

Modeling Calcium Cycling in the Heart: Progress, Pitfalls, and Challenges

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction

2. RyR Models

3. Single CRU and CRU Network Models


4. Modeling SR Ca Cycling in Single-Cell Action Potential Models
5. Modeling Mitochondrial Ca Cycling and Energetics in Action Potential Models
6. Modeling Ca-Dependent Signaling
7. Tissue-Scale Modeling for Spatiotemporal Ca and Voltage Dynamics
8. Challenges for Future Modeling
- (1)
- The first gap is to simulate sparks from a high spatial resolution CRU model to a non-spatial CRU model. As shown in the high spatial resolution models [27,28,32] (see also Figure 3B), Ca spark dynamics are spatiotemporal and depend on the spatial distribution of the RyRs in the dyadic space. Moreover, Maltsev et al. [64] showed that the occurrence of Ca sparks harnesses the Ising phase transition in a 2D array of RyRs. On the other hand, the majority of the single CRU and CRU network models ignore these spatiotemporal properties by using a RyR cluster without spatial placement of RyRs. How to correctly capture the properties of the phase transition and large fluctuations of the spatiotemporal system using a lower-resolution or non-spatial model needs to be investigated.
- (2)
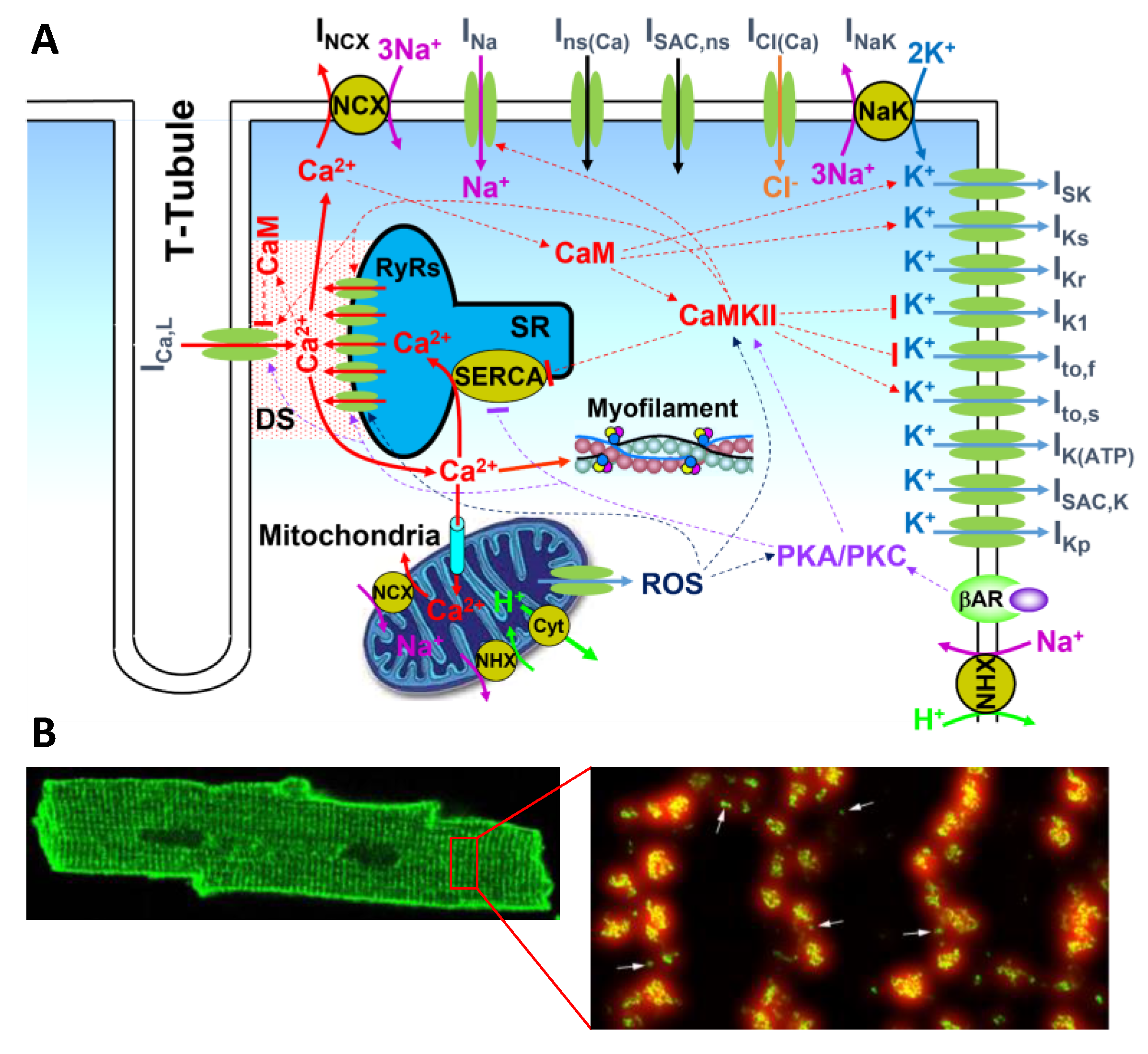
- The second gap is from CRUs (sparks) to a low-dimensional whole-cell model (Ca waves and whole-cell Ca transient). So far, using the 3D cell model for large tissue and whole-heart simulations is computationally nontrivial. Low-dimensional representations of the cell are preferred. As shown extensively by experiments and simulations (e.g., Figure 4), there is a hierarchy of Ca dynamics: quarks, sparks, spark clusters, mini-waves, persistent waves, and whole-call oscillations. How to develop a low-dimensional model to embrace these dynamics is a nontrivial challenge. As mentioned above, when Ca is low or normal, the differential equation describing the SR Ca release by Shiferaw et al. is a correct approach. At very high Ca where SR Ca release is synchronous, the global RyR model is appropriate. However, neither of the two modeling approaches can capture the Ca dynamics in the intermediate Ca range. As shown in Figure 4 (see Nivala et al. [57]), a second-order phase transition occurs for the transition from independent individual spark dynamics to the whole-cell oscillations, i.e., a critical phenomenon exists. How to model the dynamics at this phase transition using a low-dimensional representation is unknown since when criticality occurs, the dynamics is intrinsically high-dimensional. Another challenge is how to model complex 3D T-tubular structures in low-dimensional models. For example, T-tubules are disrupted in failing ventricular myocytes [108,109,110], which can cause nontrivial changes in both Ca cycling and action potential dynamics [68,72]. Correctly modeling these effects is essential for tissue-scale modeling of heart failure. Furthermore, when Ca cycling dynamics becomes spatiotemporal, their effects on Ca-dependent ionic currents and signaling, such as ICa,L and INCX as well as Ca-activated potassium currents (Figure 1A), and thus on the action potential dynamics may be nontrivial [125]. How to model these effects using a low-dimensional model is another issue needed to be concerned.
- (3)
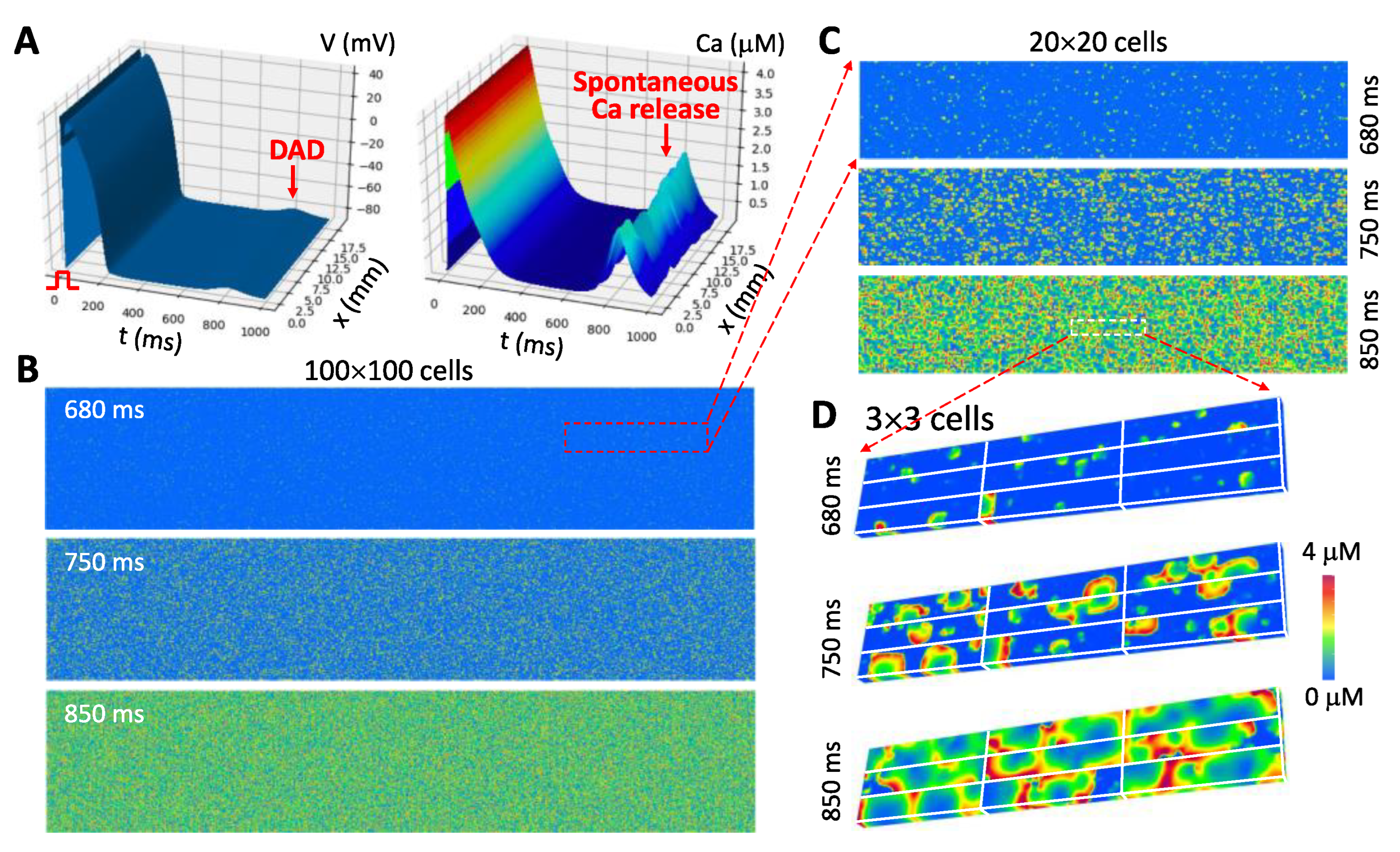
- The third gap is from a single cell to a syncytium (tissue and whole heart). For tissue or whole-heart simulations, cardiac tissue is treated as a syncytium with discretization in computer simulations being typically from Δx = Δy = Δz = 0.1 to 0.5 mm [3,153,154,155]. The dimension of a typical myocyte is 0.15 × 0.03 × 0.015 mm3, which indicates that one “computational cell” is equivalent to 15 to 1500 real cells. Under normal conditions in which conduction is fast and Ca is mainly determined by ICa,L (so that Ca is synchronized by ICa,L in different cells), this type of discretization is appropriate. However, under diseased conditions, such as ischemia and heart failure, cells are weakly coupled and the Na current is attenuated, and thus the action potential conduction in tissue is slow. For slow conduction, a discretization at the cell size is more appropriate [156,157]. Moreover, when spatiotemporal Ca dynamics, such as waves and alternans, occur in the cell, these dynamics may be dyssynchronous from cell to cell [158] (see Figure 5), and thus a resolution of the cell size may be required for investigating these dynamics. Large tissue or whole-heart simulations with a resolution at the cell size scale is computationally nontrivial.
- (4)
- (5)
- Although stochastic Markovian models are used to simulate single ion channel dynamics, they are still phenomenological models describing the transitions of an ion channel among different hypothetical states. Molecular dynamics simulations of single ion channels at the atomic scale are more accurate representations, which can provide parameter information for transition rates of Markovian models used in cell and tissue simulations, linking the molecular scale effects of gene mutations or drugs to cellular and tissue scale behaviors [162,163]. However, molecular dynamics simulation is computationally tedious [164], and it is still a major challenge to use it for the development and validation of Markovian models.
9. Conclusions
Funding
Institutional Review Board Statement
Conflicts of Interest
References
- Noble, D. Modeling the heart--from genes to cells to the whole organ. Science 2002, 295, 1678–1682. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Henriquez, C.S. A Brief History of Tissue Models For Cardiac Electrophysiology. IEEE Trans. Biomed. Eng. 2014, 61, 1457–1465. [Google Scholar] [CrossRef] [PubMed]
- Trayanova, N.A. Whole-heart modeling: Applications to cardiac electrophysiology and electromechanics. Circ. Res. 2011, 108, 113–128. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Colman, M.A.; Alvarez-Lacalle, E.; Echebarria, B.; Sato, D.; Sutanto, H.; Heijman, J. Multi-scale computational modeling of spatial calcium handling from nanodomain to whole-heart: Overview and perspectives. Front. Physiol. 2022, 13, 836622. [Google Scholar] [CrossRef]
- Qu, Z.; Hu, G.; Garfinkel, A.; Weiss, J.N. Nonlinear and stochastic dynamics in the heart. Phys. Rep. 2014, 543, 61–162. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hodgkin, A.L.; Huxley, A.F. A Quantitative Description of Membrane Current and Its Application to Conduction and Excitation in Nerve. J. Physiol. 1952, 117, 500–544. [Google Scholar] [CrossRef] [PubMed]
- Bers, D.M. Excitation-Contraction Coupling and Cardiac Contractile Force, 2nd ed.; Kluwer Academic Publishers: Dordrecht, The Netherlands, 2001. [Google Scholar]
- Soeller, C.; Crossman, D.; Gilbert, R.; Cannell, M.B. Analysis of ryanodine receptor clusters in rat and human cardiac myocytes. Proc. Natl. Acad. Sci. USA 2007, 104, 14958–14963. [Google Scholar] [CrossRef] [Green Version]
- Baddeley, D.; Jayasinghe, I.D.; Lam, L.; Rossberger, S.; Cannell, M.B.; Soeller, C. Optical single-channel resolution imaging of the ryanodine receptor distribution in rat cardiac myocytes. Proc. Natl. Acad. Sci. USA 2009, 106, 22275–22280. [Google Scholar] [CrossRef] [Green Version]
- Hiess, F.; Vallmitjana, A.; Wang, R.; Cheng, H.; ter Keurs, H.E.D.J.; Chen, J.; Hove-Madsen, L.; Benitez, R.; Chen, S.R.W. Distribution and Function of Cardiac Ryanodine Receptor Clusters in Live Ventricular Myocytes. J. Biol. Chem. 2015, 290, 20477–20487. [Google Scholar] [CrossRef] [Green Version]
- Camacho, P.; Lechleiter, J.D. Increased frequency of calcium waves in Xenopus laevis oocytes that express a calcium-ATPase. Science 1993, 260, 226–229. [Google Scholar] [CrossRef]
- Lipp, P.; Niggli, E. Microscopic spiral waves reveal positive feedback in subcellular calcium signaling. Biophys. J. 1993, 65, 2272–2276. [Google Scholar] [CrossRef] [Green Version]
- Skupin, A.; Kettenmann, H.; Winkler, U.; Wartenberg, M.; Sauer, H.; Tovey, S.C.; Taylor, C.W.; Falcke, M. How does intracellular Ca2+ oscillate: By chance or by the clock? Biophys. J. 2008, 94, 2404–2411. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bootman, M.D.; Berridge, M.J.; Lipp, P. Cooking with calcium: The recipes for composing global signals from elementary events. Cell 1997, 91, 367–373. [Google Scholar] [CrossRef] [Green Version]
- Marchant, J.S.; Parker, I. Role of elementary Ca2+ puffs in generating repetitive Ca2+ oscillations. EMBO J. 2001, 20, 65–76. [Google Scholar] [CrossRef] [Green Version]
- Cheng, H.; Lederer, M.R.; Lederer, W.J.; Cannell, M.B. Calcium sparks and [Ca2+]i waves in cardiac myocytes. Am. J. Physiol. 1996, 270, C148–C159. [Google Scholar] [CrossRef] [PubMed]
- Wier, W.G.; ter Keurs, H.E.; Marban, E.; Gao, W.D.; Balke, C.W. Ca2+ ‘sparks’ and waves in intact ventricular muscle resolved by confocal imaging. Circ. Res. 1997, 81, 462–469. [Google Scholar] [CrossRef]
- Lukyanenko, V.; Gyorke, S. Ca2+ sparks and Ca2+ waves in saponin-permeabilized rat ventricular myocytes. J. Physiol. 1999, 521, 575–585. [Google Scholar] [CrossRef]
- Thurley, K.; Tovey, S.C.; Moenke, G.; Prince, V.L.; Meena, A.; Thomas, A.P.; Skupin, A.; Taylor, C.W.; Falcke, M. Reliable encoding of stimulus intensities within random sequences of intracellular Ca2+ spikes. Sci. Signal 2014, 7, ra59. [Google Scholar] [CrossRef] [Green Version]
- Cheng, H.; Lederer, W.J. Calcium Sparks. Physiol. Rev. 2008, 88, 1491–1545. [Google Scholar] [CrossRef] [Green Version]
- Cheng, H.; Lederer, W.J.; Cannell, M.B. Calcium sparks: Elementary events underlying excitation-contraction coupling in heart muscle. Science 1993, 262, 740–744. [Google Scholar] [CrossRef]
- Qu, Z.; Weiss, J.N. Mechanisms of Ventricular Arrhythmias: From Molecular Fluctuations to Electrical Turbulence. Annu. Rev. Physiol. 2015, 77, 29–55. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiang, D.; Wang, R.; Xiao, B.; Kong, H.; Hunt, D.J.; Choi, P.; Zhang, L.; Chen, S.R. Enhanced store overload-induced Ca2+ release and channel sensitivity to luminal Ca2+ activation are common defects of RyR2 mutations linked to ventricular tachycardia and sudden death. Circ. Res. 2005, 97, 1173–1181. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Priori, S.G.; Chen, S.R.W. Inherited Dysfunction of Sarcoplasmic Reticulum Ca2+ Handling and Arrhythmogenesis. Circ. Res. 2011, 108, 871–883. [Google Scholar] [CrossRef] [Green Version]
- Laver, D.R. Regulation of the RyR channel gating by Ca2+ and Mg2+. Biophys. Rev. 2018, 10, 1087–1095. [Google Scholar] [CrossRef] [PubMed]
- Sobie, E.A.; Dilly, K.W.; dos Santos Cruz, J.; Lederer, W.J.; Jafri, M.S. Termination of cardiac Ca2+ sparks: An investigative mathematical model of calcium-induced calcium release. Biophys. J. 2002, 83, 59–78. [Google Scholar] [CrossRef] [Green Version]
- Cannell, M.B.; Kong, C.H.; Imtiaz, M.S.; Laver, D.R. Control of sarcoplasmic reticulum Ca2+ release by stochastic RyR gating within a 3D model of the cardiac dyad and importance of induction decay for CICR termination. Biophys. J. 2013, 104, 2149–2159. [Google Scholar] [CrossRef] [Green Version]
- Walker, M.A.; Williams, G.S.B.; Kohl, T.; Lehnart, S.E.; Jafri, M.S.; Greenstein, J.L.; Lederer, W.J.; Winslow, R.L. Superresolution Modeling of Calcium Release in the Heart. Biophys. J. 2014, 107, 3018–3029. [Google Scholar] [CrossRef] [Green Version]
- Stern, M.D.; Song, L.S.; Cheng, H.; Sham, J.S.; Yang, H.T.; Boheler, K.R.; Rios, E. Local control models of cardiac excitation-contraction coupling. A possible role for allosteric interactions between ryanodine receptors. J. Gen. Physiol. 1999, 113, 469–489. [Google Scholar] [CrossRef] [Green Version]
- Stern, M.D.; Cheng, H. Putting out the fire: What terminates calcium-induced calcium release in cardiac muscle? Cell Calcium 2004, 35, 591–601. [Google Scholar] [CrossRef]
- Stern, M.D.; Rios, E.; Maltsev, V.A. Life and death of a cardiac calcium spark. J. Gen. Physiol. 2013, 142, 257–274. [Google Scholar] [CrossRef]
- Laver, D.R.; Kong, C.H.T.; Imtiaz, M.S.; Cannell, M.B. Termination of calcium-induced calcium release by induction decay: An emergent property of stochastic channel gating and molecular scale architecture. J. Mol. Cell. Cardiol. 2013, 54, 98–100. [Google Scholar] [CrossRef] [PubMed]
- Winslow, R.L.; Greenstein, J.L. Extinguishing the sparks. Biophys. J. 2013, 104, 2115–2117. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Song, Z.; Karma, A.; Weiss, J.N.; Qu, Z. Long-Lasting Sparks: Multi-Metastability and Release Competition in the Calcium Release Unit Network. PLoS Comput. Biol. 2016, 12, e1004671. [Google Scholar] [CrossRef] [PubMed]
- Shannon, T.R.; Guo, T.; Bers, D.M. Ca2+ Scraps: Local Depletions of Free [Ca2+] in Cardiac Sarcoplasmic Reticulum During Contractions Leave Substantial Ca2+ Reserve. Circ. Res. 2003, 93, 40–45. [Google Scholar] [CrossRef] [Green Version]
- Zima, A.V.; Picht, E.; Bers, D.M.; Blatter, L.A. Termination of cardiac Ca2+ sparks: Role of intra-SR [Ca2+], release flux, and intra-SR Ca2+ diffusion. Circ. Res. 2008, 103, e105–e115. [Google Scholar] [CrossRef] [Green Version]
- Picht, E.; Zima, A.V.; Shannon, T.R.; Duncan, A.M.; Blatter, L.A.; Bers, D.M. Dynamic calcium movement inside cardiac sarcoplasmic reticulum during release. Circ. Res. 2011, 108, 847–856. [Google Scholar] [CrossRef] [Green Version]
- Stern, M.D.; Pizarro, G.; Rios, E. Local control model of excitation-contraction coupling in skeletal muscle. J. Gen. Physiol. 1997, 110, 415–440. [Google Scholar] [CrossRef] [Green Version]
- Shannon, T.R.; Wang, F.; Puglisi, J.; Weber, C.; Bers, D.M. A mathematical treatment of integrated Ca dynamics within the ventricular myocyte. Biophys. J. 2004, 87, 3351–3371. [Google Scholar] [CrossRef] [Green Version]
- Restrepo, J.G.; Weiss, J.N.; Karma, A. Calsequestrin-mediated mechanism for cellular calcium transient alternans. Biophys. J. 2008, 95, 3767–3789. [Google Scholar] [CrossRef] [Green Version]
- Keizer, J.; Levine, L. Ryanodine receptor adaptation and Ca2+-induced Ca2+ release-dependent Ca2+ oscillations. Biophys. J. 1996, 71, 3477–3487. [Google Scholar] [CrossRef]
- Greenstein, J.L.; Winslow, R.L. An integrative model of the cardiac ventricular myocyte incorporating local control of Ca(2+) release. Biophys. J. 2002, 83, 2918–2945. [Google Scholar] [CrossRef] [Green Version]
- Greene, D.A.; Shiferaw, Y. Mechanistic link between CaM-RyR2 interactions and the genesis of cardiac arrhythmia. Biophys. J. 2021, 120, 1469–1482. [Google Scholar] [CrossRef]
- Wang, K.; Tu, Y.; Rappel, W.J.; Levine, H. Excitation-contraction coupling gain and cooperativity of the cardiac ryanodine receptor: A modeling approach. Biophys. J. 2005, 89, 3017–3025. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Groff, J.R.; Smith, G.D. Ryanodine Receptor Allosteric Coupling and the Dynamics of Calcium Sparks. Biophys. J. 2008, 95, 135–154. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, W.; Wasserstrom, J.A.; Shiferaw, Y. Role of coupled gating between cardiac ryanodine receptors in the genesis of triggered arrhythmias. Am. J. Physiol. Heart Circ. Physiol. 2009, 297, H171–H180. [Google Scholar] [CrossRef] [Green Version]
- Dixon, R.E.; Navedo, M.F.; Binder, M.D.; Santana, L.F. Mechanisms and physiological implications of cooperative gating of clustered ion channels. Physiol. Rev. 2022, 102, 1159–1210. [Google Scholar] [CrossRef]
- Xiao, R.P.; Valdivia, H.H.; Bogdanov, K.; Valdivia, C.; Lakatta, E.G.; Cheng, H. The immunophilin FK506-binding protein modulates Ca2+ release channel closure in rat heart. J. Physiol. 1997, 500, 343–354. [Google Scholar] [CrossRef] [Green Version]
- Zima, A.V.; Picht, E.; Bers, D.M.; Blatter, L.A. Partial inhibition of sarcoplasmic reticulum ca release evokes long-lasting ca release events in ventricular myocytes: Role of luminal ca in termination of ca release. Biophys. J. 2008, 94, 1867–1879. [Google Scholar] [CrossRef] [Green Version]
- Picht, E.; DeSantiago, J.; Blatter, L.A.; Bers, D.M. Cardiac alternans do not rely on diastolic sarcoplasmic reticulum calcium content fluctuations. Circ. Res. 2006, 99, 740–748. [Google Scholar] [CrossRef] [Green Version]
- Shkryl, V.M.; Maxwell, J.T.; Domeier, T.L.; Blatter, L.A. Refractoriness of sarcoplasmic reticulum Ca release determines Ca alternans in atrial myocytes. Am. J. Physiol. Heart Circ. Physiol. 2012, 302, H2310–H2320. [Google Scholar] [CrossRef]
- Rovetti, R.; Cui, X.; Garfinkel, A.; Weiss, J.N.; Qu, Z. Spark-induced sparks as a mechanism of intracellular calcium alternans in cardiac myocytes. Circ. Res. 2010, 106, 1582–1591. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qu, Z.; Liu, M.B.; Nivala, M. A unified theory of calcium alternans in ventricular myocytes. Sci. Rep. 2016, 6, 35625. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhong, X.; Sun, B.; Vallmitjana, A.; Mi, T.; Guo, W.; Ni, M.; Wang, R.; Guo, A.; Duff, H.J.; Gillis, A.M.; et al. Suppression of ryanodine receptor function prolongs Ca2+ release refractoriness and promotes cardiac alternans in intact hearts. Biochem. J. 2016, 473, 3951–3964. [Google Scholar] [CrossRef] [Green Version]
- Sun, B.; Wei, J.; Zhong, X.; Guo, W.; Yao, J.; Wang, R.; Vallmitjana, A.; Benitez, R.; Hove-Madsen, L.; Chen, S.R.W. The cardiac ryanodine receptor, but not sarcoplasmic reticulum Ca2+-ATPase, is a major determinant of Ca2+ alternans in intact mouse hearts. J. Biol. Chem. 2018, 293, 13650–13661. [Google Scholar] [CrossRef] [Green Version]
- Boyden, P.A.; Pu, J.; Pinto, J.; Keurs, H.E. Ca2+ transients and Ca2+ waves in purkinje cells: Role in action potential initiation. Circ. Res. 2000, 86, 448–455. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nivala, M.; Ko, C.Y.; Nivala, M.; Weiss, J.N.; Qu, Z. Criticality in intracellular calcium signaling in cardiac myocytes. Biophys. J. 2012, 102, 2433–2442. [Google Scholar] [CrossRef] [Green Version]
- Nivala, M.; Ko, C.Y.; Nivala, M.; Weiss, J.N.; Qu, Z. The Emergence of Subcellular Pacemaker Sites for Calcium Waves and Oscillations. J. Physiol. 2013, 591, 5305–5320. [Google Scholar] [CrossRef] [PubMed]
- Song, Z.; Qu, Z.; Karma, A. Stochastic initiation and termination of calcium-mediated triggered activity in cardiac myocytes. Proc. Natl. Acad. Sci. USA 2017, 114, E270–E279. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, N.; Denegri, M.; Dun, W.; Boncompagni, S.; Lodola, F.; Protasi, F.; Napolitano, C.; Boyden, P.A.; Priori, S.G. Abnormal Propagation of Calcium Waves and Ultrastructural Remodeling in Recessive Catecholaminergic Polymorphic Ventricular Tachycardia. Circ. Res. 2013, 113, 142–152. [Google Scholar] [CrossRef] [Green Version]
- Stern, M.D. Theory of excitation-contraction coupling in cardiac muscle. Biophys. J. 1992, 63, 497–517. [Google Scholar] [CrossRef]
- Smith, G.D.; Keizer, J.E.; Stern, M.D.; Lederer, W.J.; Cheng, H. A simple numerical model of calcium spark formation and detection in cardiac myocytes. Biophys. J. 1998, 75, 15–32. [Google Scholar] [CrossRef] [Green Version]
- Hake, J.; Edwards, A.G.; Yu, Z.; Kekenes-Huskey, P.M.; Michailova, A.P.; McCammon, J.A.; Holst, M.J.; Hoshijima, M.; McCulloch, A.D. Modeling cardiac calcium sparks in a three-dimensional reconstruction of a calcium release unit. J. Physiol. 2012, 590, 4403–4422. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maltsev, A.V.; Maltsev, V.A.; Stern, M.D. Clusters of calcium release channels harness the Ising phase transition to confine their elementary intracellular signals. Proc. Natl. Acad. Sci. USA 2017, 114, 7525–7530. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gaur, N.; Rudy, Y. Multiscale modeling of calcium cycling in cardiac ventricular myocyte: Macroscopic consequences of microscopic dyadic function. Biophys. J. 2011, 100, 2904–2912. [Google Scholar] [CrossRef] [Green Version]
- Nivala, M.; de Lange, E.; Rovetti, R.; Qu, Z. Computational modeling and numerical methods for spatiotemporal calcium cycling in ventricular myocytes. Front. Physiol. 2012, 3, 114. [Google Scholar] [CrossRef] [Green Version]
- Colman, M.A.; Pinali, C.; Trafford, A.W.; Zhang, H.; Kitmitto, A. A computational model of spatio-temporal cardiac intracellular calcium handling with realistic structure and spatial flux distribution from sarcoplasmic reticulum and t-tubule reconstructions. PLoS Comput. Biol. 2017, 13, e1005714. [Google Scholar] [CrossRef] [Green Version]
- Song, Z.; Liu, M.B.; Qu, Z. Transverse tubular network structures in the genesis of intracellular calcium alternans and triggered activity in cardiac cells. J. Mol. Cell. Cardiol. 2018, 114, 288–299. [Google Scholar] [CrossRef]
- Hoang-Trong, T.M.; Ullah, A.; Lederer, W.J.; Jafri, M.S. A Stochastic Spatiotemporal Model of Rat Ventricular Myocyte Calcium Dynamics Demonstrated Necessary Features for Calcium Wave Propagation. Membranes 2021, 11, 989. [Google Scholar] [CrossRef]
- Stanley, H.E. Introduction to Phase Transitions and Critical Phenomena; Oxford University Press: London, UK, 1971. [Google Scholar]
- Stanley, H.E. Scaling, universality, and renormalization: Three pillars of modern critical phenomena. Rev. Mod. Phys. 1999, 71, S358–S366. [Google Scholar] [CrossRef] [Green Version]
- Nivala, M.; Song, Z.; Weiss, J.N.; Qu, Z. T-tubule disruption promotes calcium alternans in failing ventricular myocytes: Mechanistic insights from computational modeling. J. Mol. Cell. Cardiol. 2015, 79, 32–41. [Google Scholar] [CrossRef]
- Noble, D. A modification of the Hodgkin-Huxley equations applicable to Purkinje fibre action and pace-maker potentials. J. Physiol. 1962, 160, 317–352. [Google Scholar] [CrossRef] [PubMed]
- Beeler, G.W.; Reuter, H. Reconstruction of the action potential of ventricular myocardial fibres. J. Physiol. 1977, 268, 177–210. [Google Scholar] [CrossRef] [PubMed]
- Luo, C.H.; Rudy, Y. A model of the ventricular cardiac action potential: Depolarization, repolarization, and their interaction. Circ. Res. 1991, 68, 1501–1526. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- DiFrancesco, D.; Noble, D. A model of cardiac electrical activity incorporating ionic pumps and concentration changes. Philos. Trans. R. Soc. Lond. (Biol.) 1985, 307, 779–790. [Google Scholar]
- Fabiato, A. Calcium-induced release of calcium from the cardiac sarcoplasmic reticulum. Am. J. Physiol. 1983, 245, C1–C14. [Google Scholar] [CrossRef]
- Hilgemann, D.W.; Noble, D. Excitation-contraction coupling and extracellular calcium transients in rabbit atrium: Reconstruction of basic cellular mechanisms. Proc. R. Soc. Lond. B Biol. Sci. 1987, 230, 163–205. [Google Scholar]
- Jafri, M.S.; Rice, J.J.; Winslow, R.L. Cardiac Ca2+ dynamics: The roles of ryanodine receptor adaptation and sarcoplasmic reticulum load. Biophys. J. 1998, 74, 1149–1168. [Google Scholar] [CrossRef] [Green Version]
- Maltsev, V.A.; Lakatta, E.G. Synergism of coupled subsarcolemmal Ca2+ clocks and sarcolemmal voltage clocks confers robust and flexible pacemaker function in a novel pacemaker cell model. Am. J. Physiol. Heart Circ. Physiol. 2009, 296, H594–H615. [Google Scholar] [CrossRef] [Green Version]
- Shannon, T.R.; Wang, F.; Bers, D.M. Regulation of Cardiac Sarcoplasmic Reticulum Ca Release by Luminal [Ca] and Altered Gating Assessed with a Mathematical Model. Biophys. J. 2005, 89, 4096–4110. [Google Scholar] [CrossRef] [Green Version]
- Luo, C.H.; Rudy, Y. A dynamical model of the cardiac ventricular action potential: I. simulations of ionic currents and concentration changes. Circ. Res. 1994, 74, 1071–1096. [Google Scholar] [CrossRef] [Green Version]
- Faber, G.M.; Rudy, Y. Action potential and contractility changes in [Na(+)](i) overloaded cardiac myocytes: A simulation study. Biophys. J. 2000, 78, 2392–2404. [Google Scholar] [CrossRef] [Green Version]
- Chudin, E.; Goldhaber, J.; Garfinkel, A.; Weiss, J.; Kogan, B. Intracellular Ca2+ dynamics and the stability of ventricular tachycardia. Biophys. J. 1999, 77, 2930–2941. [Google Scholar] [CrossRef]
- Fox, J.J.; McHarg, J.L.; Gilmour, R.F. Ionic mechanism of electrical alternans. Am. J. Physiol. Heart Circ. Physiol. 2002, 282, H516–H530. [Google Scholar] [CrossRef]
- ten Tusscher, K.H.; Noble, D.; Noble, P.J.; Panfilov, A.V. A model for human ventricular tissue. Am. J. Physiol. Heart Circ. Physiol. 2004, 286, H1573–H1589. [Google Scholar] [CrossRef] [PubMed]
- Hund, T.J.; Rudy, Y. Rate dependence and regulation of action potential and calcium transient in a canine cardiac ventricular cell model. Circulation 2004, 110, 3168–3174. [Google Scholar] [CrossRef] [Green Version]
- Stewart, P.; Aslanidi, O.V.; Noble, D.; Noble, P.J.; Boyett, M.R.; Zhang, H. Mathematical models of the electrical action potential of Purkinje fibre cells. Philos. Trans. A Math. Phys. Eng. Sci. 2009, 367, 2225–2255. [Google Scholar] [CrossRef]
- Livshitz, L.M.; Rudy, Y. Regulation of Ca2+ and electrical alternans in cardiac myocytes: Role of CAMKII and repolarizing currents. Am. J. Physiol. Heart Circ. Physiol. 2007, 292, H2854–H2866. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- O’Hara, T.; Virag, L.; Varro, A.; Rudy, Y. Simulation of the undiseased human cardiac ventricular action potential: Model formulation and experimental validation. PLoS Comput. Biol. 2011, 7, e1002061. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shiferaw, Y.; Watanabe, M.A.; Garfinkel, A.; Weiss, J.N.; Karma, A. Model of intracellular calcium cycling in ventricular myocytes. Biophys. J. 2003, 85, 3666–3686. [Google Scholar] [CrossRef] [Green Version]
- Mahajan, A.; Shiferaw, Y.; Sato, D.; Baher, A.; Olcese, R.; Xie, L.-H.; Yang, M.-J.; Chen, P.-S.; Restrepo, J.G.; Karma, A.; et al. A rabbit ventricular action potential model replicating cardiac dynamics at rapid heart rates. Biophys. J. 2008, 94, 392–410. [Google Scholar] [CrossRef] [Green Version]
- Bassani, J.W.; Yuan, W.; Bers, D.M. Fractional SR Ca release is regulated by trigger Ca and SR Ca content in cardiac myocytes. Am. J. Physiol. 1995, 268, C1313–C1319. [Google Scholar] [CrossRef] [PubMed]
- Shannon, T.R.; Ginsburg, K.S.; Bers, D.M. Potentiation of fractional sarcoplasmic reticulum calcium release by total and free intra-sarcoplasmic reticulum calcium concentration. Biophys. J. 2000, 78, 334–343. [Google Scholar] [CrossRef]
- Hinch, R.; Greenstein, J.L.; Tanskanen, A.J.; Xu, L.; Winslow, R.L. A simplified local control model of calcium-induced calcium release in cardiac ventricular myocytes. Biophys. J. 2004, 87, 3723–3736. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Williams, G.S.; Huertas, M.A.; Sobie, E.A.; Jafri, M.S.; Smith, G.D. Moment closure for local control models of calcium-induced calcium release in cardiac myocytes. Biophys. J. 2008, 95, 1689–1703. [Google Scholar] [CrossRef] [Green Version]
- Williams, G.S.B.; Huertas, M.A.; Sobie, E.A.; Jafri, M.S.; Smith, G.D. A Probability Density Approach to Modeling Local Control of Calcium-Induced Calcium Release in Cardiac Myocytes. Biophys. J. 2007, 92, 2311–2328. [Google Scholar] [CrossRef] [Green Version]
- Huertas, M.A.; Smith, G.D.; Gyorke, S. Ca2+ alternans in a cardiac myocyte model that uses moment equations to represent heterogeneous junctional SR Ca2+. Biophys. J. 2010, 99, 377–387. [Google Scholar] [CrossRef] [Green Version]
- Shiferaw, Y.; Aistrup, G.L.; Wasserstrom, J.A. Synchronization of triggered waves in atrial tissue. Biophys. J. 2018, 115, 1130–1141. [Google Scholar] [CrossRef] [Green Version]
- Marchena, M.; Echebarria, B. Computational Model of Calcium Signaling in Cardiac Atrial Cells at the Submicron Scale. Front. Physiol. 2018, 9, 1760. [Google Scholar] [CrossRef] [Green Version]
- Marchena, M.; Echebarria, B. Influence of the tubular network on the characteristics of calcium transients in cardiac myocytes. PLoS ONE 2020, 15, e0231056. [Google Scholar] [CrossRef]
- Sutanto, H.; van Sloun, B.; Schönleitner, P.; van Zandvoort, M.A.M.J.; Antoons, G.; Heijman, J. The Subcellular Distribution of Ryanodine Receptors and L-Type Ca2+ Channels Modulates Ca2+-Transient Properties and Spontaneous Ca2+-Release Events in Atrial Cardiomyocytes. Front. Physiol. 2018, 9, 1108. [Google Scholar] [CrossRef]
- Vagos, M.R.; Arevalo, H.; Heijman, J.; Schotten, U.; Sundnes, J. A Novel Computational Model of the Rabbit Atrial Cardiomyocyte with Spatial Calcium Dynamics. Front. Physiol. 2020, 11, 556156. [Google Scholar] [CrossRef] [PubMed]
- Maltsev, A.V.; Yaniv, Y.; Stern, M.D.; Lakatta, E.G.; Maltsev, V.A. RyR-NCX-SERCA Local Cross-Talk Ensures Pacemaker Cell Function at Rest and During the Fight-or-Flight Reflex. Circ. Res. 2013, 113, e94–e100. [Google Scholar] [CrossRef] [PubMed]
- Maltsev, A.V.; Maltsev, V.A.; Stern, M.D. Stabilization of diastolic calcium signal via calcium pump regulation of complex local calcium releases and transient decay in a computational model of cardiac pacemaker cell with individual release channels. PLoS Comput. Biol. 2017, 13, e1005675. [Google Scholar] [CrossRef] [Green Version]
- Dibb, K.M.; Clarke, J.D.; Horn, M.A.; Richards, M.A.; Graham, H.K.; Eisner, D.A.; Trafford, A.W. Characterization of an Extensive Transverse Tubular Network in Sheep Atrial Myocytes and its Depletion in Heart Failure. Circ. Heart Fail. 2009, 2, 482–489. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brandenburg, S.; Kohl, T.; Williams, G.S.B.; Gusev, K.; Wagner, E.; Rog-Zielinska, E.A.; Hebisch, E.; Dura, M.; Didié, M.; Gotthardt, M.; et al. Axial tubule junctions control rapid calcium signaling in atria. J. Clin. Investig. 2016, 126, 3999–4015. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lyon, A.R.; MacLeod, K.T.; Zhang, Y.; Garcia, E.; Kanda, G.K.; Lab, M.J.; Korchev, Y.E.; Harding, S.E.; Gorelik, J. Loss of T-tubules and other changes to surface topography in ventricular myocytes from failing human and rat heart. Proc. Natl. Acad. Sci. USA 2009, 106, 6854–6859. [Google Scholar] [CrossRef] [Green Version]
- Song, L.S.; Sobie, E.A.; McCulle, S.; Lederer, W.J.; Balke, C.W.; Cheng, H. Orphaned ryanodine receptors in the failing heart. Proc. Natl. Acad. Sci. USA 2006, 103, 4305–4310. [Google Scholar] [CrossRef] [Green Version]
- Seidel, T.; Navankasattusas, S.; Ahmad, A.A.; Diakos, N.A.; Xu, W.D.; Tristani-Firouzi, M.; Bonios, M.; Taleb, I.; Li, D.Y.; Selzman, C.H.; et al. Sheet-Like Remodeling of the Transverse Tubular System in Human Heart Failure Impairs Excitation-Contraction Coupling and Functional Recovery by Mechanical Unloading. Circulation 2017, 135, 1632–1645. [Google Scholar] [CrossRef] [Green Version]
- Chen, W.; Aistrup, G.; Wasserstrom, J.A.; Shiferaw, Y. A mathematical model of spontaneous calcium release in cardiac myocytes. Am. J. Physiol. Heart Circ. Physiol. 2011, 300, H1794–H1805. [Google Scholar] [CrossRef]
- Gaeta, S.A.; Bub, G.; Abbott, G.W.; Christini, D.J. Dynamical mechanism for subcellular alternans in cardiac myocytes. Circ. Res. 2009, 105, 335–342. [Google Scholar] [CrossRef] [Green Version]
- Sato, D.; Bers, D.M.; Shiferaw, Y. Formation of spatially discordant alternans due to fluctuations and diffusion of calcium. PLoS ONE 2014, 8, e85365. [Google Scholar] [CrossRef] [Green Version]
- Li, Q.; O’Neill, S.C.; Tao, T.; Li, Y.; Eisner, D.; Zhang, H. Mechanisms by which cytoplasmic calcium wave propagation and alternans are generated in cardiac atrial myocytes lacking T-tubules-insights from a simulation study. Biophys. J. 2012, 102, 1471–1482. [Google Scholar] [CrossRef] [PubMed]
- Tao, T.; O’Neill, S.C.; Diaz, M.E.; Li, Y.T.; Eisner, D.A.; Zhang, H. Alternans of cardiac calcium cycling in a cluster of ryanodine receptors: A simulation study. Am. J. Physiol. Heart Circ. Physiol. 2008, 295, H598–H609. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lou, Q.; Belevych, A.E.; Radwanski, P.B.; Liu, B.; Kalyanasundaram, A.; Knollmann, B.C.; Fedorov, V.V.; Gyorke, S. Alternating membrane potential/calcium interplay underlies repetitive focal activity in a genetic model of calcium-dependent atrial arrhythmias. J. Physiol. 2015, 593, 1443–1458. [Google Scholar] [CrossRef]
- Belevych, A.E.; Terentyev, D.; Terentyeva, R.; Ho, H.T.; Gyorke, I.; Bonilla, I.M.; Carnes, C.A.; Billman, G.E.; Gyorke, S. Shortened Ca2+ signaling refractoriness underlies cellular arrhythmogenesis in a postinfarction model of sudden cardiac death. Circ. Res. 2012, 110, 569–577. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ko, C.Y.; Liu, M.B.; Song, Z.; Qu, Z.; Weiss, J.N. Multiscale Determinants of Delayed Afterdepolarization Amplitude in Cardiac Tissue. Biophys. J. 2017, 112, 1949–1961. [Google Scholar] [CrossRef] [Green Version]
- Qu, Z.; Xie, L.-H.; Olcese, R.; Karagueuzian, H.S.; Chen, P.-S.; Garfinkel, A.; Weiss, J.N. Early afterdepolarizations in cardiac myocytes: Beyond reduced repolarization reserve. Cardiovasc. Res. 2013, 99, 6–15. [Google Scholar] [CrossRef] [Green Version]
- Priori, S.G.; Corr, P.B. Mechanisms underlying early and delayed afterdepolarizations induced by catecholamines. Am. J. Physiol. 1990, 258, H1796–H1805. [Google Scholar] [CrossRef]
- Volders, P.G.; Vos, M.A.; Szabo, B.; Sipido, K.R.; de Groot, S.H.; Gorgels, A.P.; Wellens, H.J.; Lazzara, R. Progress in the understanding of cardiac early afterdepolarizations and torsades de pointes: Time to revise current concepts. Cardiovasc. Res. 2000, 46, 376–392. [Google Scholar] [CrossRef] [Green Version]
- Choi, B.R.; Burton, F.; Salama, G. Cytosolic Ca2+ triggers early afterdepolarizations and Torsade de Pointes in rabbit hearts with type 2 long QT syndrome. J. Physiol. 2002, 543, 615–631. [Google Scholar] [CrossRef]
- Zhao, Z.; Wen, H.; Fefelova, N.; Allen, C.; Baba, A.; Matsuda, T.; Xie, L.H. Revisiting the ionic mechanisms of early afterdepolarizations in cardiomyocytes: Predominant by Ca waves or Ca currents? Am. J. Physiol. Heart Circ. Physiol. 2012, 302, H1636–H1644. [Google Scholar] [CrossRef] [PubMed]
- Song, Z.; Ko, C.Y.; Nivala, M.; Weiss, J.N.; Qu, Z. Calcium-Voltage Coupling in the Genesis of Early and Delayed Afterdepolarizations in Cardiac Myocytes. Biophys. J. 2015, 108, 1908–1921. [Google Scholar] [CrossRef]
- Zhong, M.; Rees, C.M.; Terentyev, D.; Choi, B.-R.; Koren, G.; Karma, A. NCX-Mediated Subcellular Ca2+ Dynamics Underlying Early Afterdepolarizations in LQT2 Cardiomyocytes. Biophys. J. 2018, 115, 1019–1032. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wilson, D.; Ermentrout, B.; Němec, J.; Salama, G. A model of cardiac ryanodine receptor gating predicts experimental Ca2+-dynamics and Ca2+-triggered arrhythmia in the long QT syndrome. Chaos Interdiscip. J. Nonlinear Sci. 2017, 27, 093940. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, X.; Song, Z.; Qu, Z. Determinants of early afterdepolarization properties in ventricular myocyte models. PLoS Comput. Biol. 2018, 14, e1006382. [Google Scholar] [CrossRef] [Green Version]
- Brown, D.A.; O’Rourke, B. Cardiac mitochondria and arrhythmias. Cardiovasc. Res. 2010, 88, 241–249. [Google Scholar] [CrossRef] [Green Version]
- Ruiz-Meana, M.; Fernandez-Sanz, C.; Garcia-Dorado, D. The SR–mitochondria interaction: A new player in cardiac pathophysiology. Cardiovasc. Res. 2010, 88, 30–39. [Google Scholar] [CrossRef] [Green Version]
- Cortassa, S.; Aon, M.A.; Marban, E.; Winslow, R.L.; O’Rourke, B. An integrated model of cardiac mitochondrial energy metabolism and calcium dynamics. Biophys. J. 2003, 84, 2734–2755. [Google Scholar] [CrossRef] [Green Version]
- Cortassa, S.; Aon, M.A.; O’Rourke, B.; Jacques, R.; Tseng, H.J.; Marban, E.; Winslow, R.L. A computational model integrating electrophysiology, contraction, and mitochondrial bioenergetics in the ventricular myocyte. Biophys. J. 2006, 91, 1564–1589. [Google Scholar] [CrossRef] [Green Version]
- Zhou, L.; Cortassa, S.; Wei, A.-C.; Aon, M.A.; Winslow, R.L.; O’Rourke, B. Modeling Cardiac Action Potential Shortening Driven by Oxidative Stress-Induced Mitochondrial Oscillations in Guinea Pig Cardiomyocytes. Biophys. J. 2009, 97, 1843–1852. [Google Scholar] [CrossRef] [Green Version]
- Matsuoka, S.; Sarai, N.; Jo, H.; Noma, A. Simulation of ATP metabolism in cardiac excitation-contraction coupling. Prog. Biophys. Mol. Biol. 2004, 85, 279–299. [Google Scholar] [CrossRef] [PubMed]
- Takeuchi, A.; Kim, B.; Matsuoka, S. The mitochondrial Na+-Ca2+ exchanger, NCLX, regulates automaticity of HL-1 cardiomyocytes. Sci. Rep. 2013, 3, 2766. [Google Scholar] [CrossRef]
- Aon, M.A.; Cortassa, S.; O’Rourke, B. Percolation and criticality in a mitochondrial network. Proc. Natl. Acad. Sci. USA 2004, 101, 4447–4452. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nivala, M.; Korge, P.; Nivala, M.; Weiss, J.N.; Qu, Z. Linking flickering to waves and whole-cell oscillations in a mitochondrial network model. Biophys. J. 2011, 101, 2102–2111. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pandey, V.; Xie, L.H.; Qu, Z.; Song, Z. Mitochondrial depolarization promotes calcium alternans: Mechanistic insights from a ventricular myocyte model. PLoS Comput. Biol. 2021, 17, e1008624. [Google Scholar] [CrossRef] [PubMed]
- Song, Z.; Xie, L.-H.; Weiss, J.N.; Qu, Z. A spatiotemporal ventricular myocyte model incorporating mitochondrial calcium cycling. Biophys. J. 2019, 117, 2349–2360. [Google Scholar] [CrossRef]
- Xie, A.; Song, Z.; Liu, H.; Zhou, A.; Shi, G.; Wang, Q.; Gu, L.; Liu, M.; Xie, L.H.; Qu, Z.; et al. Mitochondrial Ca2+ Influx Contributes to Arrhythmic Risk in Nonischemic Cardiomyopathy. J. Am. Heart Assoc. 2018, 7, e007805. [Google Scholar] [CrossRef] [Green Version]
- Pandey, V.; Xie, L.-H.; Qu, Z.; Song, Z. Mitochondrial Contributions in the Genesis of Delayed Afterdepolarizations in Ventricular Myocytes. Front. Physiol. 2021, 12, 1631. [Google Scholar] [CrossRef]
- Bers, D.M. Calcium cycling and signaling in cardiac myocytes. Annu. Rev. Physiol. 2008, 70, 23–49. [Google Scholar] [CrossRef] [Green Version]
- Saucerman, J.J.; Brunton, L.L.; Michailova, A.P.; McCulloch, A.D. Modeling beta-adrenergic control of cardiac myocyte contractility in silico. J. Biol. Chem. 2003, 278, 47997–48003. [Google Scholar] [CrossRef] [Green Version]
- Xie, Y.; Grandi, E.; Puglisi, J.L.; Sato, D.; Bers, D.M. beta-adrenergic stimulation activates early afterdepolarizations transiently via kinetic mismatch of PKA targets. J. Mol. Cell. Cardiol. 2013, 58, 153–161. [Google Scholar] [CrossRef] [Green Version]
- Hashambhoy, Y.L.; Greenstein, J.L.; Winslow, R.L. Role of CaMKII in RyR leak, EC coupling and action potential duration: A computational model. J. Mol. Cell. Cardiol. 2010, 49, 617–624. [Google Scholar] [CrossRef] [PubMed]
- Hashambhoy, Y.L.; Winslow, R.L.; Greenstein, J.L. CaMKII-induced shift in modal gating explains L-type Ca(2+) current facilitation: A modeling study. Biophys. J. 2009, 96, 1770–1785. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Foteinou, P.T.; Greenstein, J.L.; Winslow, R.L. Mechanistic Investigation of the Arrhythmogenic Role of Oxidized CaMKII in the Heart. Biophys. J. 2015, 109, 838–849. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, M.B.; de Lange, E.; Garfinkel, A.; Weiss, J.N.; Qu, Z. Delayed afterdepolarizations generate both triggers and a vulnerable substrate promoting reentry in cardiac tissue. Heart Rhythm 2015, 12, 2115–2124. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Campos, F.O.; Shiferaw, Y.; Prassl, A.J.; Boyle, P.M.; Vigmond, E.J.; Plank, G. Stochastic spontaneous calcium release events trigger premature ventricular complexes by overcoming electrotonic load. Cardiovasc. Res. 2015, 107, 175–183. [Google Scholar] [CrossRef] [Green Version]
- Colman, M.A. Arrhythmia mechanisms and spontaneous calcium release: Bi-directional coupling between re-entrant and focal excitation. PLoS Comput. Biol. 2019, 15, e1007260. [Google Scholar] [CrossRef] [Green Version]
- Colman, M.A.; Holmes, M.; Whittaker, D.G.; Jayasinghe, I.; Benson, A.P. Multi-scale approaches for the simulation of cardiac electrophysiology: I—Sub-cellular and stochastic calcium dynamics from cell to organ. Methods 2020, 185, 49–59. [Google Scholar] [CrossRef] [PubMed]
- Greene, D.A.; Kaboudian, A.; Wasserstrom, J.A.; Fenton, F.H.; Shiferaw, Y. Voltage-mediated mechanism for calcium wave synchronization and arrhythmogenesis in atrial tissue. Biophys. J. 2022, 121, 383–395. [Google Scholar] [CrossRef] [PubMed]
- Walker, M.A.; Gurev, V.; Rice, J.J.; Greenstein, J.L.; Winslow, R.L. Estimating the probabilities of rare arrhythmic events in multiscale computational models of cardiac cells and tissue. PLoS Comput. Biol. 2017, 13, e1005783. [Google Scholar] [CrossRef] [Green Version]
- Xie, F.; Qu, Z.; Yang, J.; Baher, A.; Weiss, J.N.; Garfinkel, A. A simulation study of the effects of cardiac anatomy in ventricular fibrillation. J. Clin. Investig. 2004, 113, 686–693. [Google Scholar] [PubMed] [Green Version]
- Ten Tusscher, K.H.; Hren, R.; Panfilov, A.V. Organization of ventricular fibrillation in the human heart. Circ. Res. 2007, 100, e87–e101. [Google Scholar] [CrossRef] [PubMed]
- Liu, M.B.; Vandersickel, N.; Panfilov, A.V.; Qu, Z. R-from-T as a common mechanism of arrhythmia initiation in long QT syndromes. Circ. Arrhythm. Electrophysiol. 2019, 12, e007571. [Google Scholar] [CrossRef] [PubMed]
- Keener, J.P. The effects of discrete gap junction coupling on propagation in myocardium. J. Theor. Biol. 1991, 148, 49–82. [Google Scholar] [CrossRef]
- Qu, Z.; Karagueuzian, H.S.; Garfinkel, A.; Weiss, J.N. Effects of Na+ channel and cell coupling abnormalities on vulnerability to reentry: A simulation study. Am. J. Physiol. Heart Circ. Physiol. 2004, 286, H1310–H1321. [Google Scholar] [CrossRef] [Green Version]
- Huang, C.; Song, Z.; Qu, Z. Synchronization of spatially discordant voltage and calcium alternans in cardiac tissue. Phys. Rev. E 2022, 106, 024406. [Google Scholar] [CrossRef]
- Greenstein, J.L.; Winslow, R.L. Integrative systems models of cardiac excitation-contraction coupling. Circ. Res. 2011, 108, 70–84. [Google Scholar] [CrossRef] [Green Version]
- Rice, J.J.; Jafri, M.S.; Winslow, R.L. Modeling short-term interval-force relations in cardiac muscle. Am. J. Physiol. Heart Circ. Physiol. 2000, 278, H913–H931. [Google Scholar] [CrossRef]
- Rice, J.J.; Winslow, R.L.; Hunter, W.C. Comparison of putative cooperative mechanisms in cardiac muscle: Length dependence and dynamic responses. Am. J. Physiol. 1999, 276, H1734–H1754. [Google Scholar] [CrossRef]
- Silva, J.R.; Pan, H.; Wu, D.; Nekouzadeh, A.; Decker, K.F.; Cui, J.; Baker, N.A.; Sept, D.; Rudy, Y. A multiscale model linking ion-channel molecular dynamics and electrostatics to the cardiac action potential. Proc. Natl. Acad. Sci. USA 2009, 106, 11102–11106. [Google Scholar] [CrossRef] [Green Version]
- Yang, P.C.; DeMarco, K.R.; Aghasafari, P.; Jeng, M.T.; Dawson, J.R.D.; Bekker, S.; Noskov, S.Y.; Yarov-Yarovoy, V.; Vorobyov, I.; Clancy, C.E. A Computational Pipeline to Predict Cardiotoxicity: From the Atom to the Rhythm. Circ. Res. 2020, 126, 947–964. [Google Scholar] [CrossRef] [PubMed]
- Jensen, M.Ø.; Borhani, D.W.; Lindorff-Larsen, K.; Maragakis, P.; Jogini, V.; Eastwood, M.P.; Dror, R.O.; Shaw, D.E. Principles of conduction and hydrophobic gating in K+ channels. Proc. Natl. Acad. Sci. USA 2010, 107, 5833–5838. [Google Scholar] [CrossRef] [PubMed]
- Qu, Z.; Garfinkel, A.; Weiss, J.N.; Nivala, M. Multi-scale modeling in biology: How to bridge the gaps between scales? Prog. Biophys. Mol. Biol. 2011, 107, 21–31. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fox, R.F. Stochastic versions of the Hodgkin-Huxley equations. Biophys. J. 1997, 72, 2068–2074. [Google Scholar] [CrossRef] [Green Version]
- Keener, J.P. Invariant manifold reductions for Markovian ion channel dynamics. J. Math. Biol. 2009, 58, 447–457. [Google Scholar] [CrossRef] [Green Version]
- Landaw, J.; Qu, Z. Bifurcations Caused by Feedback between Voltage and Intracellular Ion Concentrations in Ventricular Myocytes. Phys. Rev. Lett. 2019, 123, 218101. [Google Scholar] [CrossRef]
- Qu, Z.; Shiferaw, Y.; Weiss, J.N. Nonlinear dynamics of cardiac excitation-contraction coupling: An iterated map study. Phys. Rev. E 2007, 75, 011927. [Google Scholar] [CrossRef]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Qu, Z.; Yan, D.; Song, Z. Modeling Calcium Cycling in the Heart: Progress, Pitfalls, and Challenges. Biomolecules 2022, 12, 1686. https://doi.org/10.3390/biom12111686
Qu Z, Yan D, Song Z. Modeling Calcium Cycling in the Heart: Progress, Pitfalls, and Challenges. Biomolecules. 2022; 12(11):1686. https://doi.org/10.3390/biom12111686
Chicago/Turabian StyleQu, Zhilin, Dasen Yan, and Zhen Song. 2022. "Modeling Calcium Cycling in the Heart: Progress, Pitfalls, and Challenges" Biomolecules 12, no. 11: 1686. https://doi.org/10.3390/biom12111686