
The Role of Ca2+ Sparks in Force Frequency Relationships in Guinea Pig Ventricular Myocytes


Abstract
:1. Introduction
2. Materials and Methods
2.1. Computational Model Development
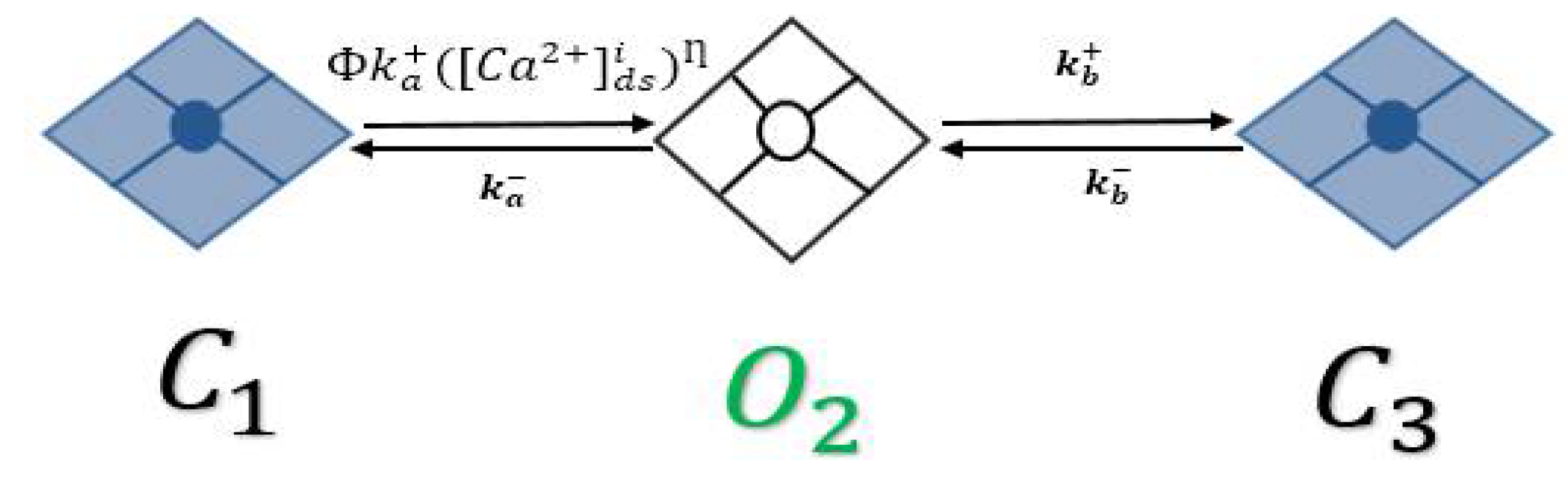
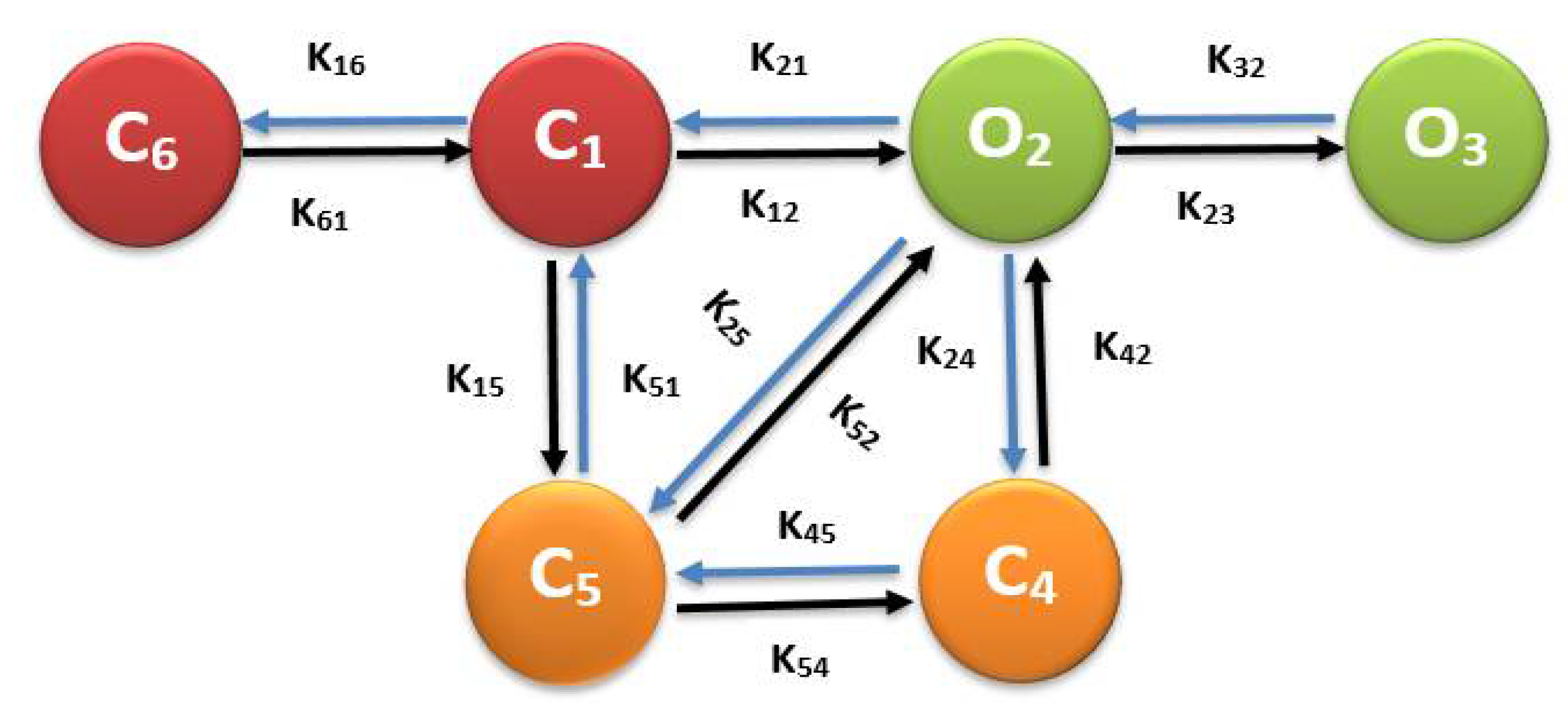
2.2. RyR Model
2.3. L-Type Ca2+ Channel Model
2.4. Simulation Frameworks
2.5. Numerical Methods
3. Results
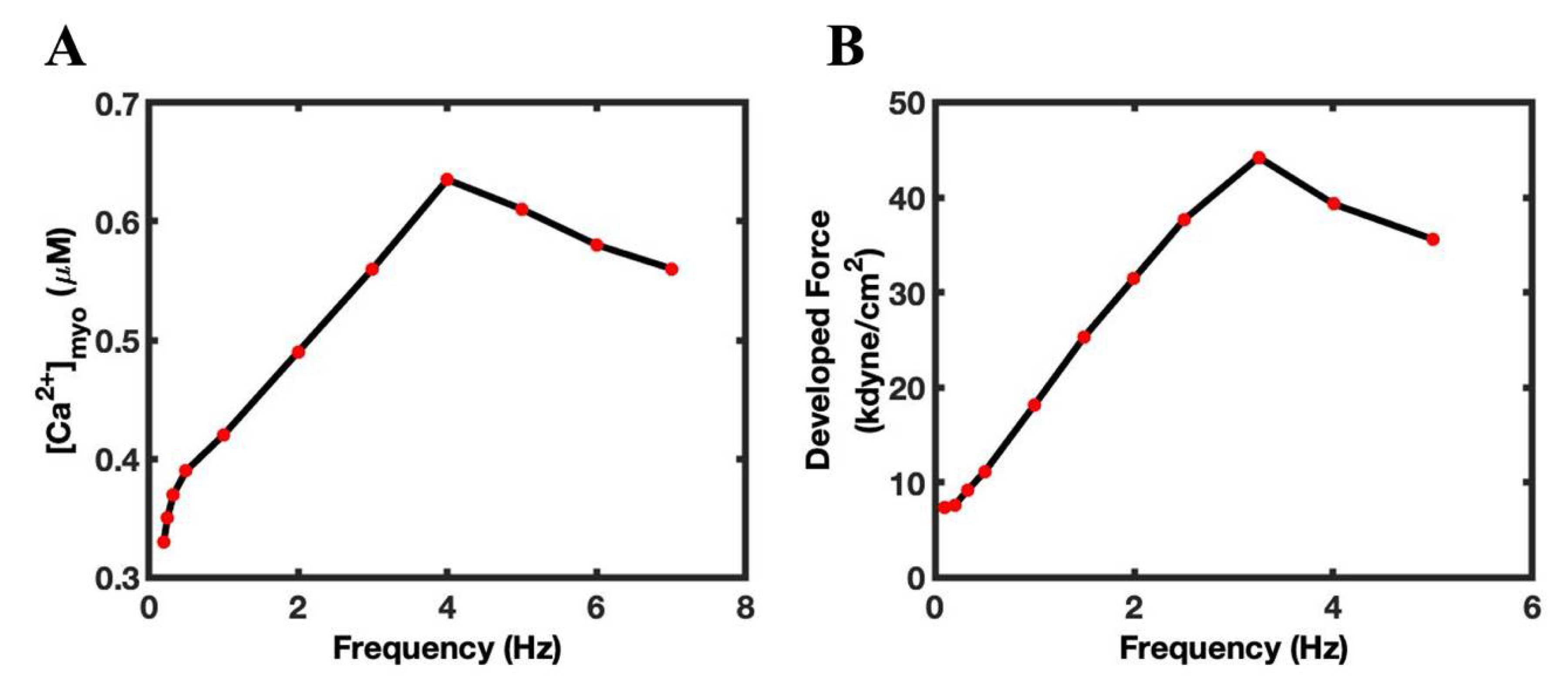
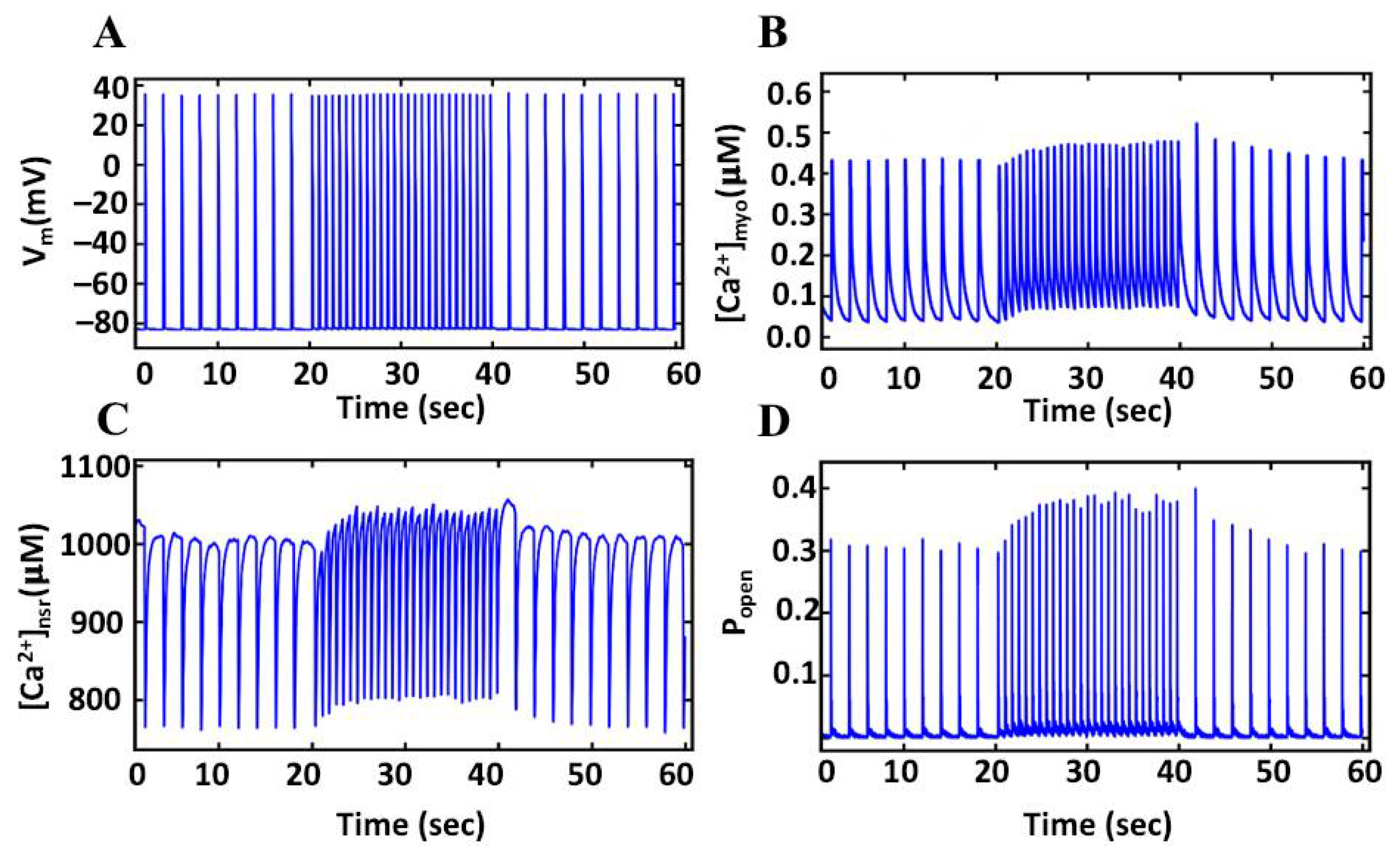
3.1. Ca2+ Transient FFR Curves
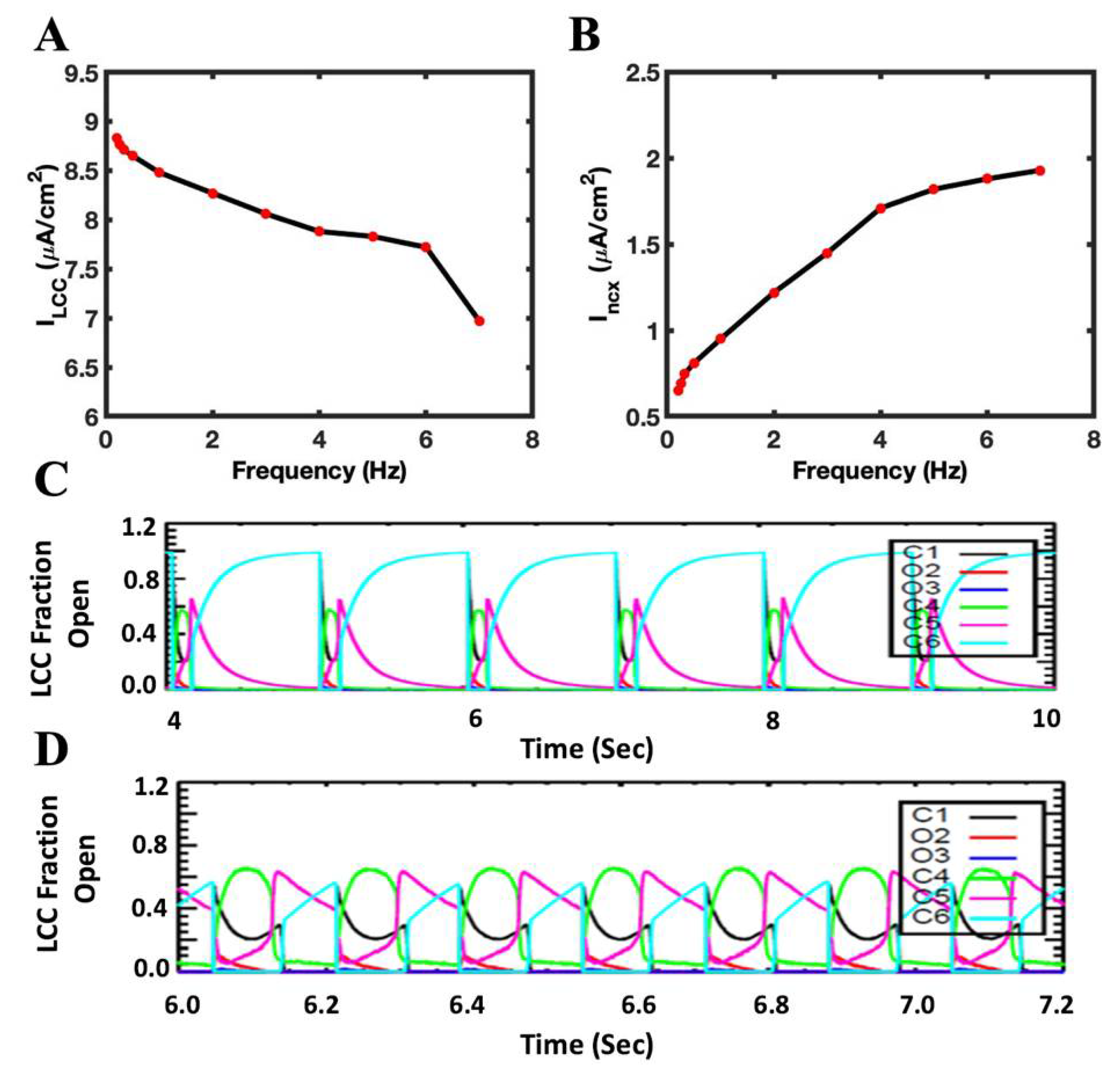
3.2. L-Type Current Decreases and Incx Current Increases with the Rapid Pacing
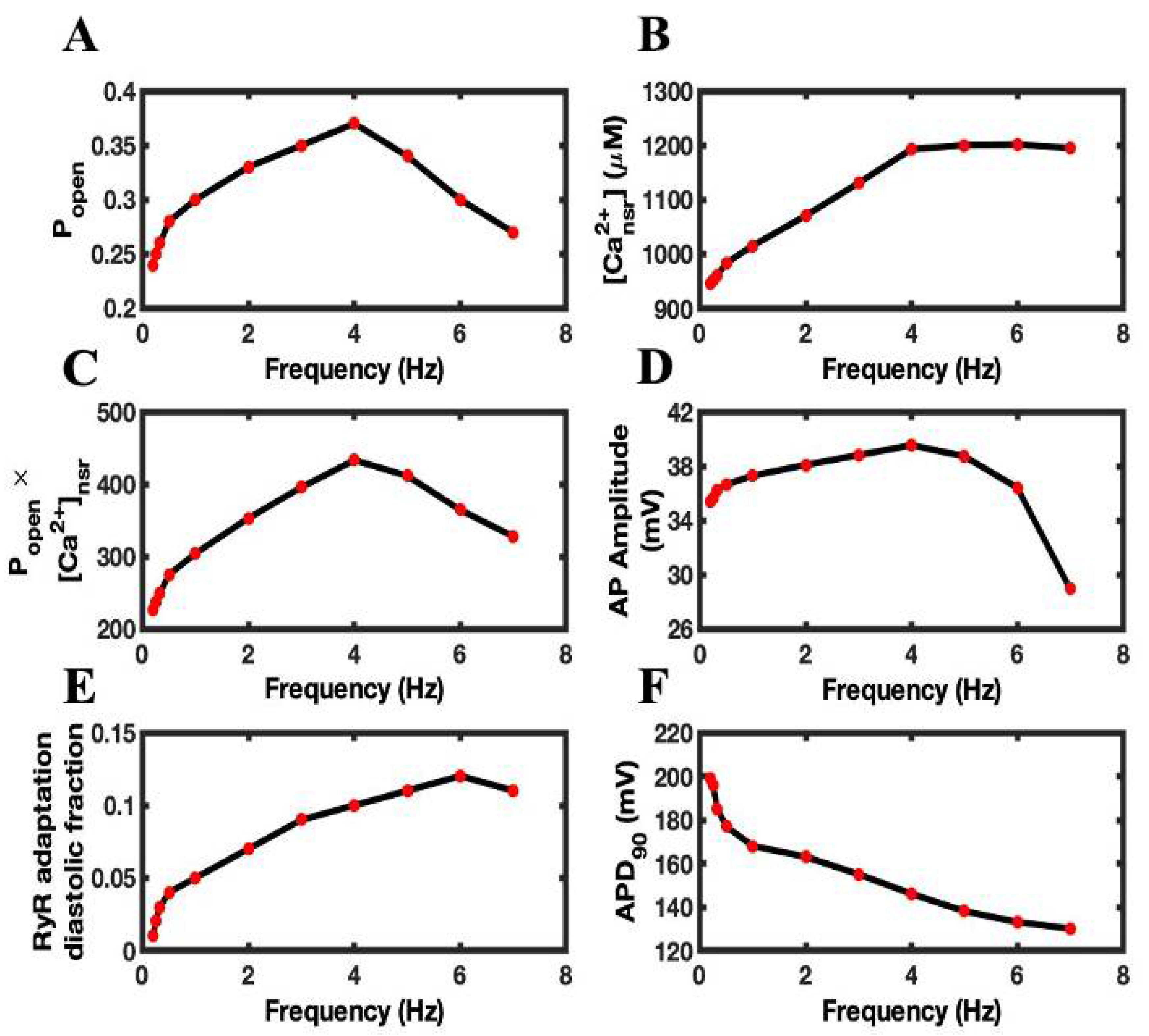
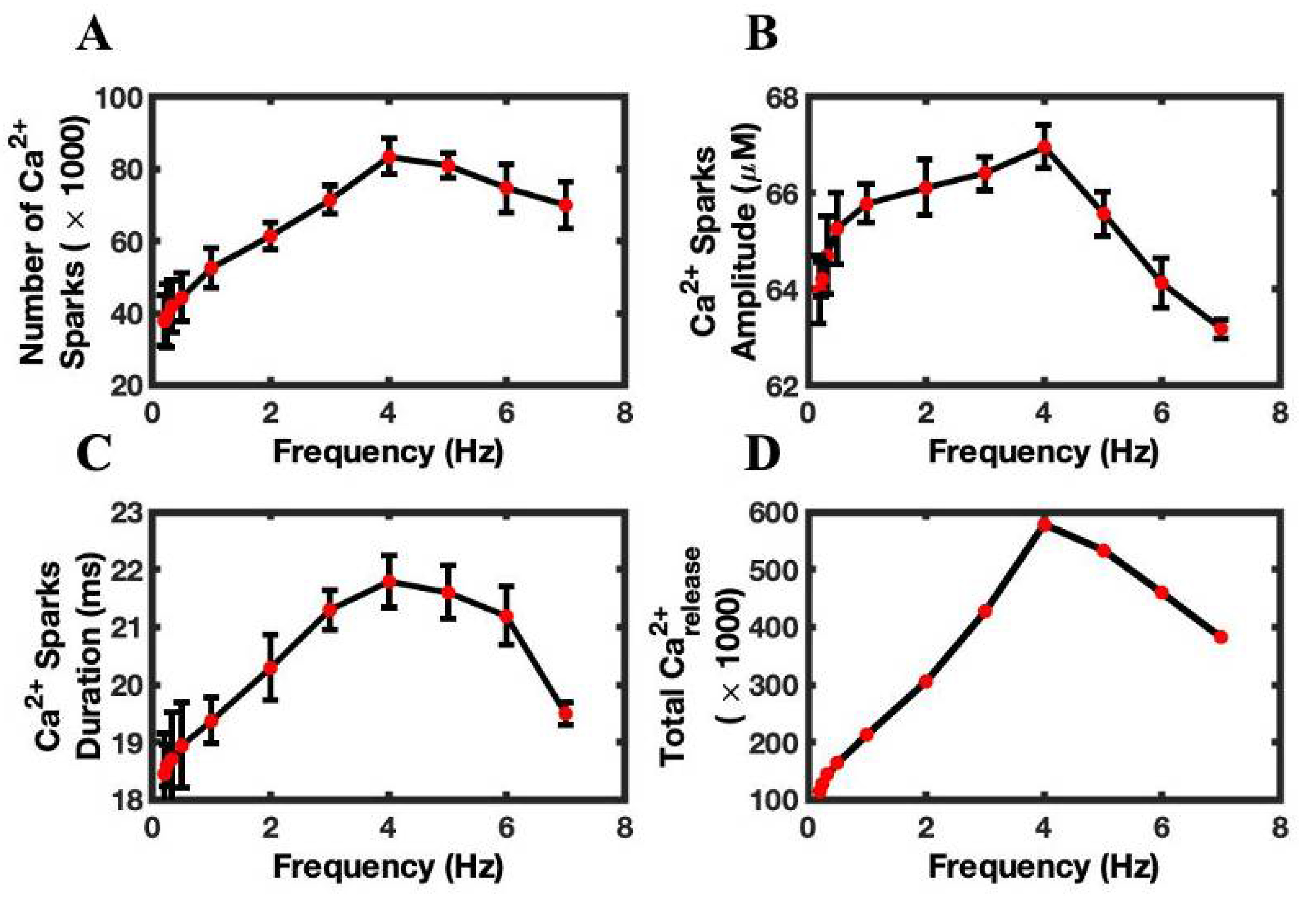
3.3. Calcium Sparks Are the Subcellular Mechanisms of FFR
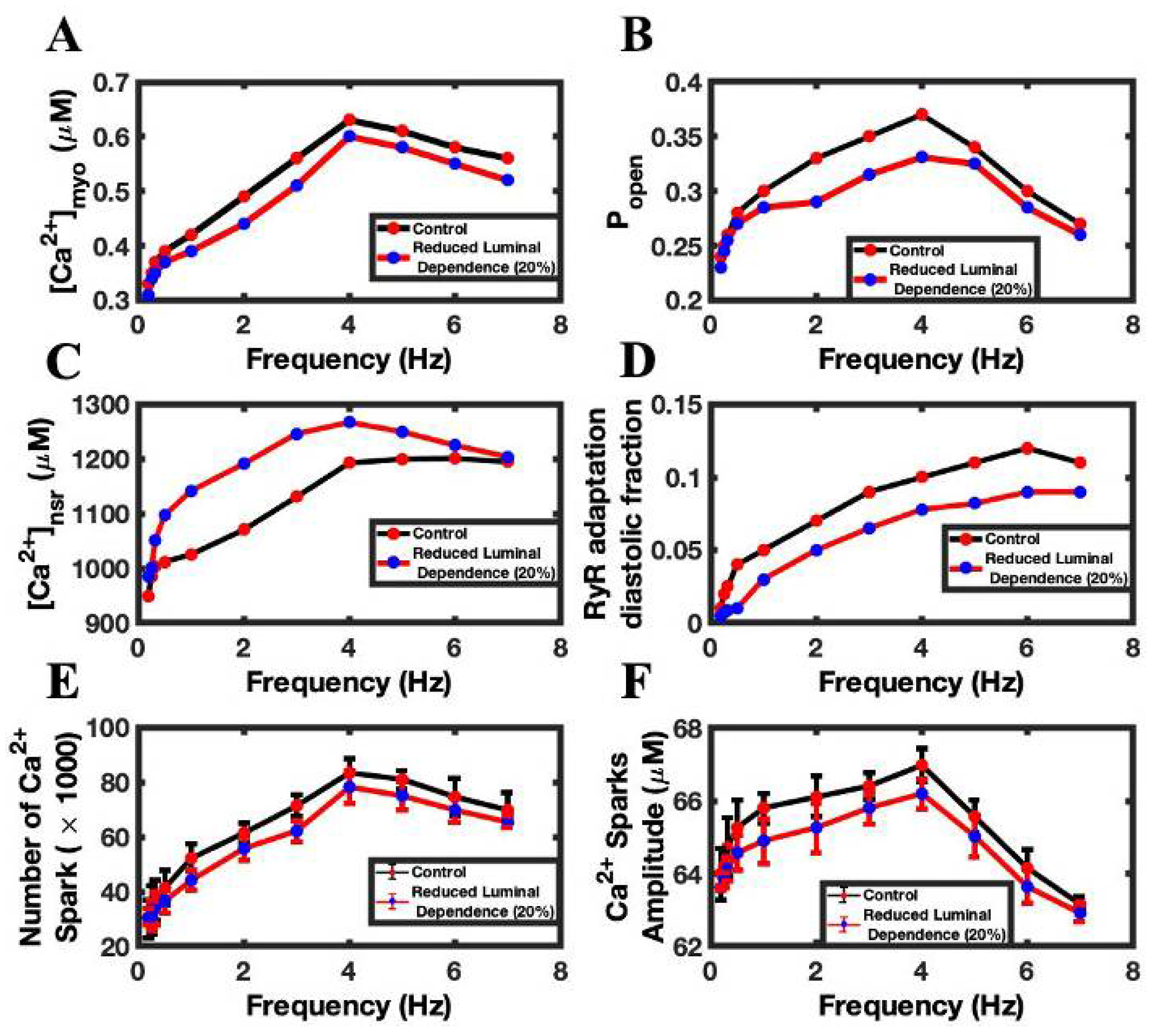
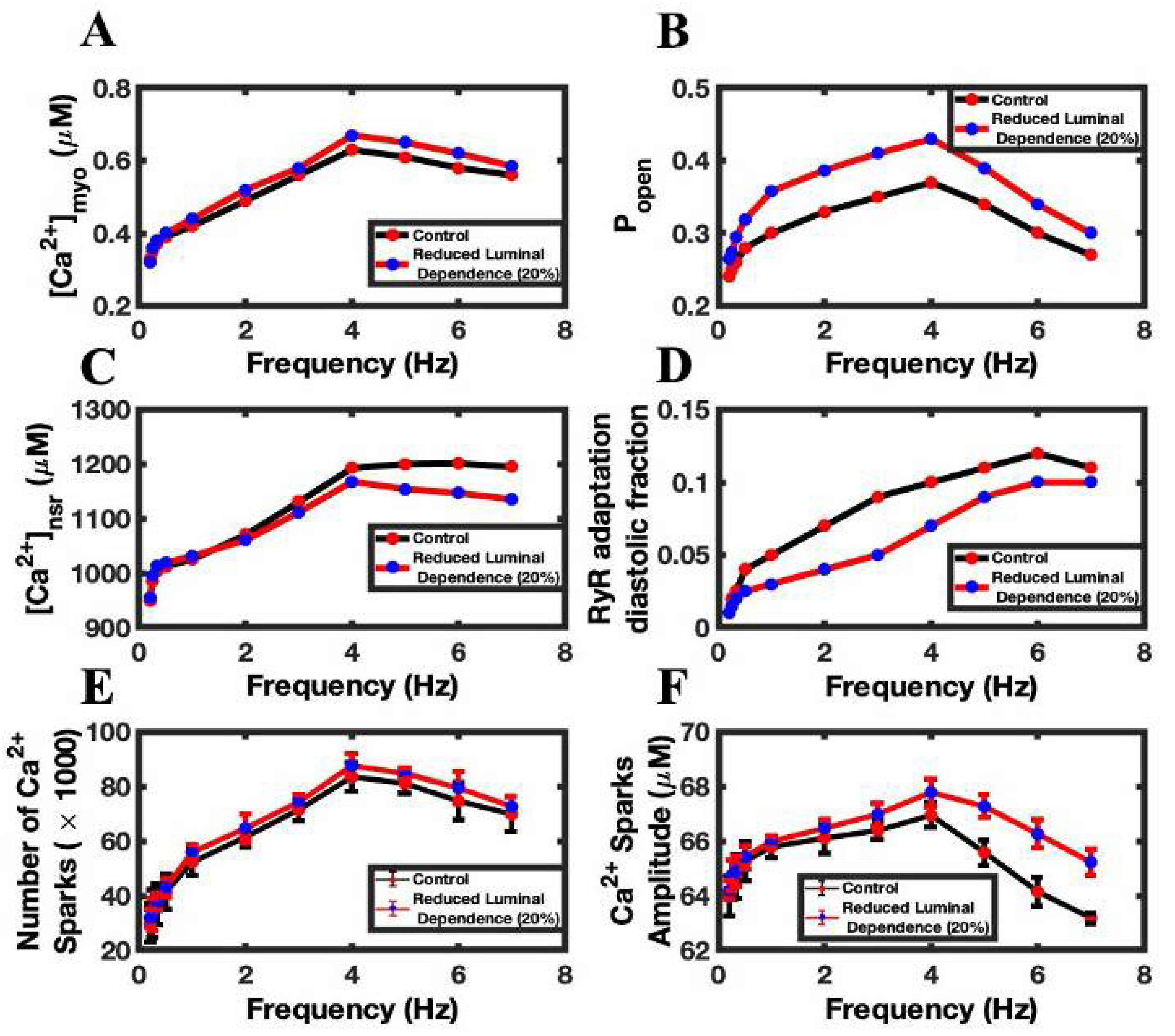
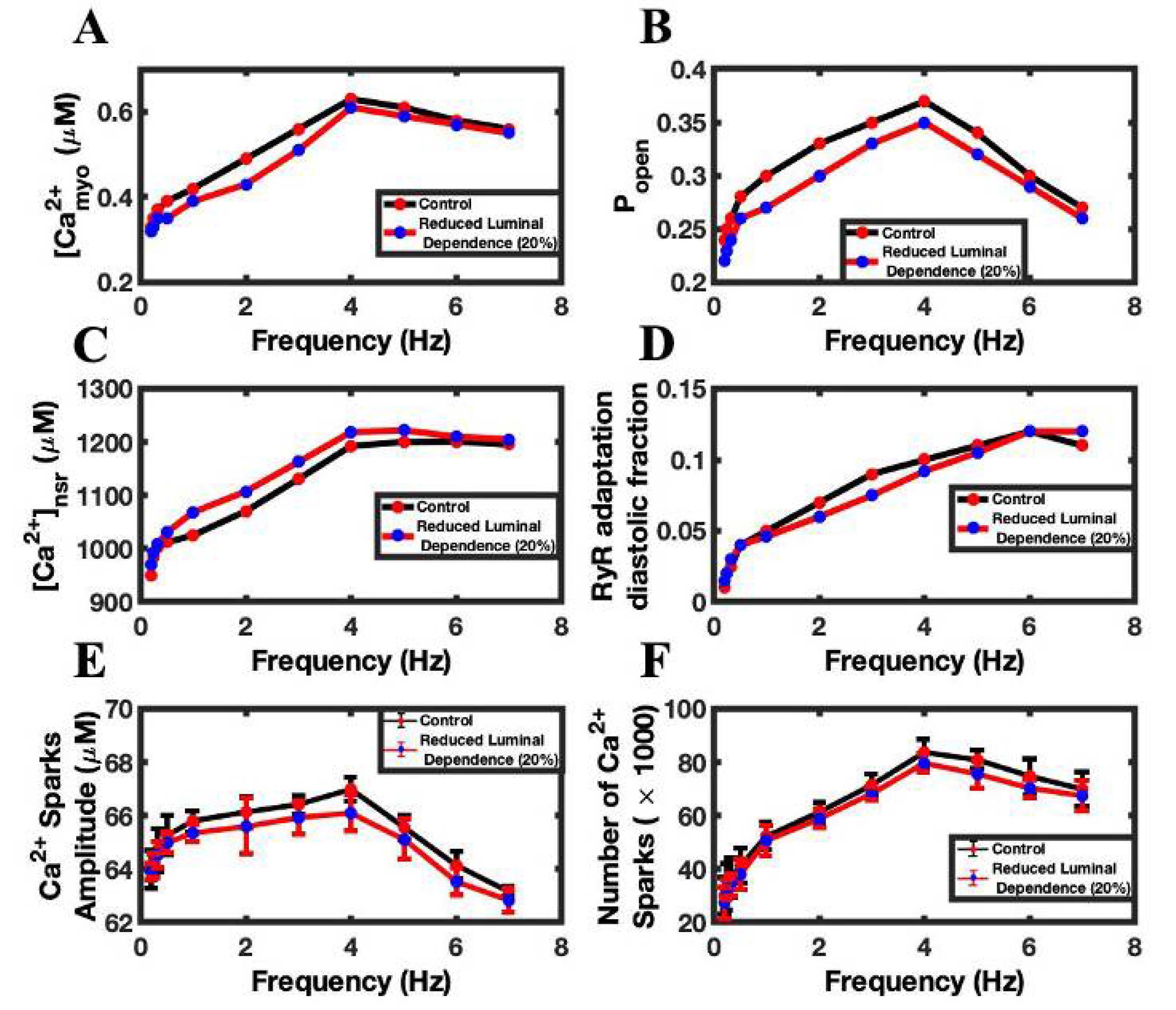
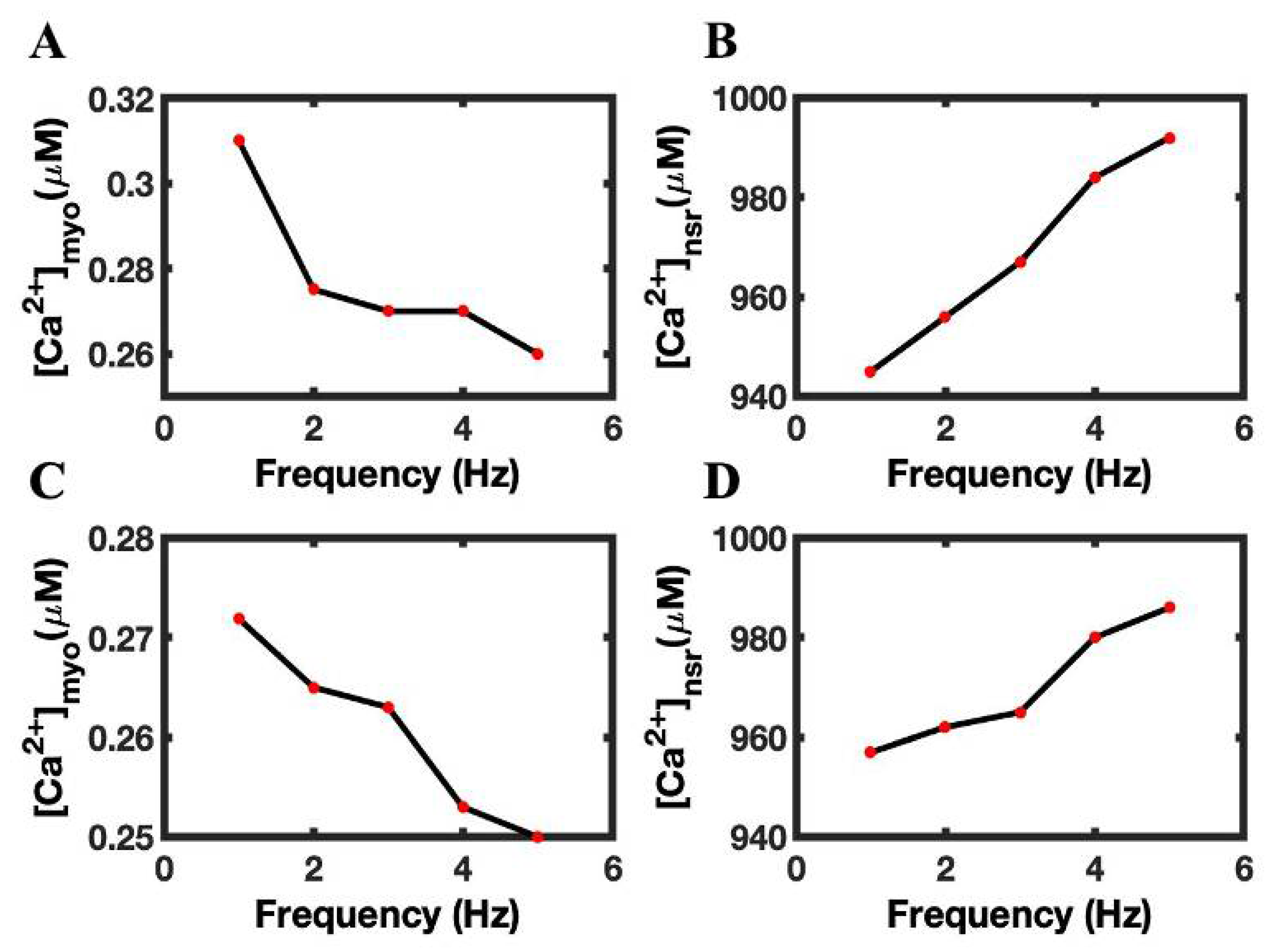
3.4. Luminal Dependence and SR Ca2+ Play Major Role in FFR
3.5. Adaptation Brings Negative Feedback Mechanism to the Open Probability of RyR2
3.6. The Role of RyR2 Opening Rate Constant in FFR
3.7. The Role of L-Type Ca2+ Channels and NCX in FFR
3.8. Pacing Protocols in FFR
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Appendix A
A.1. Calcium Release
A.2. Ryanodine Receptor Type-2 Model
A.3. L-Type Ca2+ Channel Model
A.4. Na+ Channel Model
A.5. K+ Channel Models
A.6. Sarcolemmal Pumps/Exchangers
A.7. Background Currents
A.8. Non-Specific Ca2+-Activated Currents
A.9. Sarcoplasmic Reticulum Ion Pumps
A.10. Buffering
A.11. Membrane Potential

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Variable | Value | Unit |
|---|---|---|
| [Ca]myo | 0.095489 | µM |
| [Ca]nsr | 1.00891 × 103 | µM |
| [Na]i | 1.129 × 104 | µM |
| [K]i | 1.416 × 105 | µM |
| mNa | 0.0015 | Unitless |
| hNa | 0.9849 | Unitless |
| jNa | 0.9905 | Unitless |
| aKss | 0.002 | Unitless |
| inKss | 10 | Unitless |
| aKtof | 0.0021 | Unitless |
| inKtof | 1.0 | Unitless |
| aKtos | 2.0530697 × 10−4 | Unitless |
| inKtos | 0.9994 | Unitless |
| 5 | s−1 | |
| 1 | s−1 |
| Variable | Value | Unit |
|---|---|---|
| Faraday constant (F) | 9.6485 × 104 | C/mol |
| Universal gas constant (R) | 8.314 × 103 | mJ/(mol.K) |
| Temperature (T) | 310 | K |
| [Na]o | 1.40 × 105 | µM |
| [Ca]o | 2.0 × 103 | µM |
| [K]o | 5.7 × 103 | µM |
| Cell volume (Vcell) | 36 | pL |
| Myoplasmic volume (Vmyo) | 25.84 | pL |
| Network SR volume () | 2.098 | pL |
| Junctional SR volume () | 1.82 × 10−1 | pL |
| Subspace volume () | 3.80 × 10−1 | pL |
| Concentration [SERCA] | 395 | µM |
| Kd,myo | 880 | µM |
| Kd,sr | 1900 | µM |
| RyR release rate () | 89.56 | s−1 |
| Percent nj-RyR | 0.05 | Unitless |
| Transfer rate from subspace | ||
| to bulk myoplasm () | 250 | 1/sec |
| Refill rate from nSR | ||
| to jSR () | 2.4 | s−1 |
| NCX maximum | ||
| current density () | 1600 | µA/µF |
| Kd,ncxNa | 8750 | µA/µF |
| Kd,ncxCa | 1380 | µA/µF |
| PMCA maximum | ||
| current density () | 0.95 | µA/µF |
| Kd,pmca | 0.5 | µA/µF |
| Na/K maximum | ||
| current density () | 2.0 | µA/µF |
| Variable | Value | Unit |
|---|---|---|
| [CaM]T | 24.0 | µM |
| [SRm]T | 47.0 | µM |
| [Trpn]T | 140.0 | µM |
| [SL]T | 200.0 | µM |
| 2.4 | µM−1s−1 | |
| 380.0 | s−1 | |
| 115.0 | µM−1s−1 | |
| 100.0 | s−1 | |
| 2.37 | µM−1s−1 | |
| 0.032 | s−1 | |
| 115.0 | µM−1s−1 | |
| 1000.0 | s−1 |
References
- Santana, L.F.; Cheng, E.P.; Lederer, W.J. How does the shape of the cardiac action potential control calcium signaling and contraction in the heart? J. Mol. Cell. Cardiol. 2010, 49, 901–903. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morii, I.; Kihara, Y.; Konishi, T.; Inubushi, T.; Sasayama, S. Mechanism of the negative force-frequency relatishionship in physiological intact rat ventricular myocardium studies by intracellular Ca2+ monitor with indo-1 and by 31P-nuclear magnetic resonance spectroscopy. Jpn. Circ. J. 1996, 60, 593–602. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Endoh, M. Force frequency relationship in intact mammalian ventricular myocardium: Physiological and paraphysiological relevance. Eur. J. Pharmacol. 2004, 500, 73–86. [Google Scholar] [CrossRef] [PubMed]
- Joulin, O.; Marechaux, S.; Hassoun, S.; Montaigne, D.; Lancel, S.; Neviere, R. Cardiac force-frequency relationship and frequency-dependent acceleration of relaxation are impaired in LPS-treated rats. Crit. Care 2009, 13, R14. [Google Scholar] [CrossRef] [Green Version]
- Godier-Furnémont, A.F.G.; Tibucry, M.; Wagner, E.; Dewenter, M.; Lammle, S.; El-Armouche, A.; Lehnart, S.E.; Vunjak-Novakovic, G.; Zimmermann, W.-H. Physiologic force-frequency in engineered heart muscle by electromechanical stimulation. Biomaterials 2015, 60, 82–91. [Google Scholar] [CrossRef] [Green Version]
- Schotten, U.; Greiser, M.; Braun, V.; Karlein, C.; Schoendube, F. Effect of volatile anesthetics on the force-frequency relation in human ventricular myocardium. The role of the sarcoplasmic reticulum calcium-release channel. Anesthesiology 2001, 95, 1160–1168. [Google Scholar] [CrossRef]
- Buckley, N.M.; Penefsky, Z.J.; Litwak, R.S. Comparative force-frequency relationships in human and other mammalian ventricular myocardium. Pflug. Arch. Eur. J. Physiol. 1972, 332, 259–270. [Google Scholar] [CrossRef]
- Gwathmey, J.K.; Slawsky, M.T.; Hajjar, R.J.; Briggs, G.M.; Morgan, J.P. Role of intracellular calcium handling in force-interval relationships of human ventricular myocardium. J. Clin. Investig. 1990, 85, 1599–1613. [Google Scholar] [CrossRef] [Green Version]
- Driedzic, W.R.; Gesser, H. Ca2+ protection from negative inotropic effect of contraction frequency on teleost hearts. J. Comp. Physiol. B 1985, 156, 135–142. [Google Scholar] [CrossRef]
- El-Sayed, M.F.; Abu-Mara, E.; Badr, A. Effects of changes in temperature on force-frequency relationship in the heart of catfish (Clarias gairepinus). J. Basic Appl. Zool. 2012, 65, 274–281. [Google Scholar] [CrossRef]
- Keen, J.E.; Vazon, D.M.; Farrell, A.P.; Tibbits, G.F. Effect of temperature and temperature acclimation on the ryanodine sensitivity of the trout myocardium. J. Comp. Physiol. B 1994, 164, 438–443. [Google Scholar] [CrossRef]
- Namekata, I.; Takeda, K.; Moriwaki, R.; Kazama, A.; Sato, A.; Tanaka, H.; Shigenobu, K. Role of sodium-calcium exchanger in excitation-contraction coupling of mouse myocardium. J. Pharmacol. Sci. 2004, 92, 272. [Google Scholar]
- Narayan, P.; McCune, S.A.; Robitaille, P.M.; Hohl, C.M.; Altschuld, R.A. Mechanical alternans and the force-frequency relationship in failing rat hearts. J. Mol. Cell. Cardiol. 1995, 27, 523–530. [Google Scholar] [CrossRef] [Green Version]
- Rumberger, E.; Riechel, H. The force-frequency relationship: A comparative study between warm and cold-bloded animals. Pflug. Arch. 1972, 332, 206–217. [Google Scholar] [CrossRef]
- Shiels, H.A.; Farrell, A.P. The effect of temperature and adrenaline on the relative importance of the sarcoplasmic reticulum in contributing calcium to force development in isolated ventricular trabeculae from rainbow trout. J. Exp. Biol. 1997, 200, 1607–1621. [Google Scholar] [CrossRef]
- Bohm, M.; la Rosee, K.; Schmidt, U.; Schulz, R.; Schwinger, R.H.; Erdmann, E. Force-frequency relation and inotropic stimulation in the nonfailing and failing human myocardium: Implications for the medical treatment of heart failure. J. Clin. Investig. 1992, 70, 471–475. [Google Scholar]
- Bers, D. Excitation-Contraction Coupling and Cardiac Contractile Force; Kluwer Academic: Dordrecht, The Netherlands, 2001. [Google Scholar]
- Katz, A.M. Heart Failure Pathophysiology, Molecular Biology and Clinical Management; Lippcott Williams and Wilkins: Philadelphia, PA, USA, 2000. [Google Scholar]
- Hasefus, G.; Holubarsch, C.; Hermann, H.P.; Astheimer, K.; Pieske, B.; Just, H. Influence of force frequency relationship on haemodynamics and left ventricular function in patients with non-failing hearts and in patients with dilated cardiomyopathy. Eur. Heart J. 1994, 15, 164–170. [Google Scholar] [CrossRef]
- Davies, C.; Davia, K.; Bennett, J.G.; Pepper, J.R.; Poole-Wilson, P.A.; Harding, S.E. Reduced contraction and altered frequency response of isolated ventricular myocyte from patients with heart failure. Circulation 1995, 92, 2540–2549. [Google Scholar] [CrossRef]
- Kurihara, S.; Allen, D.G. Intracellualr Ca2+ transients and relaxation in mammalian cardiac muscle. Jpn. Circ. J. 1982, 46, 39–43. [Google Scholar] [CrossRef]
- Lompre, A.M.; Anger, M.; Levitsky, D. Sarco(endo)plasmic reticulum calcium pumps in the cardiovascular system: Function and gene expression. J. Mol. Cell. Cardiol. 1994, 26, 1109–1121. [Google Scholar] [CrossRef]
- Ross, J.; Miura, T.; Kambayashi, M.; Eising, G.P.; Ryu, K. Adrenergic control of the force-frequency relation. Circulation 1995, 92, 2327–2332. [Google Scholar] [CrossRef] [PubMed]
- Bers, D.M. Cardiac excitation-contraction coupling. Nature 2002, 415, 198–205. [Google Scholar] [CrossRef] [PubMed]
- Monasky, M.M.; Jansen, P.M. The positive force-frequency relationship is maintained in absence of sarcoplasmic reticulum function in rabbit, but not in rat myocardium. J. Comp. Physiol. B 2009, 179, 469–479. [Google Scholar] [CrossRef] [PubMed]
- Bassani, J.W.; Bassani, R.A.; Bers, D.M. Relaxation in rabbit and rat cardiac cells: Species-dependent differences in cellular mechanisms. J. Physiol. 1994, 476, 279–293. [Google Scholar] [CrossRef]
- Orchard, C.H.; Lakatta, E.G. Intracellualr calcium transients and developed tensions in rat heart muscle: A mechanism for the negative interval-strength relationship. J. Gen. Physiol. 1985, 86, 637–651. [Google Scholar] [CrossRef]
- Jafri, M.S.; Rice, J.J.; Winslow, R.L. Cardiac Ca2+ Dynamics: The roles of ryanodine receptor adaptation and sarcoplasmic reticulum load. Biophys. J. 1998, 74, 1149–1168. [Google Scholar] [CrossRef] [Green Version]
- Williams, G.S.; Chikando, A.C.; Tuan, H.T.; Sobie, E.A.; Lederer, W.J.; Jafri, M.S. Dynamics of Calcium sparks and calcium leak in the heart. Biophys. J. 2011, 101, 1287–1296. [Google Scholar] [CrossRef] [Green Version]
- Hoang-Trong, M.T.; Ullah, A.; Lederer, W.J.; Jafri, M.S. Cardiac Alternans Occurs through the Synergy of Voltage- and Calcium-Dependent Mechanisms. Membranes 2021, 11, 794. [Google Scholar] [CrossRef]
- Hoang-Trong, T.M.; Ullah, A.; Lederer, W.J.; Jafri, M.S. A Stochastic Spatiotemporal Model of Rat Ventricular Myocyte Calcium Dynamics Demonstrated Necessary Features for Calcium Wave Propagation. Membranes 2021, 11, 989. [Google Scholar] [CrossRef]
- Luo, C.H.; Rudy, Y. A model of the ventricular cardiac action potential. Depolarization, repolarization, and their interaction. Circ. Res. 1991, 68, 1501–1526. [Google Scholar] [CrossRef] [Green Version]
- Luo, C.H.; Rudy, Y. A dynamic model of the ventricular cardiac action potential. I. Simulations of ionic current and concentration changes. Circ. Res. 1994, 74, 1070–1096. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luo, C.H.; Rudy, Y. A dynamic model of the cardiac ventricular action potential. II. Afterdepolarizations, triggred activity, and potentiation. Circ. Res. 1994, 74, 1097–1113. [Google Scholar] [CrossRef] [PubMed]
- Wagner, E.; Lauterbach, M.A.; Kohl, T.; Westphal, V.; Williams, G.S.B.; Steinbrecher, J.H.; Strreich, J.-H.; Korff, B.; Tuan, H.-T.M.; Hagen, B.; et al. Stimulation emission depletion live-cell super-resolution imaging shows proliferative remodeling of T-tubule membrane structures after myocardial infraction. Circ. Res. 2012, 111, 402–414. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, L.; Fan, J.-S.; Clark, J.W.; Palade, P.T. A model of the L-type Ca2+ channel in rat ventricular myocytes: Ion selectivity and inactivation mechanisms. J. Physiol. 2000, 15529, 139–158. [Google Scholar] [CrossRef] [PubMed]
- Morotti, S.; Grandi, E.; Summa, A.; Ginsburg, K.S.; Bers, D.M. Theoretical study of L-type Ca2+ current inactivation kinetics during action potential repolarization and early afterdepolarizations. J. Physiol. 2012, 590, 4465–4481. [Google Scholar] [CrossRef] [PubMed]
- Fabiato, A. Simulated calcium current can both cause calcium loading in and trigger calcium release from the sarcoplasmic reticulumof a skinned canine cardiac Purkinje cell. J. Gen. Physiol. 1985, 85, 291–320. [Google Scholar] [CrossRef] [Green Version]
- Jafri, M.S.; Hoang-Trong, M.; Williams, G.S. Methods and System for Utilizing Markov Chain Monte Carlo Simulations. U.S. Patent 9009095, 14 April 2015. [Google Scholar]
- Takagi, S.; Kihara, Y.; Toyoda, F.; Morita, T.; Sasayama, S.; Mitsuiye, T. Cold acclimation of guinea pig depressed contraction of cardiac papillary muscle. Am. J. Physiol. 1999, 276, R923–R928. [Google Scholar] [CrossRef]
- Kubalova, Z. Inactivation of l-type calcium channels in cardiomyocytes. Experimental and theoretical approaches. Gen. Physiol. Biophys. 2003, 22, 441–454. [Google Scholar]
- Lukyanenko, V.S.G. Ca2+ sparks and Ca2+ waves in saponin-permeable rat ventricular myocytes. J. Physiol. 1999, 521, 575–585. [Google Scholar] [CrossRef]
- Izu, L.T.; Wier, W.; Blake, W. Theoretical Analysis of the Ca2+ spark amplitude distribution. Biophys. J. 1998, 75, 1144–1162. [Google Scholar] [CrossRef] [Green Version]
- Sun, Y.B.; Irving, M. The molecular basis of the steep force-calcium relation in heart muscle. J. Mol. Cell. Cardiol. 2010, 48, 859–865. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Szigligeti, P.; Pankucsi, C.; Banyasz, T.; Varro, A.; Nanasi, P.P. Action potential duration and force frequency relationship in isolated rabbit, guinea pig and rat cardiac muscle. J. Comp. Physiol. B 1996, 166, 150–155. [Google Scholar] [CrossRef] [PubMed]
- Varian, K.D.; Janssen, P.M.L. Frequency dependent acceleration of relaxation involves decreased myofilament calcium sensitivity. Am. J. Physiol. Heart Circ. Physiol. 2007, 292, H2212–H2219. [Google Scholar] [CrossRef] [PubMed]
- Janssen, P.M.L.; Periasamy, M. Determinants of frequency dependant contraction and relaxation of mammalian myocardium. J. Mol. Cell. Cardiol. 2007, 43, 523–531. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Valdivia, H.H.; Kaplan, J.H.; Ellis-Davies, G.C.; Lederer, J.W. Rapid adaptation of cardiac ryanodine receptors: Modulation of Mg2+ and phosphorylation. Science 1995, 267, 1997–2000. [Google Scholar] [CrossRef] [Green Version]
- Gyroke, S.; Fill, M. Ryanodine receptor adaptational mechanism of Ca2+-induced Ca2+ release in heart. Science 1993, 260, 807–809. [Google Scholar] [CrossRef]
- Puglisi, J.L.; Negroni, J.A.; Chen-Izu, Y.; Bers, D.M. The force-frequency relationship: Insights from mathematical modeling. Adv. Physiol. Educ. 2013, 37, 28–34. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Chen, M.S.; Liu, H.C.; Xiao, J.H.; Wang, J.L. The relationship between frequency dependence of action potential duration and the expression of TRPC3 in rabbit ventricular myocyte. Cell. Physiol. Biochem. 2014, 33, 646–656. [Google Scholar] [CrossRef]
- Hattori, Y.; Toyama, J.; Kodama, I. Cytosolic calcium staircase in ventricular myocytes isolated from guinea pigs and rats. Cardiovasc. Res. 1991, 25, 622–629. [Google Scholar] [CrossRef]
- Fill, M.; Gillespie, D. Ryanodine Receptor Open Times Are Determined in the Closed State. Biophys. J. 2018, 115, 1160–1165. [Google Scholar] [CrossRef] [Green Version]
- Sobie, E.A.; Dilly, K.W.; Cruz, J.S.; Lederer, W.J.; Jafri, M.S. Termination of cardiac Ca2+ sparks: An investgative mathematical model of calcium-induced calcium release. Biophys. J. 2002, 86, 3329–3331. [Google Scholar] [CrossRef] [Green Version]
- Mulieri, L.A.; Hasenfuss, G.; Leavitt, B.; Allen, P.D.; Albert, N.R. Altered myocardial force-frequency relation in human heart failure. Circulation 1992, 85, 1743–1750. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eisner, D.A.; Trafford, A.W. Heart failure and the ryanodine receptor. Circ. Res. 2002, 91, 979–981. [Google Scholar] [CrossRef] [PubMed]
- Marx, S.O.; Reiken, S.; Hisamatsu, Y.; Jayaraman, T.; Burkhoff, D.; Rosembit, N.; Marks, A.R. PKA phosphorylation dissociates FKBP12.6 from the calcium release channel (ryanodine receptor): Defective regulation in failing hearts. Cell 2000, 101, 365–376. [Google Scholar] [CrossRef] [Green Version]
- Pieske, B.; Maier, L.S.; Bers, D.M.; Hasenfus, G. Ca2+ handling and sarcoplasmic reticulum Ca2+ content in isolated failing and nonfailing human myocardium. Circ. Res. 1999, 85, 38–46. [Google Scholar] [CrossRef] [Green Version]
- Winslow, R.L.; Rice, J.; Jafri, S. Modeling the cellular basis of altered excitation-contraction coupling in heart failure. Prog. Biophys. Mol. Biol. 1998, 69, 497–514. [Google Scholar] [CrossRef]
- Hobai, I.A.; O’Rourke, B. Decreased sarcoplasmic reticulum calcium content is responsible for defective excitation-contraction coupling in canine heart failure. Circulation 2001, 103, 1577–1584. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Groff, J.R.; Smith, G.D. Ryanodine receptor allosteric coupling and the dynamics of calcium sparks. Biophys. J. 2008, 95, 135–154. [Google Scholar] [CrossRef] [Green Version]
- Tran, K.; Smith, N.P.; Loiselle, D.S.; Crampin, E.J. A Thermodynamic Model of the Cardiac Sarcoplasmic/Endoplasmic Ca2+ (SERCA) Pump. Biophys. J. 2009, 96, 2029–2042. [Google Scholar] [CrossRef]











Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Paudel, R.; Jafri, M.S.; Ullah, A. The Role of Ca2+ Sparks in Force Frequency Relationships in Guinea Pig Ventricular Myocytes. Biomolecules 2022, 12, 1577. https://doi.org/10.3390/biom12111577
Paudel R, Jafri MS, Ullah A. The Role of Ca2+ Sparks in Force Frequency Relationships in Guinea Pig Ventricular Myocytes. Biomolecules. 2022; 12(11):1577. https://doi.org/10.3390/biom12111577
Chicago/Turabian StylePaudel, Roshan, Mohsin Saleet Jafri, and Aman Ullah. 2022. "The Role of Ca2+ Sparks in Force Frequency Relationships in Guinea Pig Ventricular Myocytes" Biomolecules 12, no. 11: 1577. https://doi.org/10.3390/biom12111577