
Structural Studies of Piperine Inclusion Complexes in Native and Derivative β-Cyclodextrins



Abstract
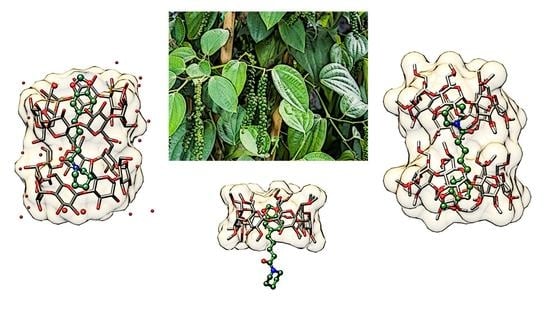
:
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Phase Solubility Study
2.3. Single-Crystal Preparation
2.4. X-ray Diffraction Experiments
2.5. Computational Methods
3. Results
3.1. Phase Solubility Analysis
3.2. The Crystal Structure of PN/β-CD
3.3. The Crystal Structure of PN/DM-β-CD
3.4. The Crystal Structure of PN/TM-β-CD
3.5. MDs Trajectories Analysis
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding

Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Butt, M.S.; Pasha, I.; Sultan, M.T.; Randhawa, M.A.; Saeed, F.; Ahmed, W. Black Pepper and Health Claims: A Comprehensive Treatise. Crit. Rev. Food Sci. Nutr. 2013, 53, 875–886. [Google Scholar] [CrossRef] [PubMed]
- Wattanathorn, J.; Chonpathompikunlert, P.; Muchimapura, S.; Priprem, A.; Tankamnerdthai, O. Piperine, the Potential Functional Food for Mood and Cognitive Disorders. Food Chem. Toxicol. 2008, 46, 3106–3110. [Google Scholar] [CrossRef] [PubMed]
- Shityakov, S.; Bigdelian, E.; Hussein, A.A.; Hussain, M.B.; Tripathi, Y.C.; Khan, M.U.; Shariati, M.A. Phytochemical and Pharmacological Attributes of Piperine: A Bioactive Ingredient of Black Pepper. Eur. J. Med. Chem. 2019, 176, 149–161. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mohanraj, V.; Aravindan, B.; Jayaprakash, C.; Thenmozhi, M. In silico drug design and extraction of piperine an inhibitor for fernesyltransferase in Cryptococcus neoformans. World J. Pharm. Res. 2014, 3, 1107–1120. [Google Scholar]
- Chonpathompikunlert, P.; Yoshitomi, T.; Han, J.; Isoda, H.; Nagasaki, Y. The Use of Nitroxide Radical-Containing Nanoparticles Coupled with Piperine to Protect Neuroblastoma SH-SY5Y Cells from Aβ-Induced Oxidative Stress. Biomaterials 2011, 32, 8605–8612. [Google Scholar] [CrossRef]
- Chonpathompikunlert, P.; Wattanathorn, J.; Muchimapura, S. Piperine, the Main Alkaloid of Thai Black Pepper, Protects against Neurodegeneration and Cognitive Impairment in Animal Model of Cognitive Deficit like Condition of Alzheimer’s Disease. Food Chem. Toxicol. 2010, 48, 798–802. [Google Scholar] [CrossRef]
- Liu, J.; Chen, M.; Wang, X.; Wang, Y.; Duan, C.; Gao, G.; Lu, L.; Wu, X.; Wang, X.; Yang, H. Piperine Induces Autophagy by Enhancing Protein Phosphotase 2A Activity in a Rotenone-Induced Parkinson’s Disease Model. Oncotarget 2016, 7, 60823–60843. [Google Scholar] [CrossRef] [Green Version]
- Stojanović-Radić, Z.; Pejčić, M.; Dimitrijević, M.; Aleksić, A.; V. Anil Kumar, N.; Salehi, B.; C. Cho, W.; Sharifi-Rad, J. Piperine-A Major Principle of Black Pepper: A Review of Its Bioactivity and Studies. Appl. Sci. 2019, 9, 4270. [Google Scholar] [CrossRef] [Green Version]
- Sriwiriyajan, S.; Tedasen, A.; Lailerd, N.; Boonyaphiphat, P.; Nitiruangjarat, A.; Deng, Y.; Graidist, P. Anticancer and Cancer Prevention Effects of Piperine-Free Piper Nigrum Extract On. Cancer Prev. Res. 2016, 9, 74–82. [Google Scholar] [CrossRef] [Green Version]
- Rather, R.A.; Bhagat, M. Cancer Chemoprevention and Piperine: Molecular Mechanisms and Therapeutic Opportunities. Front. Cell Dev. Biol. 2018, 6, 10. [Google Scholar] [CrossRef] [Green Version]
- Tiwari, A.; Mahadik, K.R.; Gabhe, S.Y. Piperine: A Comprehensive Review of Methods of Isolation, Purification, and Biological Properties. Med. Drug Discov. 2020, 7, 100027. [Google Scholar] [CrossRef]
- Alshehri, S.; Haq, N.; Shakeel, F. Solubility, Molecular Interactions and Mixing Thermodynamic Properties of Piperine in Various Pure Solvents at Different Temperatures. J. Mol. Liq. 2018, 250, 63–70. [Google Scholar] [CrossRef]
- Hashimoto, K.; Yaoi, T.; Koshiba, H.; Yoshida, T.; Maoka, T.; Fujiwara, Y.; Yamamoto, Y.; Mori, K. Photochemical Isomerization of Piperine, a Pungent Constituent in Pepper. Food Sci. Technol. Int. Tokyo 1996, 2, 24–29. [Google Scholar] [CrossRef] [Green Version]
- Carneiro, S.B.; Costa Duarte, F.Í.; Heimfarth, L.; Siqueira Quintans, J.d.S.; Quintans-Júnior, L.J.; Veiga Júnior, V.F.d.; Neves de Lima, Á.A. Cyclodextrin−Drug Inclusion Complexes: In Vivo and In Vitro Approaches. Int. J. Mol. Sci. 2019, 20, 642. [Google Scholar] [CrossRef] [Green Version]
- Cheirsilp, B.; Rakmai, J. Inclusion Complex Formation of Cyclodextrin with Its Guest and Their Applications. Biol. Eng. Med. 2017, 2, 1–6. [Google Scholar] [CrossRef] [Green Version]
- Matencio, A.; Caldera, F.; Cecone, C.; López-Nicolás, J.M.; Trotta, F. Cyclic Oligosaccharides as Active Drugs, an Updated Review. Pharmaceuticals 2020, 13, 281. [Google Scholar] [CrossRef]
- Gadade, D.D.; Pekamwar, S.S. Cyclodextrin Based Nanoparticles for Drug Delivery and Theranostics. Adv. Pharm. Bull. 2020, 10, 166–183. [Google Scholar] [CrossRef] [Green Version]
- Ezawa, T.; Inoue, Y.; Tunvichien, S.; Suzuki, R.; Kanamoto, I. Changes in the Physicochemical Properties of Piperine/Cyclodextrin Due to the Formation of Inclusion Complexes. Int. J. Med. Chem. 2016, 2016, 9. [Google Scholar] [CrossRef] [Green Version]
- Ezawa, T.; Inoue, Y.; Murata, I.; Takao, K.; Sugita, Y.; Kanamoto, I. Characterization of the Dissolution Behavior of Piperine/Cyclodextrins Inclusion Complexes. AAPS PharmSciTech 2018, 19, 923–933. [Google Scholar] [CrossRef]
- Ezawa, T.; Inoue, Y.; Murata, I.; Takao, K.; Sugita, Y.; Kanamoto, I. Evaluation of the Molecular State of Piperine in Cyclodextrin Complexes by Near-Infrared Spectroscopy and Solid-State Fluorescence Measurements. Int. J. Med. Chem. 2019, 2019, 14. [Google Scholar] [CrossRef]
- Quilaqueo, M.; Millao, S.; Luzardo-Ocampo, I.; Campos-Vega, R.; Acevedo, F.; Shene, C.; Rubilar, M. Inclusion of Piperine in β-Cyclodextrin Complexes Improves Their Bioaccessibility and in Vitro Antioxidant Capacity. Food Hydrocoll. 2019, 91, 143–152. [Google Scholar] [CrossRef]
- Debnath, S.; Mishra, J. Understanding the Intrinsic Fluorescence of Piperine in Microheterogeneous Media: Partitioning and Loading Studies. New J. Chem. 2020, 44, 8317–8324. [Google Scholar] [CrossRef]
- Ezawa, T.; Inagaki, Y.; Kashiwaba, K.; Matsumoto, N.; Moteki, H.; Murata, I.; Inoue, Y.; Kimura, M.; Ogihara, M.; Kanamoto, I. Solubility of Piperine and Its Inclusion Complexes in Biorelevant Media and Their Effect on Attenuating Mouse Ileum Contractions. ACS Omega 2021, 6, 6953–6964. [Google Scholar] [CrossRef]
- Liu, K.; Liu, H.; Li, Z.; Li, W.; Li, L. In Vitro Dissolution Study on Inclusion Complex of Piperine with Ethylenediamine-β-Cyclodextrin. J. Incl. Phenom. Macrocycl. Chem. 2020, 96, 233–243. [Google Scholar] [CrossRef]
- Hatziagapiou, K.; Bethanis, K.; Koniari, E.; Christoforides, E.; Nikola, O.; Andreou, A.; Mantzou, A.; Chrousos, G.P.; Kanaka-Gantenbein, C.; Lambrou, G.I. Biophysical Studies and In Vitro Effects of Tumor Cell Lines of Cannabidiol and Its Cyclodextrin Inclusion Complexes. Pharmaceutics 2022, 14, 706. [Google Scholar] [CrossRef] [PubMed]
- Higuchi, T.; Connors, K.A. Phase Solubility Techniques. Adv. Anal. Chem. Instrum. 1965, 4, 117–212. Available online: http://www.oalib.com/references/7163685 (accessed on 14 July 2016).
- Sheldrick, G.M. SAINT; Version 8.37; Bruker AXS Inc.: Madison, WI, USA, 2013. [Google Scholar]
- Sheldrick, G.M. SADABS; Bruker AXS Inc.: Madison, WI, USA, 2012. [Google Scholar]
- Sheldrick, G.M. TWINABS; Bruker AXS Inc.: Madison, WI, USA, 2012. [Google Scholar]
- Sheldrick, G.M. Experimental Phasing with SHELXC/D/E: Combining Chain Tracing with Density Modification. Acta Crystallogr. Sect. D 2010, 66, 479–485. [Google Scholar] [CrossRef] [Green Version]
- Sheldrick, G.M. Crystal Structure Refinement with SHELXL. Acta Crystallogr. Sect. C 2015, 71, 3–8. [Google Scholar] [CrossRef] [Green Version]
- Hübschle, C.B.; Sheldrick, G.M.; Dittrich, B. ShelXle: A Qt Graphical User Interface for SHELXL. J. Appl. Crystallogr. 2011, 44, 1281–1284. [Google Scholar] [CrossRef] [Green Version]
- Schüttelkopf, A.W.; van Aalten, D.M.F. PRODRG: A Tool for High-Throughput Crystallography of Protein–Ligand Complexes. Acta Crystallogr. Sect. D Biol. Crystallogr. 2004, 60, 1355–1363. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thorn, A.; Dittrich, B.; Sheldrick, G.M. Enhanced Rigid-Bond Restraints. Acta Crystallogr. Sect. A: Found. Crystallogr. 2012, 68, 448–451. [Google Scholar] [CrossRef] [Green Version]
- Macrae, C.F.; Bruno, I.J.; Chisholm, J.A.; Edgington, P.R.; McCabe, P.; Pidcock, E.; Rodriguez-Monge, L.; Taylor, R.; van de Streek, J.; Wood, P.A. Mercury CSD 2.0–New Features for the Visualization and Investigation of Crystal Structures. J. Appl. Crystallogr. 2008, 41, 466–470. [Google Scholar] [CrossRef]
- Schrödinger, LLC. The PyMOL Molecular Graphics System, Version 1.8; Schrödinger, LLC: New York, NY, USA, 2015. [Google Scholar]
- Dolomanov, O.V.; Bourhis, L.J.; Gildea, R.J.; Howard, J.A.K.; Puschmann, H. OLEX2: A Complete Structure Solution, Refinement and Analysis Program. J. Appl. Crystallogr. 2009, 42, 339–341. [Google Scholar] [CrossRef]
- Trott, O.; Olson, A.J. AutoDock Vina: Improving the Speed and Accuracy of Docking with a New Scoring Function, Efficient Optimization, and Multithreading. J. Comput. Chem. 2009, 31, 455–461. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jadhav, P. Piperine-Hydroxy Acid-Cyclodextrin Inclusion Complexes; Antioxidant, Antiinflammatory, and Stability Studies: PART II. AJP 2021, 15, 130–140. [Google Scholar] [CrossRef]
- Pedretti, A.; Mazzolari, A.; Gervasoni, S.; Fumagalli, L.; Vistoli, G. The VEGA Suite of Programs: An Versatile Platform for Cheminformatics and Drug Design Projects. Bioinformatics 2021, 37, 1174–1175. [Google Scholar] [CrossRef] [PubMed]
- Gordon, M.S.; Schmidt, M.W. Chapter 41-Advances in Electronic Structure Theory: GAMESS a Decade Later. In Theory and Applications of Computational Chemistry; Dykstra, C.E., Frenking, G., Kim, K.S., Scuseria, G.E., Eds.; Elsevier: Amsterdam, The Netherlands, 2005; pp. 1167–1189. ISBN 978-0-444-51719-7. [Google Scholar]
- Salomon-Ferrer, R.; Case, D.A.; Walker, R.C. An Overview of the Amber Biomolecular Simulation Package. Wiley Interdiscip. Rev. Comput. Mol. Sci. 2012, 3, 198–210. [Google Scholar] [CrossRef]
- Cezard, C.; Trivelli, X.; Aubry, F.; Djedaini-Pilard, F.; Dupradeau, F.-Y. Molecular Dynamics Studies of Native and Substituted Cyclodextrins in Different Media: 1. Charge Derivation and Force Field Performances. Phys. Chem Chem Phys. 2011, 13, 15103–15121. [Google Scholar] [CrossRef]
- Kirschner, K.N.; Yongye, A.B.; Tschampel, S.M.; Gonzalez-Outeirino, J.; Daniels, C.R.; Foley, B.L.; Woods, R.J. GLYCAM06: A Generalizable Biomolecular Force Field. Carbohydrates. J. Comput. Chem. 2008, 29, 622–655. [Google Scholar] [CrossRef] [Green Version]
- Mark, P.; Nilsson, L. Structure and Dynamics of the TIP3P, SPC, and SPC/E Water Models at 298 K. J. Phys. Chem. A 2001, 105, 9954–9960. [Google Scholar] [CrossRef]
- Roe, D.R.; Cheatham, T.E. 3rd PTRAJ and CPPTRAJ: Software for Processing and Analysis of Molecular Dynamics Trajectory Data. J. Chem. Theory Comput. 2013, 9, 3084–3095. [Google Scholar] [CrossRef]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD: Visual Molecular Dynamics. J. Mol. Graph. 1996, 14. 33–38, 27–28. [Google Scholar] [CrossRef] [PubMed]
- Genheden, S.; Ryde, U. The MM/PBSA and MM/GBSA Methods to Estimate Ligand-Binding Affinities. Expert Opin. Drug Discov. 2015, 10, 449–461. [Google Scholar] [CrossRef] [PubMed]
- Miller, B.R., 3rd; McGee, T.D.J.; Swails, J.M.; Homeyer, N.; Gohlke, H.; Roitberg, A.E. MMPBSA.Py: An Efficient Program for End-State Free Energy Calculations. J. Chem. Theory Comput. 2012, 8, 3314–3321. [Google Scholar] [CrossRef] [PubMed]
- Wang, E.; Sun, H.; Wang, J.; Wang, Z.; Liu, H.; Zhang, J.Z.H.; Hou, T. End-Point Binding Free Energy Calculation with MM/PBSA and MM/GBSA: Strategies and Applications in Drug Design. Chem. Rev. 2019, 119, 9478–9508. [Google Scholar] [CrossRef]
- Wang, J.; Hou, T.; Xu, X. Recent Advances in Free Energy Calculations with a Combination of Molecular Mechanics and Continuum Models. Curr. Comput. -Aided Drug Des. 2006, 2, 287–306. [Google Scholar] [CrossRef] [Green Version]
- Gilson, M.K.; Honig, B. Calculation of the Total Electrostatic Energy of a Macromolecular System: Solvation Energies, Binding Energies, and Conformational Analysis. Proteins Struct. Funct. Bioinform. 1988, 4, 7–18. [Google Scholar] [CrossRef]
- Loftsson, T.; Másson, M.; Sigurjónsdóttir, J.F. Methods to Enhance the Complexation Efficiency of Cylodextrins. S.T.P. Pharma Sci. 1999, 9, 237–242. [Google Scholar]
- Ceborska, M. Structural Investigation of the β-Cyclodextrin Complexes with Linalool and Isopinocampheol–Influence of Monoterpenes Cyclicity on the Host–Guest Stoichiometry. Chem. Phys. Lett. 2016, 651, 192–197. [Google Scholar] [CrossRef]
- Ogawa, N.; Nagase, H.; Loftsson, T.; Endo, T.; Takahashi, C.; Kawashima, Y.; Ueda, H.; Yamamoto, H. Crystallographic and Theoretical Studies of an Inclusion Complex of β-Cyclodextrin with Fentanyl. Int. J. Pharm. 2017, 531, 588–594. [Google Scholar] [CrossRef]
- Bethanis, K.; Christoforides, E.; Tsorteki, F.; Fourtaka, K.; Mentzafos, D. Structural Studies of the Inclusion Compounds of α-Naphthaleneacetic Acid in Heptakis(2,6-Di-O-Methyl)-β-Cyclodextrin and Heptakis(2,3,6-Tri-O-Methyl)-β-Cyclodextrin by X-Ray Crystallography and Molecular Dynamics. J. Incl. Phenom. Macrocycl. Chem. 2018, 92, 157–171. [Google Scholar] [CrossRef]
- Christoforides, E.; Fourtaka, K.; Andreou, A.; Bethanis, K. X-ray Crystallography and Molecular Dynamics Studies of the Inclusion Complexes of Geraniol in β-Cyclodextrin, Heptakis (2,6-Di-O-Methyl)-β-Cyclodextrin and Heptakis (2,3,6-Tri-O-Methyl)-β-Cyclodextrin. J. Mol. Struct. 2020, 1202, 127350. [Google Scholar] [CrossRef]
- Bonnet, P.; Jaime, C.; Morin-Allory, L. α-, β-, and γ-Cyclodextrin Dimers. Molecular Modeling Studies by Molecular Mechanics and Molecular Dynamics Simulations. J. Org. Chem. 2001, 66, 689–692. [Google Scholar] [CrossRef] [PubMed]
- Bethanis, K.; Christoforides, E.; Andreou, A.; Eliopoulos, E. Molecular Symmetry of Permethylated β-Cyclodextrins upon Complexation. Symmetry 2022, 14, 2214. [Google Scholar] [CrossRef]
- Alshehri, S.; Imam, S.S.; Hussain, A.; Altamimi, M.A. Formulation of Piperine Ternary Inclusion Complex Using β CD and HPMC: Physicochemical Characterization, Molecular Docking, and Antimicrobial Testing. Processes 2020, 8, 1450. [Google Scholar] [CrossRef]
- Saokham, P.; Muankaew, C.; Jansook, P.; Loftsson, T. Solubility of Cyclodextrins and Drug/Cyclodextrin Complexes. Molecules 2018, 23, 1161. [Google Scholar] [CrossRef] [Green Version]
- Loftsson, T.; Hreinsdóttir, D.; Másson, M. Evaluation of Cyclodextrin Solubilization of Drugs. Int. J. Pharm. 2005, 302, 18–28. [Google Scholar] [CrossRef]
- Loftsson, T.; Hreinsdóttir, D.; Másson, M. The Complexation Efficiency. J. Incl. Phenom. Macrocycl. Chem. 2007, 57, 545–552. [Google Scholar] [CrossRef]
- Wenz, G.; Han, B.-H.; Müller, A. Cyclodextrin Rotaxanes and Polyrotaxanes. Chem. Rev. 2006, 106, 782–817. [Google Scholar] [CrossRef]
- Anconi, C.; Soares Nascimento Junior, C.; Almeida, W.; Santos, H. Theoretical Study of α-CD Based [3] Pseudorotaxanes: The Role Played by Threadlike Polymer on the Stability of Cyclodextrin Dimers. J. Braz. Chem. Soc. -JBCS 2008, 19, 1033–1040. [Google Scholar] [CrossRef]
- Christoforides, E.; Papaioannou, A.; Bethanis, K. Crystal Structure of the Inclusion Complex of Cholesterol in β-Cyclodextrin and Molecular Dynamics Studies. Beilstein J. Org. Chem. 2018, 14, 838–848. [Google Scholar] [CrossRef] [PubMed]
- Dos Santos, C.; Buera, M.P.; Mazzobre, M.F. Phase Solubility Studies and Stability of Cholesterol/Beta-Cyclodextrin Inclusion Complexes. J. Sci Food Agric. 2011, 91, 2551–2557. [Google Scholar] [CrossRef] [PubMed]
- Imam, S.S.; Alshehri, S.; Alzahrani, T.A.; Hussain, A.; Altamimi, M.A. Formulation and Evaluation of Supramolecular Food-Grade Piperine HP β CD and TPGS Complex: Dissolution, Physicochemical Characterization, Molecular Docking, In Vitro Antioxidant Activity, and Antimicrobial Assessment. Molecules 2020, 25, 4716. [Google Scholar] [CrossRef] [PubMed]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| PN/β-CD | PN/DM-β-CD | PN/TM-β-CD | |
|---|---|---|---|
| Crystal data | |||
| Chemical formula | 2(C42H70O35)C17H19NO3· 21.5(H2O) | C56H98O35·0.5(C17H19NO3) | 2(C63H112O35)C17H19NO3 |
| Mr | 2902.32 | 1474.00 | 3141.35 |
| Crystal system, space group | Triclinic, P1 | Monoclinic, P21 | Monoclinic, P21 |
| Temperature (K) | 100 | 120 | 100 |
| a, b, c (Å) | 15.435 (2), 15.452 (2), 15.502 (2) | 15.364 (4), 10.3253 (16), 25.006 (5) | 20.8000 (14), 14.7391 (10), 27.0968 (18) |
| β (°) | 104.761 (5), 100.770 (5), 104.207 (6) | 106.552 (16) | 96.214 (4) |
| V (Å3) | 3341.9 (6) | 3802.4 (13) | 8258.3 (10) |
| Z | 1 | 2 | 2 |
| Radiation type | Cu Ka | Cu Ka | Cu Ka |
| μ (mm−1) | 1.14 | 0.90 | 0.86 |
| Crystal size (mm3) | 0.5 × 0.4 × 0.2 | 0.8 × 0.12 × 0.1 | 0.22 × 0.16 × 0.08 |
| Data collection | |||
| Diffractometer | Bruker APEX-II | Bruker APEX-II | Bruker APEX-II |
| Absorption correction | Multi-scan TWINABS—Bruker AXS scaling for twinned crystals—Version 2012/1 | Multi-scan SADABS2016/2—Bruker AXS area detector scaling and absorption correction | Multi-scan SADABS2016/2—Bruker AXS area detector scaling and absorption correction |
| Tmin, Tmax | 0.607, 0.796 | 0.352, 0.752 | 0.582, 0.75 |
| No. of measured, independent and observed [I > 2σ(I)] reflections | 16,593 (domain 1) 17,055 (domain 2) 17,875 (composites) 21,234, 21,234, 20,381 | 58,942, 12,341, 10,665 | 88,314, 13,469, 11,219 |
| Rint | 0.05 | 0.072 | 0.089 |
| (sin θ/λ)max (Å−1) | 0.581 | 0.582 | 0.465 |
| Refinement | |||
| R[F2 > 2σ(F2)], wR(F2), S | 0.076, 0.203, 1.02 | 0.100, 0.271, 1.06 | 0.118, 0.316, 1.04 |
| No. of reflections | 21,234 | 12,341 | 13,469 |
| No. of parameters | 1688 | 1044 | 1224 |
| No. of restraints | 209 | 77 | 213 |
| H-atom treatment | H-atom parameters constrained | H-atom parameters constrained | H-atom parameters constrained |
| Δρmax, Δρmin (e Å−3) | 0.74, −0.48 | 0.48, −0.45 | 0.67, −0.55 |
| Complexes | Type | Linear Equation | R2 | Kc (M−1) | CE (%) |
|---|---|---|---|---|---|
| PN/β-CD | Bs | y = 0.0652x + 0.0378 (for linear portion) | 0.9939 | 1800 ± 300 | 7.0 ± 0.4 |
| PN/RM-β-CD | AL- | y = 0.1562x − 0.3348 | 0.9988 | 4900 ± 500 | 18.5 ± 0.3 |
| PN/HP-β-CD | AL- | y = 0.1148x − 0.3337 | 0.9968 | 3400 ± 300 | 13.0 ± 0.3 |
| PN1/ β-CD | PN2/ β-CD | PN/ DM-β-CD | PN/ TM-β-CD | PN_i.m.1/ HP-β-CD | PN_i.m.2/ HP-β-CD | |
|---|---|---|---|---|---|---|
| ΔEvdW | −51.70 ± 4.08 | −50.79 ± 4.27 | −51.16 ± 3.01 | −53.41 ± 3.60 | −27.15 ± 2.24 | −26.23 ± 2.60 |
| ΔEele | −10.41 ± 4.64 | −9.66 ± 4.18 | −11.09 ± 4.08 | −6.43 ± 2.58 | −5.79 ± 3.15 | −7.00 ± 3.16 |
| ΔEMM a | −62.11 ± 6.25 | −60.45 ± 6.45 | −62.25 ± 5.36 | −59.84 ± 4.84 | −32.94 ± 3.86 | −33.27 ± 3.90 |
| ΔGGB | 26.68 ± 3.91 | 25.31 ± 4.01 | 26.43 ± 3.84 | 27.16 ± 3.00 | 23.64 ± 3.68 | 25.43 ± 3.28 |
| ΔGnonpolar | −5.44 ± 0.30 | −5.33 ± 0.25 | −5.49 ± 0.20 | −5.37 ± 0.23 | −3.28 ± 0.25 | −3.29 ± 0.34 |
| ΔGsolvation b | 21.24 ± 3.84 | 19.99 ± 3.90 | 20.93 ± 3.83 | 21.78 ± 2.93 | 20.35 ± 3.59 | 22.16 ± 3.12 |
| ΔH c | −40.87 ± 4.60 | −40.47 ± 4.56 | −41.31 ± 2.94 | −38.06 ± 3.20 | −12.58 ± 2.08 | −11.11 ± 2.29 |
| T·ΔS d | −20.43 ± 3.76 | −19.93 ± 3.14 | −16.51 ± 4.16 | −17.61 ± 3.78 | −15.63 ± 1.48 | −16.31 ± 1.71 |
| ΔGbinding e | −20.43 ± 5.94 | −20.53 ± 5.53 | −24.80 ± 5.10 | −20.45 ± 4.95 | +3.04 ± 2.69 | +5.20 ± 2.86 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Christoforides, E.; Andreou, A.; Papaioannou, A.; Bethanis, K. Structural Studies of Piperine Inclusion Complexes in Native and Derivative β-Cyclodextrins. Biomolecules 2022, 12, 1762. https://doi.org/10.3390/biom12121762
Christoforides E, Andreou A, Papaioannou A, Bethanis K. Structural Studies of Piperine Inclusion Complexes in Native and Derivative β-Cyclodextrins. Biomolecules. 2022; 12(12):1762. https://doi.org/10.3390/biom12121762
Chicago/Turabian StyleChristoforides, Elias, Athena Andreou, Andreas Papaioannou, and Kostas Bethanis. 2022. "Structural Studies of Piperine Inclusion Complexes in Native and Derivative β-Cyclodextrins" Biomolecules 12, no. 12: 1762. https://doi.org/10.3390/biom12121762