J.A. Chemoprevention of B-cell lymphomas by inhibition of the Myc target spermidine synthase. Cancer Prev. Res. 2010, 3, 140–147. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, L.; Liu, L.; Rao, J.N.; Esmaili, A.; Strauch, E.D.; Bass, B.L.; Wang, J.Y. JunD stabilization results in inhibition of normal intestinal epithelial cell growth through P21 after polyamine depletion. Gastroenterology 2002, 123, 764–779. [Google Scholar] [CrossRef] [PubMed]
- Lutz, W.; Stohr, M.; Schurmann, J.; Wenzel, A.; Lohr, A.; Schwab, M. Conditional expression of N-myc in human neuroblastoma cells increases expression of alpha-prothymosin and ornithine decarboxylase and accelerates progression into S-phase early after mitogenic stimulation of quiescent cells. Oncogene 1996, 13, 803–812. [Google Scholar] [PubMed]
- Gamble, L.D.; Purgato, S.; Murray, J.; Xiao, L.; Yu, D.M.T.; Hanssen, K.M.; Giorgi, F.M.; Carter, D.R.; Gifford, A.J.; Valli, E.; et al. Inhibition of polyamine synthesis and uptake reduces tumor progression and prolongs survival in mouse models of neuroblastoma. Sci. Transl. Med. 2019, 11, eaau1099. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, X.; Pearson, A.; Lunec, J. The MYCN oncoprotein as a drug development target. Cancer Lett. 2003, 197, 125–130. [Google Scholar] [CrossRef]
- Wagner, A.J.; Meyers, C.; Laimins, L.A.; Hay, N. c-Myc induces the expression and activity of ornithine decarboxylase. Cell Growth Differ. 1993, 4, 879–883. [Google Scholar]
- Hogarty, M.D.; Norris, M.D.; Davis, K.; Liu, X.; Evageliou, N.F.; Hayes, C.S.; Pawel, B.; Guo, R.; Zhao, H.; Sekyere, E.; et al. ODC1 is a critical determinant of MYCN oncogenesis and a therapeutic target in neuroblastoma. Cancer Res. 2008, 68, 9735–9745. [Google Scholar] [CrossRef] [Green Version]
- Dwivedi, R.S.; Wang, L.J.; Mirkin, B.L. S-adenosylmethionine synthetase is overexpressed in murine neuroblastoma cells resistant to nucleoside analogue inhibitors of S-adenosylhomocysteine hydrolase: A novel mechanism of drug resistance. Cancer Res. 1999, 59, 1852–1856. [Google Scholar]
- Fernandez, P.C.; Frank, S.R.; Wang, L.; Schroeder, M.; Liu, S.; Greene, J.; Cocito, A.; Amati, B. Genomic targets of the human c-Myc protein. Genes Dev. 2003, 17, 1115–1129. [Google Scholar] [CrossRef] [Green Version]
- Evageliou, N.F.; Hogarty, M.D. Disrupting polyamine homeostasis as a therapeutic strategy for neuroblastoma. Clin. Cancer Res 2009, 15, 5956–5961. [Google Scholar] [CrossRef] [Green Version]
- Evageliou, N.F.; Haber, M.; Vu, A.; Laetsch, T.W.; Murray, J.; Gamble, L.D.; Cheng, N.C.; Liu, K.; Reese, M.; Corrigan, K.A.; et al. Polyamine Antagonist Therapies Inhibit Neuroblastoma Initiation and Progression. Clin. Cancer Res. 2016, 22, 4391–4404. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pirnes-Karhu, S.; Jantunen, E.; Mantymaa, P.; Mustjoki, S.; Alhonen, L.; Uimari, A. Spermidine/spermine N(1)-acetyltransferase activity associates with white blood cell count in myeloid leukemias. Exp. Hematol. 2014, 42, 574–580. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Ruan, P.; Zhao, Y.; Li, X.; Wang, J.; Wu, X.; Liu, T.; Wang, S.; Hou, J.; Li, W.; et al. Spermidine/spermine N1-acetyltransferase regulates cell growth and metastasis via AKT/beta-catenin signaling pathways in hepatocellular and colorectal carcinoma cells. Oncotarget 2017, 8, 1092–1109. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tormey, D.C.; Waalkes, T.P.; Ahmann, D.; Gehrke, C.W.; Zumwatt, R.W.; Snyder, J.; Hansen, H. Biological markers in breast carcinoma. I. Incidence of abnormalities of CEA, HCG, three polyamines, and three minor nucleosides. Cancer 1975, 35, 1095–1100. [Google Scholar] [CrossRef]
- Liu, R.; Li, P.; Bi, C.W.; Ma, R.; Yin, Y.; Bi, K.; Li, Q. Plasma N-acetylputrescine, cadaverine and 1,3-diaminopropane: Potential biomarkers of lung cancer used to evaluate the efficacy of anticancer drugs. Oncotarget 2017, 8, 88575–88585. [Google Scholar] [CrossRef] [Green Version]
- Xu, H.; Liu, R.; He, B.; Bi, C.W.; Bi, K.; Li, Q. Polyamine Metabolites Profiling for Characterization of Lung and Liver Cancer Using an LC-Tandem MS Method with Multiple Statistical Data Mining Strategies: Discovering Potential Cancer Biomarkers in Human Plasma and Urine. Molecules 2016, 21, 1040. [Google Scholar] [CrossRef] [Green Version]
- Kato, M.; Onishi, H.; Matsumoto, K.; Motoshita, J.; Tsuruta, N.; Higuchi, K.; Katano, M. Prognostic significance of urine N1, N12-diacetylspermine in patients with non-small cell lung cancer. Anticancer Res. 2014, 34, 3053–3059. [Google Scholar]
- Takahashi, Y.; Sakaguchi, K.; Horio, H.; Hiramatsu, K.; Moriya, S.; Takahashi, K.; Kawakita, M. Urinary N1, N12-diacetylspermine is a non-invasive marker for the diagnosis and prognosis of non-small-cell lung cancer. Br. J. Cancer 2015, 113, 1493–1501. [Google Scholar] [CrossRef]
- Niemi, R.J.; Roine, A.N.; Hakkinen, M.R.; Kumpulainen, P.S.; Keinanen, T.A.; Vepsalainen, J.J.; Lehtimaki, T.; Oksala, N.K.; Maenpaa, J.U. Urinary Polyamines as Biomarkers for Ovarian Cancer. Int. J. Gynecol. Cancer 2017, 27, 1360–1366. [Google Scholar] [CrossRef] [Green Version]
- Dejea, C.M.; Sears, C.L. Do biofilms confer a pro-carcinogenic state? Gut Microbes 2016, 7, 54–57. [Google Scholar] [CrossRef] [Green Version]
- Johnson, C.H.; Dejea, C.M.; Edler, D.; Hoang, L.T.; Santidrian, A.F.; Felding, B.H.; Ivanisevic, J.; Cho, K.; Wick, E.C.; Hechenbleikner, E.M.; et al. Metabolism links bacterial biofilms and colon carcinogenesis. Cell Metab. 2015, 21, 891–897. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nakajima, T.; Katsumata, K.; Kuwabara, H.; Soya, R.; Enomoto, M.; Ishizaki, T.; Tsuchida, A.; Mori, M.; Hiwatari, K.; Soga, T.; et al. Urinary Polyamine Biomarker Panels with Machine-Learning Differentiated Colorectal Cancers, Benign Disease, and Healthy Controls. Int. J. Mol. Sci. 2018, 19, 756. [Google Scholar] [CrossRef] [PubMed]
- Liu, R.; Lin, X.; Li, Z.; Li, Q.; Bi, K. Quantitative metabolomics for investigating the value of polyamines in the early diagnosis and therapy of colorectal cancer. Oncotarget 2018, 9, 4583–4592. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fitzgerald, B.L.; Mahapatra, S.; Farmer, D.K.; McNeil, M.R.; Casero, R.A., Jr.; Belisle, J.T. Elucidating the Structure of N(1)-Acetylisoputreanine: A Novel Polyamine Catabolite in Human Urine. ACS Omega 2017, 2, 3921–3930. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Giskeodegard, G.F.; Bertilsson, H.; Selnaes, K.M.; Wright, A.J.; Bathen, T.F.; Viset, T.; Halgunset, J.; Angelsen, A.; Gribbestad, I.S.; Tessem, M.B. Spermine and citrate as metabolic biomarkers for assessing prostate cancer aggressiveness. PLoS ONE 2013, 8, e62375. [Google Scholar] [CrossRef] [Green Version]
- Su, X.; Wellen, K.E.; Rabinowitz, J.D. Metabolic control of methylation and acetylation. Curr. Opin. Chem. Biol. 2016, 30, 52–60. [Google Scholar] [CrossRef] [Green Version]
- Jones, P.A.; Baylin, S.B. The fundamental role of epigenetic events in cancer. Nat. Rev. Genet. 2002, 3, 415–428. [Google Scholar] [CrossRef]
- Pan, Y.; Liu, G.; Zhou, F.; Su, B.; Li, Y. DNA methylation profiles in cancer diagnosis and therapeutics. Clin. Exp. Med. 2018, 18, 1–14. [Google Scholar] [CrossRef]
- Konno, M.; Matsui, H.; Koseki, J.; Asai, A.; Kano, Y.; Kawamoto, K.; Nishida, N.; Sakai, D.; Kudo, T.; Satoh, T.; et al. Computational trans-omics approach characterised methylomic and transcriptomic involvements and identified novel therapeutic targets for chemoresistance in gastrointestinal cancer stem cells. Sci. Rep. 2018, 8, 899. [Google Scholar] [CrossRef] [Green Version]
- Payne, S.R. From discovery to the clinic: The novel DNA methylation biomarker (m)SEPT9 for the detection of colorectal cancer in blood. Epigenomics 2010, 2, 575–585. [Google Scholar] [CrossRef]
- Wu, T.; Giovannucci, E.; Welge, J.; Mallick, P.; Tang, W.Y.; Ho, S.M. Measurement of GSTP1 promoter methylation in body fluids may complement PSA screening: A meta-analysis. Br. J. Cancer 2011, 105, 65–73. [Google Scholar] [CrossRef] [Green Version]
- Chen, Y.; Li, J.; Yu, X.; Li, S.; Zhang, X.; Mo, Z.; Hu, Y. APC gene hypermethylation and prostate cancer: A systematic review and meta-analysis. Eur. J. Hum. Genet. 2013, 21, 929–935. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dietrich, D.; Hasinger, O.; Liebenberg, V.; Field, J.K.; Kristiansen, G.; Soltermann, A. DNA methylation of the homeobox genes PITX2 and SHOX2 predicts outcome in non-small-cell lung cancer patients. Diagn. Mol. Pathol. 2012, 21, 93–104. [Google Scholar] [CrossRef] [PubMed]
- Darwiche, K.; Zarogoulidis, P.; Baehner, K.; Welter, S.; Tetzner, R.; Wohlschlaeger, J.; Theegarten, D.; Nakajima, T.; Freitag, L. Assessment of SHOX2 methylation in EBUS-TBNA specimen improves accuracy in lung cancer staging. Ann. Oncol. 2013, 24, 2866–2870. [Google Scholar] [CrossRef]
- Binabaj, M.M.; Bahrami, A.; ShahidSales, S.; Joodi, M.; Joudi Mashhad, M.; Hassanian, S.M.; Anvari, K.; Avan, A. The prognostic value of MGMT promoter methylation in glioblastoma: A meta-analysis of clinical trials. J. Cell Physiol. 2018, 233, 378–386. [Google Scholar] [CrossRef]
- Wick, W.; Weller, M.; van den Bent, M.; Sanson, M.; Weiler, M.; von Deimling, A.; Plass, C.; Hegi, M.; Platten, M.; Reifenberger, G. MGMT testing--the challenges for biomarker-based glioma treatment. Nat. Rev. Neurol. 2014, 10, 372–385. [Google Scholar] [CrossRef] [Green Version]
- Yin, A.A.; Zhang, L.H.; Cheng, J.X.; Dong, Y.; Liu, B.L.; Han, N.; Zhang, X. The predictive but not prognostic value of MGMT promoter methylation status in elderly glioblastoma patients: A meta-analysis. PLoS ONE 2014, 9, e85102. [Google Scholar] [CrossRef] [Green Version]
- Imperiale, T.F.; Ransohoff, D.F.; Itzkowitz, S.H.; Levin, T.R.; Lavin, P.; Lidgard, G.P.; Ahlquist, D.A.; Berger, B.M. Multitarget stool DNA testing for colorectal-cancer screening. N. Engl. J. Med. 2014, 370, 1287–1297. [Google Scholar] [CrossRef]
- Meyer, K.D.; Saletore, Y.; Zumbo, P.; Elemento, O.; Mason, C.E.; Jaffrey, S.R. Comprehensive analysis of mRNA methylation reveals enrichment in 3’ UTRs and near stop codons. Cell 2012, 149, 1635–1646. [Google Scholar] [CrossRef] [Green Version]
- Dominissini, D.; Moshitch-Moshkovitz, S.; Schwartz, S.; Salmon-Divon, M.; Ungar, L.; Osenberg, S.; Cesarkas, K.; Jacob-Hirsch, J.; Amariglio, N.; Kupiec, M.; et al. Topology of the human and mouse m6A RNA methylomes revealed by m6A-seq. Nature 2012, 485, 201–206. [Google Scholar] [CrossRef]
- Liu, J.; Yue, Y.; Han, D.; Wang, X.; Fu, Y.; Zhang, L.; Jia, G.; Yu, M.; Lu, Z.; Deng, X.; et al. A METTL3-METTL14 complex mediates mammalian nuclear RNA N6-adenosine methylation. Nat. Chem. Biol. 2014, 10, 93–95. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vu, L.P.; Pickering, B.F.; Cheng, Y.; Zaccara, S.; Nguyen, D.; Minuesa, G.; Chou, T.; Chow, A.; Saletore, Y.; MacKay, M.; et al. The N(6)-methyladenosine (m(6)A)-forming enzyme METTL3 controls myeloid differentiation of normal hematopoietic and leukemia cells. Nat. Med. 2017, 23, 1369–1376. [Google Scholar] [CrossRef] [PubMed]
- Cai, X.; Wang, X.; Cao, C.; Gao, Y.; Zhang, S.; Yang, Z.; Liu, Y.; Zhang, X.; Zhang, W.; Ye, L. HBXIP-elevated methyltransferase METTL3 promotes the progression of breast cancer via inhibiting tumor suppressor let-7g. Cancer Lett. 2018, 415, 11–19. [Google Scholar] [CrossRef] [PubMed]
- Chen, M.; Wei, L.; Law, C.T.; Tsang, F.H.; Shen, J.; Cheng, C.L.; Tsang, L.H.; Ho, D.W.; Chiu, D.K.; Lee, J.M.; et al. RNA N6-methyladenosine methyltransferase-like 3 promotes liver cancer progression through YTHDF2-dependent posttranscriptional silencing of SOCS2. Hepatology 2018, 67, 2254–2270. [Google Scholar] [CrossRef] [Green Version]
- Nishizawa, Y.; Konno, M.; Asai, A.; Koseki, J.; Kawamoto, K.; Miyoshi, N.; Takahashi, H.; Nishida, N.; Haraguchi, N.; Sakai, D.; et al. Oncogene c-Myc promotes epitranscriptome m(6)A reader YTHDF1 expression in colorectal cancer. Oncotarget 2018, 9, 7476–7486. [Google Scholar] [CrossRef] [Green Version]
- Taketo, K.; Konno, M.; Asai, A.; Koseki, J.; Toratani, M.; Satoh, T.; Doki, Y.; Mori, M.; Ishii, H.; Ogawa, K. The epitranscriptome m6A writer METTL3 promotes chemo- and radioresistance in pancreatic cancer cells. Int. J. Oncol. 2018, 52, 621–629. [Google Scholar] [CrossRef] [Green Version]
- Konno, M.; Koseki, J.; Asai, A.; Yamagata, A.; Shimamura, T.; Motooka, D.; Okuzaki, D.; Kawamoto, K.; Mizushima, T.; Eguchi, H.; et al. Distinct methylation levels of mature microRNAs in gastrointestinal cancers. Nat. Commun. 2019, 10, 3888. [Google Scholar] [CrossRef] [Green Version]
- Cui, Q.; Shi, H.; Ye, P.; Li, L.; Qu, Q.; Sun, G.; Sun, G.; Lu, Z.; Huang, Y.; Yang, C.G.; et al. m(6)A RNA Methylation Regulates the Self-Renewal and Tumorigenesis of Glioblastoma Stem Cells. Cell Rep. 2017, 18, 2622–2634. [Google Scholar] [CrossRef]
- Schones, D.E.; Zhao, K. Genome-wide approaches to studying chromatin modifications. Nat. Rev. Genet. 2008, 9, 179–191. [Google Scholar] [CrossRef]
- van Haaften, G.; Dalgliesh, G.L.; Davies, H.; Chen, L.; Bignell, G.; Greenman, C.; Edkins, S.; Hardy, C.; O’Meara, S.; Teague, J.; et al. Somatic mutations of the histone H3K27 demethylase gene UTX in human cancer. Nat. Genet. 2009, 41, 521–523. [Google Scholar] [CrossRef] [Green Version]
- Morin, R.D.; Mendez-Lago, M.; Mungall, A.J.; Goya, R.; Mungall, K.L.; Corbett, R.D.; Johnson, N.A.; Severson, T.M.; Chiu, R.; Field, M.; et al. Frequent mutation of histone-modifying genes in non-Hodgkin lymphoma. Nature 2011, 476, 298–303. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roesch, A.; Fukunaga-Kalabis, M.; Schmidt, E.C.; Zabierowski, S.E.; Brafford, P.A.; Vultur, A.; Basu, D.; Gimotty, P.; Vogt, T.; Herlyn, M. A temporarily distinct subpopulation of slow-cycling melanoma cells is required for continuous tumor growth. Cell 2010, 141, 583–594. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kano, Y.; Konno, M.; Ohta, K.; Haraguchi, N.; Nishikawa, S.; Kagawa, Y.; Hamabe, A.; Hasegawa, S.; Ogawa, H.; Fukusumi, T.; et al. Jumonji/Arid1b (Jarid1b) protein modulates human esophageal cancer cell growth. Mol. Clin. Oncol. 2013, 1, 753–757. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ohta, K.; Haraguchi, N.; Kano, Y.; Kagawa, Y.; Konno, M.; Nishikawa, S.; Hamabe, A.; Hasegawa, S.; Ogawa, H.; Fukusumi, T.; et al. Depletion of JARID1B induces cellular senescence in human colorectal cancer. Int. J. Oncol. 2013, 42, 1212–1218. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cellarier, E.; Durando, X.; Vasson, M.P.; Farges, M.C.; Demiden, A.; Maurizis, J.C.; Madelmont, J.C.; Chollet, P. Methionine dependency and cancer treatment. Cancer Treat. Rev. 2003, 29, 489–499. [Google Scholar] [CrossRef] [PubMed]
- Sadhu, M.J.; Guan, Q.; Li, F.; Sales-Lee, J.; Iavarone, A.T.; Hammond, M.C.; Cande, W.Z.; Rine, J. Nutritional control of epigenetic processes in yeast and human cells. Genetics 2013, 195, 831–844. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shiraki, N.; Shiraki, Y.; Tsuyama, T.; Obata, F.; Miura, M.; Nagae, G.; Aburatani, H.; Kume, K.; Endo, F.; Kume, S. Methionine metabolism regulates maintenance and differentiation of human pluripotent stem cells. Cell Metab. 2014, 19, 780–794. [Google Scholar] [CrossRef] [Green Version]
- Hayes, J.D.; Dinkova-Kostova, A.T.; Tew, K.D. Oxidative Stress in Cancer. Cancer Cell 2020, 38, 167–197. [Google Scholar] [CrossRef]
- Pizzino, G.; Irrera, N.; Cucinotta, M.; Pallio, G.; Mannino, F.; Arcoraci, V.; Squadrito, F.; Altavilla, D.; Bitto, A. Oxidative Stress: Harms and Benefits for Human Health. Oxid. Med. Cell Longev. 2017, 2017, 8416763. [Google Scholar] [CrossRef] [Green Version]
- Majumdar, S.; Mukherjee, S.; Maiti, A.; Karmakar, S.; Das, A.S.; Mukherjee, M.; Nanda, A.; Mitra, C. Folic acid or combination of folic acid and vitamin B(12) prevents short-term arsenic trioxide-induced systemic and mitochondrial dysfunction and DNA damage. Environ. Toxicol. 2009, 24, 377–387. [Google Scholar] [CrossRef]
- Bagnyukova, T.V.; Powell, C.L.; Pavliv, O.; Tryndyak, V.P.; Pogribny, I.P. Induction of oxidative stress and DNA damage in rat brain by a folate/methyl-deficient diet. Brain. Res. 2008, 1237, 44–51. [Google Scholar] [CrossRef] [PubMed]
- Cano, M.J.; Ayala, A.; Murillo, M.L.; Carreras, O. Protective effect of folic acid against oxidative stress produced in 21-day postpartum rats by maternal-ethanol chronic consumption during pregnancy and lactation period. Free Radic Res. 2001, 34, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Chiarello, P.G.; Vannucchi, M.T.; Moyses Neto, M.; Vannucchi, H. Hyperhomocysteinemia and oxidative stress in hemodialysis: Effects of supplementation with folic acid. Int. J. Vitam. Nutr. Res. 2003, 73, 431–438. [Google Scholar] [CrossRef] [PubMed]
- Cavallaro, R.A.; Fuso, A.; Nicolia, V.; Scarpa, S. S-adenosylmethionine prevents oxidative stress and modulates glutathione metabolism in TgCRND8 mice fed a B-vitamin deficient diet. J. Alzheimers Dis. 2010, 20, 997–1002. [Google Scholar] [CrossRef]
- Ballatori, N.; Krance, S.M.; Notenboom, S.; Shi, S.; Tieu, K.; Hammond, C.L. Glutathione dysregulation and the etiology and progression of human diseases. Biol. Chem. 2009, 390, 191–214. [Google Scholar] [CrossRef] [Green Version]
- Pizzorno, J. Homocysteine: Friend or Foe? Integr. Med. 2014, 13, 8–14. [Google Scholar]
- Siow, Y.L.; Au-Yeung, K.K.W.; Woo, C.W.H.; Karmin, O. Homocysteine stimulates phosphorylation of NADPH oxidase p47phox and p67phox subunits in monocytes via protein kinase Cbeta activation. Biochem. J. 2006, 398, 73–82. [Google Scholar] [CrossRef] [Green Version]
- Zinno, P.; Motta, V.; Guantario, B.; Natella, F.; Roselli, M.; Bello, C.; Comitato, R.; Carminati, D.; Tidona, F.; Meucci, A.; et al. Supplementation with dairy matrices impacts on homocysteine levels and gut microbiota composition of hyperhomocysteinemic mice. Eur. J. Nutr. 2020, 59, 345–358. [Google Scholar] [CrossRef]
- Lu, H.; Samanta, D.; Xiang, L.; Zhang, H.; Hu, H.; Chen, I.; Bullen, J.W.; Semenza, G.L. Chemotherapy triggers HIF-1-dependent glutathione synthesis and copper chelation that induces the breast cancer stem cell phenotype. Proc. Natl. Acad. Sci. USA 2015, 112, E4600–E4609. [Google Scholar] [CrossRef]
- White, E. The role for autophagy in cancer. J. Clin. Investig. 2015, 125, 42–46. [Google Scholar] [CrossRef] [Green Version]
- Levine, B.; Kroemer, G. Autophagy in the pathogenesis of disease. Cell 2008, 132, 27–42. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Galluzzi, L.; Pietrocola, F.; Bravo-San Pedro, J.M.; Amaravadi, R.K.; Baehrecke, E.H.; Cecconi, F.; Codogno, P.; Debnath, J.; Gewirtz, D.A.; Karantza, V.; et al. Autophagy in malignant transformation and cancer progression. EMBO J. 2015, 34, 856–880. [Google Scholar] [CrossRef] [PubMed]
- Jiang, P.; Mizushima, N. Autophagy and human diseases. Cell Res. 2014, 24, 69–79. [Google Scholar] [CrossRef] [Green Version]
- Choi, A.M.; Ryter, S.W.; Levine, B. Autophagy in human health and disease. N. Engl. J. Med. 2013, 368, 651–662. [Google Scholar] [CrossRef]
- Liu, D.; Shaukat, Z.; Xu, T.; Denton, D.; Saint, R.; Gregory, S. Autophagy regulates the survival of cells with chromosomal instability. Oncotarget 2016, 7, 63913–63923. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Santaguida, S.; Vasile, E.; White, E.; Amon, A. Aneuploidy-induced cellular stresses limit autophagic degradation. Genes Dev. 2015, 29, 2010–2021. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stingele, S.; Stoehr, G.; Peplowska, K.; Cox, J.; Mann, M.; Storchova, Z. Global analysis of genome, transcriptome and proteome reveals the response to aneuploidy in human cells. Mol. Syst. Biol. 2012, 8, 608. [Google Scholar] [CrossRef]
- Tang, H.W.; Wang, Y.B.; Wang, S.L.; Wu, M.H.; Lin, S.Y.; Chen, G.C. Atg1-mediated myosin II activation regulates autophagosome formation during starvation-induced autophagy. EMBO J. 2011, 30, 636–651. [Google Scholar] [CrossRef] [Green Version]
- Mathew, R.; Kongara, S.; Beaudoin, B.; Karp, C.M.; Bray, K.; Degenhardt, K.; Chen, G.; Jin, S.; White, E. Autophagy suppresses tumor progression by limiting chromosomal instability. Genes Dev. 2007, 21, 1367–1381. [Google Scholar] [CrossRef]
- Karantza-Wadsworth, V.; Patel, S.; Kravchuk, O.; Chen, G.; Mathew, R.; Jin, S.; White, E. Autophagy mitigates metabolic stress and genome damage in mammary tumorigenesis. Genes Dev. 2007, 21, 1621–1635. [Google Scholar] [CrossRef] [Green Version]
- Guo, J.Y.; Chen, H.Y.; Mathew, R.; Fan, J.; Strohecker, A.M.; Karsli-Uzunbas, G.; Kamphorst, J.J.; Chen, G.; Lemons, J.M.; Karantza, V.; et al. Activated Ras requires autophagy to maintain oxidative metabolism and tumorigenesis. Genes Dev. 2011, 25, 460–470. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lock, R.; Roy, S.; Kenific, C.M.; Su, J.S.; Salas, E.; Ronen, S.M.; Debnath, J. Autophagy facilitates glycolysis during Ras-mediated oncogenic transformation. Mol. Biol. Cell 2011, 22, 165–178. [Google Scholar] [CrossRef] [PubMed]
- Nakatogawa, H.; Suzuki, K.; Kamada, Y.; Ohsumi, Y. Dynamics and diversity in autophagy mechanisms: Lessons from yeast. Nat. Rev. Mol. Cell Biol. 2009, 10, 458–467. [Google Scholar] [CrossRef] [Green Version]
- Cheng, Y.; Ren, X.; Hait, W.N.; Yang, J.M. Therapeutic targeting of autophagy in disease: Biology and pharmacology. Pharmacol. Rev. 2013, 65, 1162–1197. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Swampillai, A.L.; Salomoni, P.; Short, S.C. The role of autophagy in clinical practice. Clin. Oncol. R. Coll. Radiol. 2012, 24, 387–395. [Google Scholar] [CrossRef] [PubMed]
- Jayaram, H.; Hoelper, D.; Jain, S.U.; Cantone, N.; Lundgren, S.M.; Poy, F.; Allis, C.D.; Cummings, R.; Bellon, S.; Lewis, P.W. S-adenosyl methionine is necessary for inhibition of the methyltransferase G9a by the lysine 9 to methionine mutation on histone H3. Proc. Natl. Acad. Sci. USA 2016, 113, 6182–6187. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martinez-Chantar, M.L.; Vazquez-Chantada, M.; Garnacho, M.; Latasa, M.U.; Varela-Rey, M.; Dotor, J.; Santamaria, M.; Martinez-Cruz, L.A.; Parada, L.A.; Lu, S.C.; et al. S-adenosylmethionine regulates cytoplasmic HuR via AMP-activated kinase. Gastroenterology 2006, 131, 223–232. [Google Scholar] [CrossRef]
- Yang, A.; Jiao, Y.; Yang, S.; Deng, M.; Yang, X.; Mao, C.; Sun, Y.; Ding, N.; Li, N.; Zhang, M.; et al. Homocysteine activates autophagy by inhibition of CFTR expression via interaction between DNA methylation and H3K27me3 in mouse liver. Cell Death Dis. 2018, 9, 169. [Google Scholar] [CrossRef] [Green Version]
- Desideri, E.; Filomeni, G.; Ciriolo, M.R. Glutathione participates in the modulation of starvation-induced autophagy in carcinoma cells. Autophagy 2012, 8, 1769–1781. [Google Scholar] [CrossRef]
- Chin, R.M.; Fu, X.; Pai, M.Y.; Vergnes, L.; Hwang, H.; Deng, G.; Diep, S.; Lomenick, B.; Meli, V.S.; Monsalve, G.C.; et al. The metabolite alpha-ketoglutarate extends lifespan by inhibiting ATP synthase and TOR. Nature 2014, 510, 397–401. [Google Scholar] [CrossRef] [Green Version]
- Eisenberg, T.; Knauer, H.; Schauer, A.; Buttner, S.; Ruckenstuhl, C.; Carmona-Gutierrez, D.; Ring, J.; Schroeder, S.; Magnes, C.; Antonacci, L.; et al. Induction of autophagy by spermidine promotes longevity. Nat. Cell Biol. 2009, 11, 1305–1314. [Google Scholar] [CrossRef] [PubMed]
- Vanrell, M.C.; Cueto, J.A.; Barclay, J.J.; Carrillo, C.; Colombo, M.I.; Gottlieb, R.A.; Romano, P.S. Polyamine depletion inhibits the autophagic response modulating Trypanosoma cruzi infectivity. Autophagy 2013, 9, 1080–1093. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, S.Y.; Ko, K.S. Effects of S-Adenosylmethionine and Its Combinations With Taurine and/or Betaine on Glutathione Homeostasis in Ethanol-induced Acute Hepatotoxicity. J. Cancer Prev. 2016, 21, 164–172. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paul, B.D.; Sbodio, J.I.; Snyder, S.H. Cysteine Metabolism in Neuronal Redox Homeostasis. Trends Pharmacol. Sci. 2018, 39, 513–524. [Google Scholar] [CrossRef] [PubMed]
- Villar, V.H.; Merhi, F.; Djavaheri-Mergny, M.; Duran, R.V. Glutaminolysis and autophagy in cancer. Autophagy 2015, 11, 1198–1208. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mancilla, H.; Maldonado, R.; Cereceda, K.; Villarroel-Espindola, F.; Montes de Oca, M.; Angulo, C.; Castro, M.A.; Slebe, J.C.; Vera, J.C.; Lavandero, S.; et al. Glutathione Depletion Induces Spermatogonial Cell Autophagy. J. Cell Biochem. 2015, 116, 2283–2292. [Google Scholar] [CrossRef] [PubMed]
- Sutter, B.M.; Wu, X.; Laxman, S.; Tu, B.P. Methionine inhibits autophagy and promotes growth by inducing the SAM-responsive methylation of PP2A. Cell 2013, 154, 403–415. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gu, X.; Orozco, J.M.; Saxton, R.A.; Condon, K.J.; Liu, G.Y.; Krawczyk, P.A.; Scaria, S.M.; Harper, J.W.; Gygi, S.P.; Sabatini, D.M. SAMTOR is an S-adenosylmethionine sensor for the mTORC1 pathway. Science 2017, 358, 813–818. [Google Scholar] [CrossRef] [Green Version]
- Pietrocola, F.; Lachkar, S.; Enot, D.P.; Niso-Santano, M.; Bravo-San Pedro, J.M.; Sica, V.; Izzo, V.; Maiuri, M.C.; Madeo, F.; Marino, G.; et al. Spermidine induces autophagy by inhibiting the acetyltransferase EP300. Cell Death Differ. 2015, 22, 509–516. [Google Scholar] [CrossRef]
- Minois, N.; Carmona-Gutierrez, D.; Madeo, F. Polyamines in aging and disease. Aging 2011, 3, 716–732. [Google Scholar] [CrossRef] [Green Version]
- Zhang, H.; Alsaleh, G.; Feltham, J.; Sun, Y.; Napolitano, G.; Riffelmacher, T.; Charles, P.; Frau, L.; Hublitz, P.; Yu, Z.; et al. Polyamines Control eIF5A Hypusination, TFEB Translation, and Autophagy to Reverse B Cell Senescence. Mol. Cell 2019, 76, 110–125.e119. [Google Scholar] [CrossRef] [Green Version]
- Madeo, F.; Pietrocola, F.; Eisenberg, T.; Kroemer, G. Caloric restriction mimetics: Towards a molecular definition. Nat. Rev. Drug Discov. 2014, 13, 727–740. [Google Scholar] [CrossRef]
- Mishima, Y.; Terui, Y.; Mishima, Y.; Taniyama, A.; Kuniyoshi, R.; Takizawa, T.; Kimura, S.; Ozawa, K.; Hatake, K. Autophagy and autophagic cell death are next targets for elimination of the resistance to tyrosine kinase inhibitors. Cancer Sci. 2008, 99, 2200–2208. [Google Scholar] [CrossRef]
- Calabretta, B.; Salomoni, P. Inhibition of autophagy: A new strategy to enhance sensitivity of chronic myeloid leukemia stem cells to tyrosine kinase inhibitors. Leuk. Lymphoma 2011, 52 (Suppl. S1), 54–59. [Google Scholar] [CrossRef]
- Chen, S.; Rehman, S.K.; Zhang, W.; Wen, A.; Yao, L.; Zhang, J. Autophagy is a therapeutic target in anticancer drug resistance. Biochim. Biophys. Acta 2010, 1806, 220–229. [Google Scholar] [CrossRef]
- Sui, X.; Chen, R.; Wang, Z.; Huang, Z.; Kong, N.; Zhang, M.; Han, W.; Lou, F.; Yang, J.; Zhang, Q.; et al. Autophagy and chemotherapy resistance: A promising therapeutic target for cancer treatment. Cell Death Dis. 2013, 4, e838. [Google Scholar] [CrossRef] [Green Version]
- Manegold, C. Pemetrexed (Alimta, MTA, multitargeted antifolate, LY231514) for malignant pleural mesothelioma. Semin. Oncol. 2003, 30, 32–36. [Google Scholar] [CrossRef]
- Hertz, R.; Li, M.C.; Spencer, D.B. Effect of methotrexate therapy upon choriocarcinoma and chorioadenoma. Proc. Soc. Exp. Biol. Med. 1956, 93, 361–366. [Google Scholar] [CrossRef]
- Vousden, K.H.; Lu, X. Live or let die: The cell’s response to p53. Nat. Rev. Cancer 2002, 2, 594–604. [Google Scholar] [CrossRef]
- Start, K. Treating phenylketonuria by a phenylalanine-free diet. Prof. Care Mother Child 1998, 8, 109–110. [Google Scholar]
- Hayashi, S.; Tanaka, T.; Naito, J.; Suda, M. Dietary and hormonal regulation of serine synthesis in the rat. J. Biochem. 1975, 77, 207–219. [Google Scholar]
- Gravel, S.P.; Hulea, L.; Toban, N.; Birman, E.; Blouin, M.J.; Zakikhani, M.; Zhao, Y.; Topisirovic, I.; St-Pierre, J.; Pollak, M. Serine deprivation enhances antineoplastic activity of biguanides. Cancer Res. 2014, 74, 7521–7533. [Google Scholar] [CrossRef] [Green Version]
- Mullarky, E.; Lucki, N.C.; Beheshti Zavareh, R.; Anglin, J.L.; Gomes, A.P.; Nicolay, B.N.; Wong, J.C.; Christen, S.; Takahashi, H.; Singh, P.K.; et al. Identification of a small molecule inhibitor of 3-phosphoglycerate dehydrogenase to target serine biosynthesis in cancers. Proc. Natl. Acad. Sci. USA 2016, 113, 1778–1783. [Google Scholar] [CrossRef] [Green Version]
- Pacold, M.E.; Brimacombe, K.R.; Chan, S.H.; Rohde, J.M.; Lewis, C.A.; Swier, L.J.; Possemato, R.; Chen, W.W.; Sullivan, L.B.; Fiske, B.P.; et al. A PHGDH inhibitor reveals coordination of serine synthesis and one-carbon unit fate. Nat. Chem. Biol. 2016, 12, 452–458. [Google Scholar] [CrossRef] [Green Version]
- Chen, J.; Chung, F.; Yang, G.; Pu, M.; Gao, H.; Jiang, W.; Yin, H.; Capka, V.; Kasibhatla, S.; Laffitte, B.; et al. Phosphoglycerate dehydrogenase is dispensable for breast tumor maintenance and growth. Oncotarget 2013, 4, 2502–2511. [Google Scholar] [CrossRef] [Green Version]
- Breillout, F.; Hadida, F.; Echinard-Garin, P.; Lascaux, V.; Poupon, M.F. Decreased rat rhabdomyosarcoma pulmonary metastases in response to a low methionine diet. Anticancer Res. 1987, 7, 861–867. [Google Scholar]
- Guo, H.; Lishko, V.K.; Herrera, H.; Groce, A.; Kubota, T.; Hoffman, R.M. Therapeutic tumor-specific cell cycle block induced by methionine starvation in vivo. Cancer Res. 1993, 53, 5676–5679. [Google Scholar]
- Hoshiya, Y.; Guo, H.; Kubota, T.; Inada, T.; Asanuma, F.; Yamada, Y.; Koh, J.; Kitajima, M.; Hoffman, R.M. Human tumors are methionine dependent in vivo. Anticancer Res. 1995, 15, 717–718. [Google Scholar]
- Komninou, D.; Leutzinger, Y.; Reddy, B.S.; Richie, J.P., Jr. Methionine restriction inhibits colon carcinogenesis. Nutr. Cancer 2006, 54, 202–208. [Google Scholar] [CrossRef]
- Tan, Y.; Xu, M.; Guo, H.; Sun, X.; Kubota, T.; Hoffman, R.M. Anticancer efficacy of methioninase in vivo. Anticancer Res. 1996, 16, 3931–3936. [Google Scholar]
- Hoffman, R.M.; Hoshiya, Y.; Guo, W. Efficacy of Methionine-Restricted Diets on Cancers In Vivo. Methods Mol. Biol. 2019, 1866, 75–81. [Google Scholar] [CrossRef]
- Hoffman, R.M. Clinical Studies of Methionine-Restricted Diets for Cancer Patients. Methods Mol. Biol. 2019, 1866, 95–105. [Google Scholar] [CrossRef]
- Goseki, N.; Yamazaki, S.; Shimojyu, K.; Kando, F.; Maruyama, M.; Endo, M.; Koike, M.; Takahashi, H. Synergistic effect of methionine-depleting total parenteral nutrition with 5-fluorouracil on human gastric cancer: A randomized, prospective clinical trial. Jpn. J. Cancer Res. 1995, 86, 484–489. [Google Scholar] [CrossRef]
- Tan, Y.; Xu, M.; Tan, X.; Tan, X.; Wang, X.; Saikawa, Y.; Nagahama, T.; Sun, X.; Lenz, M.; Hoffman, R.M. Overexpression and large-scale production of recombinant L-methionine-alpha-deamino-gamma-mercaptomethane-lyase for novel anticancer therapy. Protein Expr. Purif. 1997, 9, 233–245. [Google Scholar] [CrossRef]
- Hoffman, R.M.; Tan, Y.; Li, S.; Han, Q.; Yagi, S.; Takakura, T.; Takimoto, A.; Inagaki, K.; Kudou, D. Development of Recombinant Methioninase for Cancer Treatment. Methods Mol. Biol. 2019, 1866, 107–131. [Google Scholar] [CrossRef]
- Kreis, W. Tumor therapy by deprivation of L-methionine: Rationale and results. Cancer Treat. Rep. 1979, 63, 1069–1072. [Google Scholar]
- Tisdale, M.J.; Jack, G.W.; Eridani, S. Differential sensitivity of normal and leukaemic haemopoietic cells to methionine deprivation by L-methioninase. Leuk. Res. 1983, 7, 269–277. [Google Scholar] [CrossRef]
- Hoffman, R.M.; Murakami, T.; Kawaguchi, K.; Igarashi, K.; Tan, Y.; Li, S.; Han, Q. High Efficacy of Recombinant Methioninase on Patient-Derived Orthotopic Xenograft (PDOX) Mouse Models of Cancer. Methods Mol. Biol. 2019, 1866, 149–161. [Google Scholar] [CrossRef]
- Kawaguchi, K.; Han, Q.; Li, S.; Tan, Y.; Igarashi, K.; Kiyuna, T.; Miyake, K.; Miyake, M.; Chmielowski, B.; Nelson, S.D.; et al. Targeting methionine with oral recombinant methioninase (o-rMETase) arrests a patient-derived orthotopic xenograft (PDOX) model of BRAF-V600E mutant melanoma: Implications for chronic clinical cancer therapy and prevention. Cell Cycle 2018, 17, 356–361. [Google Scholar] [CrossRef] [Green Version]
- Kawaguchi, K.; Han, Q.; Li, S.; Tan, Y.; Igarashi, K.; Murakami, T.; Unno, M.; Hoffman, R.M. Efficacy of Recombinant Methioninase (rMETase) on Recalcitrant Cancer Patient-Derived Orthotopic Xenograft (PDOX) Mouse Models: A Review. Cells 2019, 8, 410. [Google Scholar] [CrossRef] [Green Version]
- Mihich, E. Current Studies with Methylglyoxal-Bis(Guanylhydrazone). Cancer Res. 1963, 23, 1375–1389. [Google Scholar]
- Williams-Ashman, H.G.; Schenone, A. Methyl glyoxal bis(guanylhydrazone) as a potent inhibitor of mammalian and yeast S-adenosylmethionine decarboxylases. Biochem. Biophys. Res. Commun. 1972, 46, 288–295. [Google Scholar] [CrossRef]
- Pegg, A.E. S-Adenosylmethionine decarboxylase. Essays Biochem. 2009, 46, 25–45. [Google Scholar] [CrossRef] [Green Version]
- Pleshkewych, A.; Kramer, D.L.; Kelly, E.; Porter, C.W. Independence of drug action on mitochondria and polyamines in L1210 leukemia cells treated with methylglyoxal-bis(guanylhydrazone). Cancer Res. 1980, 40, 4533–4540. [Google Scholar]
- Murray-Stewart, T.R.; Woster, P.M.; Casero, R.A., Jr. Targeting polyamine metabolism for cancer therapy and prevention. Biochem. J. 2016, 473, 2937–2953. [Google Scholar] [CrossRef] [Green Version]
- Ikeguchi, Y.; Bewley, M.C.; Pegg, A.E. Aminopropyltransferases: Function, structure and genetics. J. Biochem. 2006, 139, 1–9. [Google Scholar] [CrossRef]
- Prakash, N.J.; Schechter, P.J.; Grove, J.; Koch-Weser, J. Effect of alpha-difluoromethylornithine, an enzyme-activated irreversible inhibitor of ornithine decarboxylase, on L1210 leukemia in mice. Cancer Res. 1978, 38, 3059–3062. [Google Scholar]
- Weeks, C.E.; Herrmann, A.L.; Nelson, F.R.; Slaga, T.J. alpha-Difluoromethylornithine, an irreversible inhibitor of ornithine decarboxylase, inhibits tumor promoter-induced polyamine accumulation and carcinogenesis in mouse skin. Proc. Natl. Acad. Sci. USA 1982, 79, 6028–6032. [Google Scholar] [CrossRef] [Green Version]
- Mamont, P.S.; Siat, M.; Joder-Ohlenbusch, A.M.; Bernhardt, A.; Casara, P. Effects of (2R, 5R)-6-heptyne-2,5-diamine, a potent inhibitor of L-ornithine decarboxylase, on rat hepatoma cells cultured in vitro. Eur. J. Biochem. 1984, 142, 457–463. [Google Scholar] [CrossRef]
- Seidenfeld, J. Effects of difluoromethylornithine on proliferation, polyamine content and plating efficiency of cultured human carcinoma cells. Cancer Chemother. Pharmacol. 1985, 15, 196–202. [Google Scholar] [CrossRef]
- Abeloff, M.D.; Rosen, S.T.; Luk, G.D.; Baylin, S.B.; Zeltzman, M.; Sjoerdsma, A. Phase II trials of alpha-difluoromethylornithine, an inhibitor of polyamine synthesis, in advanced small cell lung cancer and colon cancer. Cancer Treat. Rep. 1986, 70, 843–845. [Google Scholar]
- Luk, G.D.; Abeloff, M.D.; Griffin, C.A.; Baylin, S.B. Successful treatment with DL-alpha-difluoromethylornithine in established human small cell variant lung carcinoma implants in athymic mice. Cancer Res. 1983, 43, 4239–4243. [Google Scholar]
- Bassiri, H.; Benavides, A.; Haber, M.; Gilmour, S.K.; Norris, M.D.; Hogarty, M.D. Translational development of difluoromethylornithine (DFMO) for the treatment of neuroblastoma. Transl. Pediatr. 2015, 4, 226–238. [Google Scholar] [CrossRef]
- Horn, Y.; Schechter, P.J.; Marton, L.J. Phase I-II clinical trial with alpha-difluoromethylornithine--an inhibitor of polyamine biosynthesis. Eur. J. Cancer Clin. Oncol. 1987, 23, 1103–1107. [Google Scholar] [CrossRef]
- Meyskens, F.L.; Kingsley, E.M.; Glattke, T.; Loescher, L.; Booth, A. A phase II study of alpha-difluoromethylornithine (DFMO) for the treatment of metastatic melanoma. Investig. New Drugs 1986, 4, 257–262. [Google Scholar] [CrossRef]
- Burns, M.R.; Graminski, G.F.; Weeks, R.S.; Chen, Y.; O’Brien, T.G. Lipophilic lysine-spermine conjugates are potent polyamine transport inhibitors for use in combination with a polyamine biosynthesis inhibitor. J. Med. Chem. 2009, 52, 1983–1993. [Google Scholar] [CrossRef] [Green Version]
- Samal, K.; Zhao, P.; Kendzicky, A.; Yco, L.P.; McClung, H.; Gerner, E.; Burns, M.; Bachmann, A.S.; Sholler, G. AMXT-1501, a novel polyamine transport inhibitor, synergizes with DFMO in inhibiting neuroblastoma cell proliferation by targeting both ornithine decarboxylase and polyamine transport. Int. J. Cancer 2013, 133, 1323–1333. [Google Scholar] [CrossRef]
- Levin, V.A.; Hess, K.R.; Choucair, A.; Flynn, P.J.; Jaeckle, K.A.; Kyritsis, A.P.; Yung, W.K.; Prados, M.D.; Bruner, J.M.; Ictech, S.; et al. Phase III randomized study of postradiotherapy chemotherapy with combination alpha-difluoromethylornithine-PCV versus PCV for anaplastic gliomas. Clin. Cancer Res. 2003, 9, 981–990. [Google Scholar]
- Levin, V.A.; Uhm, J.H.; Jaeckle, K.A.; Choucair, A.; Flynn, P.J.; Yung, W.K.A.; Prados, M.D.; Bruner, J.M.; Chang, S.M.; Kyritsis, A.P.; et al. Phase III randomized study of postradiotherapy chemotherapy with alpha-difluoromethylornithine-procarbazine, N-(2-chloroethyl)-N’-cyclohexyl-N-nitrosurea, vincristine (DFMO-PCV) versus PCV for glioblastoma multiforme. Clin. Cancer Res. 2000, 6, 3878–3884. [Google Scholar]
- Saulnier Sholler, G.L.; Gerner, E.W.; Bergendahl, G.; MacArthur, R.B.; VanderWerff, A.; Ashikaga, T.; Bond, J.P.; Ferguson, W.; Roberts, W.; Wada, R.K.; et al. A Phase I Trial of DFMO Targeting Polyamine Addiction in Patients with Relapsed/Refractory Neuroblastoma. PLoS ONE 2015, 10, e0127246. [Google Scholar] [CrossRef] [Green Version]
- Meyskens, F.L., Jr.; Gerner, E.W. Development of difluoromethylornithine (DFMO) as a chemoprevention agent. Clin. Cancer Res. 1999, 5, 945–951. [Google Scholar] [PubMed]
- Nigro, N.D.; Bull, A.W.; Boyd, M.E. Inhibition of intestinal carcinogenesis in rats: Effect of difluoromethylornithine with piroxicam or fish oil. J. Natl. Cancer Inst. 1986, 77, 1309–1313. [Google Scholar]
- Gerner, E.W.; Meyskens, F.L., Jr.; Goldschmid, S.; Lance, P.; Pelot, D. Rationale for, and design of, a clinical trial targeting polyamine metabolism for colon cancer chemoprevention. Amino Acids 2007, 33, 189–195. [Google Scholar] [CrossRef] [Green Version]
- Tang, K.C.; Pegg, A.E.; Coward, J.K. Specific and potent inhibition of spermidine synthase by the transition-state analog, S-adenosyl-3-thio-1,8-diaminooctane. Biochem. Biophys. Res. Commun. 1980, 96, 1371–1377. [Google Scholar] [CrossRef]
- Woster, P.M.; Black, A.Y.; Duff, K.J.; Coward, J.K.; Pegg, A.E. Synthesis and biological evaluation of S-adenosyl-1,12-diamino-3-thio-9-azadodecane, a multisubstrate adduct inhibitor of spermine synthase. J. Med. Chem. 1989, 32, 1300–1307. [Google Scholar] [CrossRef]
- Porter, C.W.; Bergeron, R.J. Regulation of polyamine biosynthetic activity by spermidine and spermine analogs--a novel antiproliferative strategy. Adv. Exp. Med. Biol. 1988, 250, 677–690. [Google Scholar] [CrossRef]
- Cervelli, M.; Bellavia, G.; Fratini, E.; Amendola, R.; Polticelli, F.; Barba, M.; Federico, R.; Signore, F.; Gucciardo, G.; Grillo, R.; et al. Spermine oxidase (SMO) activity in breast tumor tissues and biochemical analysis of the anticancer spermine analogues BENSpm and CPENSpm. BMC Cancer 2010, 10, 555. [Google Scholar] [CrossRef]
- McCloskey, D.E.; Yang, J.; Woster, P.M.; Davidson, N.E.; Casero, R.A., Jr. Polyamine analogue induction of programmed cell death in human lung tumor cells. Clin. Cancer Res. 1996, 2, 441–446. [Google Scholar]
- Casero, R.A., Jr.; Mank, A.R.; Saab, N.H.; Wu, R.; Dyer, W.J.; Woster, P.M. Growth and biochemical effects of unsymmetrically substituted polyamine analogues in human lung tumor cells 1. Cancer Chemother. Pharmacol. 1995, 36, 69–74. [Google Scholar] [CrossRef]
- McCloskey, D.E.; Casero, R.A., Jr.; Woster, P.M.; Davidson, N.E. Induction of programmed cell death in human breast cancer cells by an unsymmetrically alkylated polyamine analogue. Cancer Res. 1995, 55, 3233–3236. [Google Scholar]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Islam, A.; Shaukat, Z.; Hussain, R.; Gregory, S.L. One-Carbon and Polyamine Metabolism as Cancer Therapy Targets. Biomolecules 2022, 12, 1902. https://doi.org/10.3390/biom12121902
Islam A, Shaukat Z, Hussain R, Gregory SL. One-Carbon and Polyamine Metabolism as Cancer Therapy Targets. Biomolecules. 2022; 12(12):1902. https://doi.org/10.3390/biom12121902
Chicago/Turabian StyleIslam, Anowarul, Zeeshan Shaukat, Rashid Hussain, and Stephen L. Gregory. 2022. "One-Carbon and Polyamine Metabolism as Cancer Therapy Targets" Biomolecules 12, no. 12: 1902. https://doi.org/10.3390/biom12121902