

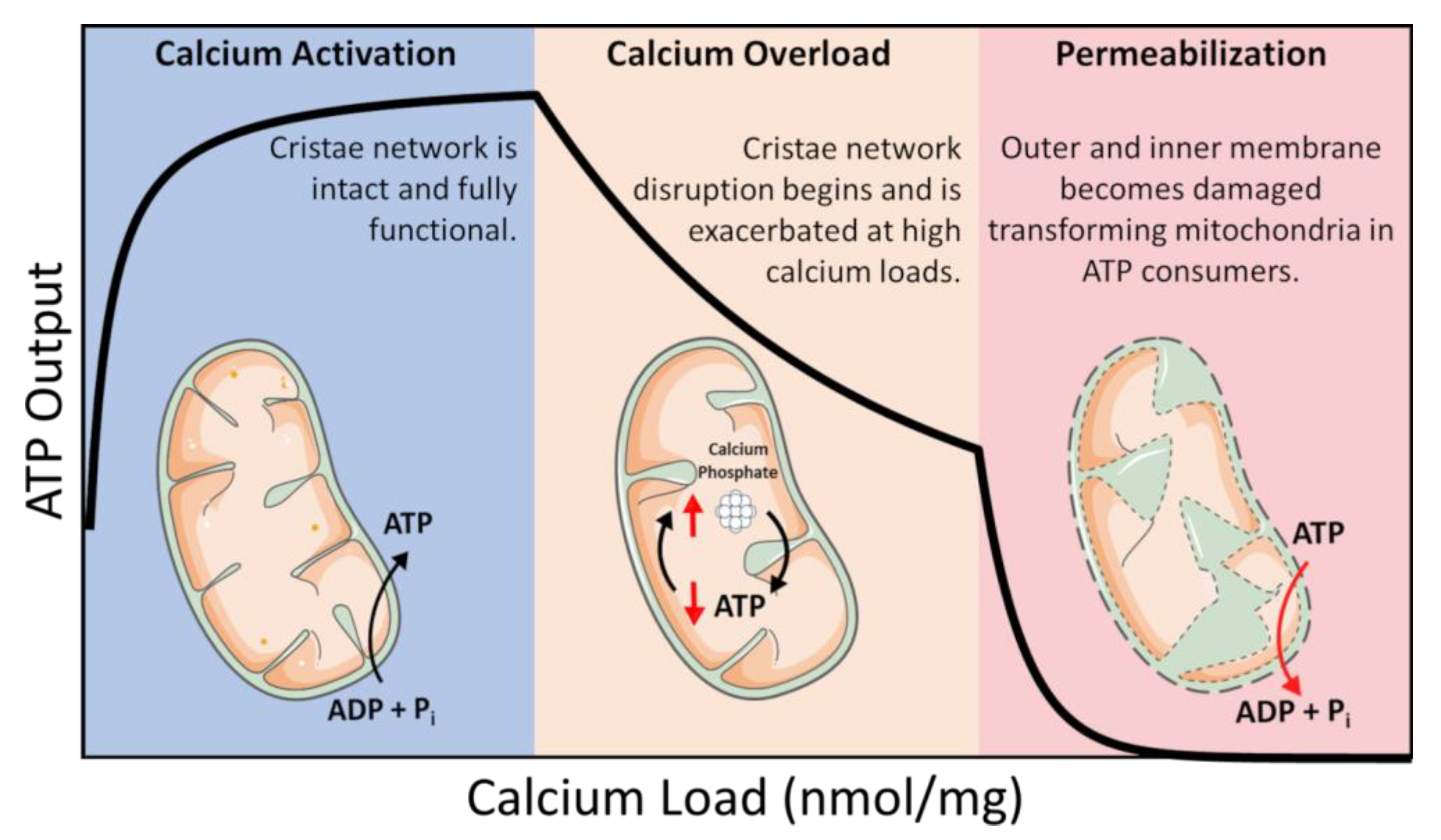
Calcium Overload and Mitochondrial Metabolism


{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction: Mitochondrial Calcium—The Good and the Bad
2. Materials and Methods
3. Calcium Homeostasis, Entry, and Exit Pathways
4. Calcium TCA and ETC
5. Mitochondrial Calcium Buffering
6. Potential Role of Annexins in Mitochondria
7. Structure/Function Axis
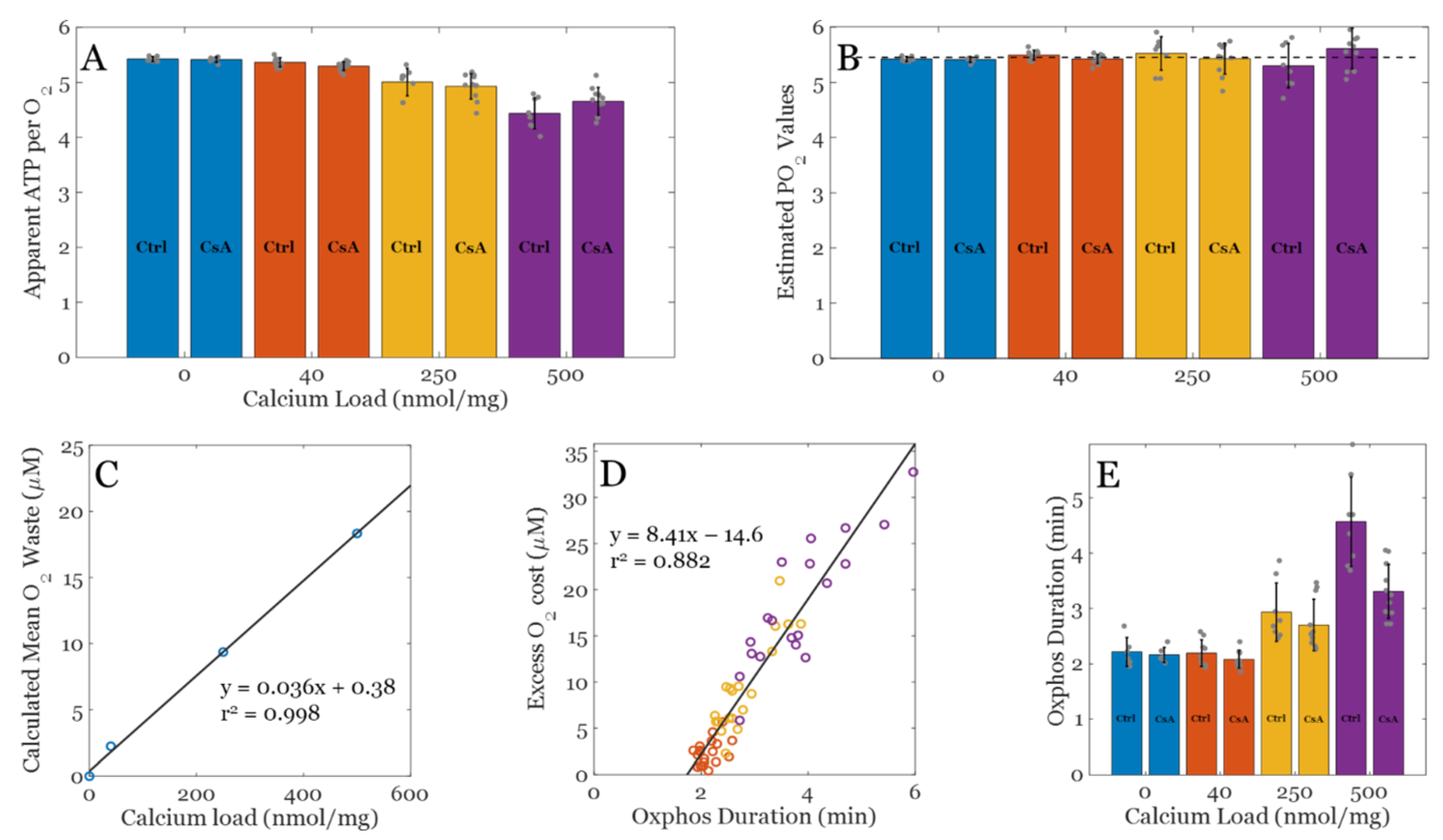
8. Oxygen Utilization in the Calcium Overloaded State
9. Concluding Remarks
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Malyala, S.; Zhang, Y.; Strubbe, J.O.; Bazil, J.N. Calcium phosphate precipitation inhibits mitochondrial energy metabolism. PLoS Comput. Biol. 2019, 15, e1006719. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Strubbe-Rivera, J.O.; Chen, J.; West, B.A.; Parent, K.N.; Wei, G.W.; Bazil, J.N. Modeling the Effects of Calcium Overload on Mitochondrial Ultrastructural Remodeling. Appl. Sci. 2021, 11, 2071. [Google Scholar] [CrossRef] [PubMed]
- Duchen, M.R. Mitochondria and calcium: From cell signalling to cell death. J. Physiol. 2000, 529, 57–68. [Google Scholar] [CrossRef] [PubMed]
- Petronilli, V.; Nicolli, A.; Costantini, P.; Colonna, R.; Bernardi, P. Regulation of the permeability transition pore, a voltage-dependent mitochondrial channel inhibited by cyclosporin A. Biochim. et Biophys. Acta 1994, 1187, 255–259. [Google Scholar] [CrossRef] [PubMed]
- Strubbe-Rivera, J.O.; Schrad, J.R.; Pavlov, E.V.; Conway, J.F.; Parent, K.N.; Bazil, J.N. The mitochondrial permeability transition phenomenon elucidated by cryo-EM reveals the genuine impact of calcium overload on mitochondrial structure and function. Sci. Rep. 2021, 11, 1–15. [Google Scholar] [CrossRef]
- Glancy, B.; Balaban, R.S. Role of Mitochondrial Ca2+ in the Regulation of Cellular Energetics. Biochemistry 2012, 51, 2959–2973. [Google Scholar] [CrossRef] [Green Version]
- Duong, Q.V.; Hoffman, A.; Zhong, K.; Dessinger, M.J.; Zhang, Y.; Bazil, J.N. Calcium overload decreases net free radical emission in cardiac mitochondria. Mitochondrion 2020, 51, 126–139. [Google Scholar] [CrossRef]
- Carafoli, E. The fateful encounter of mitochondria with calcium: How did it happen? Biochim. Biophys. Acta 2010, 1797, 595–606. [Google Scholar] [CrossRef] [Green Version]
- Varanyuwatana, P.; Halestrap, A.P. The roles of phosphate and the phosphate carrier in the mitochondrial permeability transition pore. Mitochondrion 2012, 12, 120–125. [Google Scholar] [CrossRef]
- Petronilli, V.; Cola, C.; Massari, S.; Colonna, R.; Bernardi, P. Physiological effectors modify voltage sensing by the cyclosporin A-sensitive permeability transition pore of mitochondria. J. Biol. Chem. 1993, 268, 21939–21945. [Google Scholar] [CrossRef]
- Basso, E.; Petronilli, V.; Forte, M.A.; Bernardi, P. Phosphate Is Essential for Inhibition of the Mitochondrial Permeability Transition Pore by Cyclosporin A and by Cyclophilin D Ablation. J. Biol. Chem. 2008, 283, 26307–26311. [Google Scholar] [CrossRef] [Green Version]
- Bround, M.J.; Bers, D.M.; Molkentin, J.D. A 20/20 view of ANT function in mitochondrial biology and necrotic cell death. J Mol Cell Cardiol. 2020, 144, A3–A13. [Google Scholar] [CrossRef]
- Carrer, A.; Tommasin, L.; Šileikytė, J.; Ciscato, F.; Filadi, R.; Urbani, A.; Forte, M.; Rasola, A.; Szabò, I.; Carraro, M.; et al. Defining the molecular mechanisms of the mitochondrial permeability transition through genetic manipulation of F-ATP synthase. Nat. Commun. 2021, 12, 1–12. [Google Scholar] [CrossRef]
- Halestrap, A.P. What is the mitochondrial permeability transition pore? J Mol Cell Cardiol 2009, 46, 821–831. [Google Scholar] [CrossRef]
- Bauer, T.M.; Murphy, E. Role of Mitochondrial Calcium and the Permeability Transition Pore in Regulating Cell Death. Circ. Res. 2020, 126, 280–293. [Google Scholar] [CrossRef]
- Kremer, J.R.; Mastronarde, D.N.; Mc Intosh, J.R. Computer Visualization of Three-Dimensional Image Data Using IMOD. J. Struct. Biol. 1996, 116, 71–76. [Google Scholar] [CrossRef] [Green Version]
- Alevriadou, B.R.; Patel, A.; Noble, M.; Ghosh, S.; Gohil, V.M.; Stathopulos, P.B.; Madesh, M. Molecular nature and physiological role of the mitochondrial calcium uniporter channel. Am. J. Physiol. Physiol. 2021, 320, C465–C482. [Google Scholar] [CrossRef]
- Csordás, G.; Golenár, T.; Seifert, E.L.; Kamer, K.J.; Sancak, Y.; Perocchi, F.; Moffat, C.; Weaver, D.; Perez, S.d.l.F.; Bogorad, R.; et al. MICU1 Controls Both the Threshold and Cooperative Activation of the Mitochondrial Ca2+ Uniporter. Cell Metab. 2013, 17, 976–987. [Google Scholar] [CrossRef] [Green Version]
- Belosludtsev, K.N.; Dubinin, M.V.; Belosludtseva, N.V.; Mironova, G.D. Mitochondrial Ca2+ Transport: Mechanisms, Molecular Structures, and Role in Cells. Biochemistry 2019, 84, 593–607. [Google Scholar] [CrossRef]
- De Stefani, D.; Patron, M.; Rizzuto, R. Structure and function of the mitochondrial calcium uniporter complex. Biochim. et Biophys. Acta (BBA)-Mol. Cell Res. 2015, 1853, 2006–2011. [Google Scholar] [CrossRef]
- Wei, A.-C.; Liu, T.; Winslow, R.L.; O’Rourke, B. Dynamics of matrix-free Ca2+ in cardiac mitochondria: Two components of Ca2+ uptake and role of phosphate buffering. J. Gen. Physiol. 2012, 139, 465–478. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mishra, J.; Jhun, B.S.; Hurst, S.; Csordás, G.; Sheu, S.S. The Mitochondrial Ca2+ Uniporter: Structure, Function, and Pharmacology. Handb Exp Pharm. 2017, 240, 129–156. [Google Scholar]
- Gunter, T.E.; Yule, D.I.; Gunter, K.K.; Eliseev, R.A.; Salter, J.D. Calcium and mitochondria. FEBS Lett. 2004, 567, 96–102. [Google Scholar] [CrossRef] [PubMed]
- Kirichok, Y.; Krapivinsky, G.; Clapham, D.E. The mitochondrial calcium uniporter is a highly selective ion channel. Nature 2004, 427, 360–364. [Google Scholar] [CrossRef] [PubMed]
- Garbincius, J.F.; Elrod, J.W. Mitochondrial calcium exchange in physiology and disease. Physiol. Rev. 2022, 102, 893–992. [Google Scholar] [CrossRef]
- Dash, R.K.; Beard, D.A. Analysis of cardiac mitochondrial Na+-Ca2+ exchanger kinetics with a biophysical model of mitochondrial Ca2+ handling suggests a 3:1 stoichiometry. J. Physiol. 2008, 586, 3267–3285. [Google Scholar] [CrossRef]
- Austin, S.; Mekis, R.; Mohammed, S.E.M.; Scalise, M.; Pfeiffer, C.; Galluccio, M.; Borovec, T.; Parapatics, K.; Vitko, D.; Dinhopl, N.; et al. MICSI is the Ca2+/H+ antiporter of mammalian mitochondria. BioRxiv 2021, 1–53. [Google Scholar] [CrossRef]
- Gunter, K.K.; Zuscik, M.J.; Gunter, T.E. The Na(+)-independent Ca2+ efflux mechanism of liver mitochondria is not a passive Ca2+/2H+ exchanger. J. Biol. Chem. 1991, 266, 21640–21648. [Google Scholar] [CrossRef]
- Wingrove, E.D.; Gunter, E.T. Kinetics of mitochondrial calcium transport. II. A kinetic description of the sodium-dependent calcium efflux mechanism of liver mitochondria and inhibition by ruthenium red and by tetraphenylphosphonium. J. Biol. Chem. 1986, 261, 15166–15171. [Google Scholar] [CrossRef]
- Wingrove, E.D.; Gunter, E.T. Kinetics of mitochondrial calcium transport. I. Characteristics of the sodium-independent calcium efflux mechanism of liver mitochondria. J. Biol. Chem. 1986, 261, 15159–15165. [Google Scholar] [CrossRef]
- Garbincius, J.F.; Luongo, T.S.; Jadiya, P.; Hildebrand, A.N.; Kolmetzky, D.W.; Mangold, A.S.; Roy, R.; Ibetti, J.; Nwokedi, M.; Koch, W.J.; et al. Enhanced NCLX-dependent mitochondrial Ca2+ efflux attenuates pathological remodeling in heart failure. J. Mol. Cell. Cardiol. 2022, 167, 52–66. [Google Scholar] [CrossRef]
- Drago, I.; Pizzo, P.; Pozzan, T. After half a century mitochondrial calcium in- and efflux machineries reveal themselves. EMBO J. 2011, 30, 4119–4125. [Google Scholar] [CrossRef] [Green Version]
- Pizzo, P.; Drago, I.; Filadi, R.; Pozzan, T. Mitochondrial Ca²⁺ homeostasis: Mechanism, role, and tissue specificities. Pflug. Arch 2012, 464, 3–17. [Google Scholar] [CrossRef]
- Rizzuto, R.; Pozzan, T. Microdomains of Intracellular Ca2+: Molecular Determinants and Functional Consequences. Physiol. Rev. 2006, 86, 369–408. [Google Scholar] [CrossRef]
- Bers, D.M. Altered Cardiac Myocyte Ca Regulation In Heart Failure. Physiology 2006, 21, 380–387. [Google Scholar] [CrossRef] [Green Version]
- Hale, C.C.; Bossuyt, J.; Hill, C.K.; Price, E.M.; Schulze, D.H.; Lederer, J.W.; Poljak, R.; Braden, B.C. Sodium-Calcium Exchange Crystallization. Ann. N. Y. Acad. Sci. 2006, 976, 100–102. [Google Scholar] [CrossRef]
- Boyman, L.; Lederer, W.J. How the mitochondrial calcium uniporter complex (MCUcx) works. Proc. Natl. Acad. Sci. USA 2020, 117, 22634–22636. [Google Scholar] [CrossRef]
- Williams, G.S.; Boyman, L.; Chikando, A.C.; Khairallah, R.J.; Lederer, W.J. Mitochondrial calcium uptake. Proc. Natl. Acad. Sci. USA 2013, 110, 10479–10486. [Google Scholar] [CrossRef] [Green Version]
- Szanda, G.; Koncz, P.; Várnai, P.; Spät, A. Mitochondrial Ca2+ uptake with and without the formation of high-Ca2+ microdomains. Cell Calcium 2006, 40, 527–537. [Google Scholar] [CrossRef]
- Chalmers, S.; Nicholls, D.G. The Relationship between Free and Total Calcium Concentrations in the Matrix of Liver and Brain Mitochondria. J. Biol. Chem. 2003, 278, 19062–19070. [Google Scholar] [CrossRef] [Green Version]
- Zhang, X.; Tomar, N.; Kandel, S.M.; Audi, S.H.; Cowley, A.W.; Dash, R.K. Substrate- and Calcium-Dependent Differential Regulation of Mitochondrial Oxidative Phosphorylation and Energy Production in the Heart and Kidney. Cells 2021, 11, 131. [Google Scholar] [CrossRef] [PubMed]
- Bernardi, P.; Petronilli, V. The permeability transition pore as a mitochondrial calcium release channel: A critical appraisal. J. Bioenerg. Biomembr. 1996, 28, 131–138. [Google Scholar] [CrossRef] [PubMed]
- Chinopoulos, C.; Adam-Vizi, V. Mitochondrial Ca2+ sequestration and precipitation revisited. FEBS J. 2010, 277, 3637–3651. [Google Scholar] [CrossRef] [PubMed]
- Bernardi, P. Mitochondrial Transport of Cations: Channels, Exchangers, and Permeability Transition. Physiol. Rev. 1999, 79, 1127–1155. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, N.; Li, W.-N.; Shu, W.-Q.; Lv, Y.; Jia, D.-L. Blocking the mitochondrial permeability transition pore with cyclosporine-A can restore cardioprotection of ischemic postconditioning in hypercholesterolemic rat heart. Eur. Rev. Med. Pharmacol. Sci. 2015, 19, 446–454. [Google Scholar]
- Bernardi, P. Mechanisms for Ca2+-dependent permeability transition in mitochondria. Proc. Natl. Acad. Sci. USA 2020, 117, 2743–2744. [Google Scholar] [CrossRef]
- Poburko, D.; Domingo, J.S.; Demaurex, N. Dynamic Regulation of the Mitochondrial Proton Gradient during Cytosolic Calcium Elevations. J. Biol. Chem. 2011, 286, 11672–11684. [Google Scholar] [CrossRef] [Green Version]
- Seifert, E.L.; Ligeti, E.; Mayr, J.A.; Sondheimer, N.; Hajnóczky, G. The mitochondrial phosphate carrier: Role in oxidative metabolism, calcium handling and mitochondrial disease. Biochem. Biophys. Res. Commun. 2015, 464, 369–375. [Google Scholar] [CrossRef] [Green Version]
- Nicholls, D.G.; Chalmers, S. The Integration of Mitochondrial Calcium Transport and Storage. J. Bioenerg. Biomembr. 2004, 36, 277–281. [Google Scholar] [CrossRef]
- Carafoli, E.; Patriarca, P.; Rossi, C.S. A comparative study of the role of mitochondria and the sarcoplasmic reticulum in the uptake and release of Ca++ by the rat diaphragm. J. Cell. Physiol. 1969, 74, 17–29. [Google Scholar] [CrossRef]
- Duvvuri, B.; Lood, C. Mitochondrial Calcification. Immunometabolism 2021, 3, 8. [Google Scholar] [CrossRef]
- Carafoli, E.; Rossi, C.S.; Lehninger, A.L. Uptake of adenine nucleotides by respiring mitochondria during active accumulation of Ca++ and phosphate. J. Biol. Chem. 1965, 240, 2254–2261. [Google Scholar] [CrossRef]
- Bazil, J.N.; Blomeyer, C.A.; Pradhan, R.K.; Camara, A.K.S.; Dash, R.K. Modeling the calcium sequestration system in isolated guinea pig cardiac mitochondria. J. Bioenerg. Biomembr. 2012, 45, 177–188. [Google Scholar] [CrossRef] [Green Version]
- Bandorowicz-Pikuła, J.; Woś, M.; Sekrecka-Belniak, A.; Pikuła, S. [Annexins in mitochondria]. Postep. Biochem. 2017, 62, 216–223. [Google Scholar]
- Szewczyk, A.; Pikula, S. Adenosine 5′-triphosphate: An intracellular metabolic messenger. Biochim. Biophys Acta 1998, 1365, 333–353. [Google Scholar] [CrossRef] [Green Version]
- Bandorowicz-Pikuła, J.; Buchet, R.; Pikuła, S. Annexins as nucleotide-binding proteins: Facts and speculations. BioEssays 2001, 23, 170–178. [Google Scholar] [CrossRef]
- Rescher, U.; Gerke, V. Annexins—Unique membrane binding proteins with diverse functions. J. Cell Sci. 2004, 117, 2631–2639. [Google Scholar] [CrossRef] [Green Version]
- Camors, E.; Monceau, V.; Charlemagne, D. Annexins and Ca handling in the heart. Cardiovasc. Res. 2005, 65, 793–802. [Google Scholar] [CrossRef] [Green Version]
- Gerke, V.; Creutz, C.E.; Moss, S.E. Annexins: Linking Ca2+ signalling to membrane dynamics. Nat. Rev. Mol. Cell Biol. 2005, 6, 449–461. [Google Scholar] [CrossRef]
- Chlystun, M.; Campanella, M.; Law, A.-L.; Duchen, M.R.; Fatimathas, L.; Levine, T.P.; Gerke, V.; Moss, S.E. Regulation of Mitochondrial Morphogenesis by Annexin A6. PLoS ONE 2013, 8, e53774. [Google Scholar] [CrossRef] [Green Version]
- Croissant, C.; Carmeille, R.; Brévart, C.; Bouter, A. Annexins and Membrane Repair Dysfunctions in Muscular Dystrophies. Int. J. Mol. Sci. 2021, 22, 5276. [Google Scholar] [CrossRef] [PubMed]
- Sophia, N.; Koerdt, A.P.K.A.; Volker, G. Chapter Three—Annexins and Plasma Membrane Repai. In Plasma Membrane Repair; Andrade, L.O., Ed.; Elsevier: Amsterdam, The Netherlands, 2019; pp. 43–65. [Google Scholar]
- Munn, E.A. The Structure of Mitochondria; Academic Press: London, UK; New York, NY, USA, 1974; 465 p. [Google Scholar]
- Hayden, M.R. The Mighty Mitochondria Are Unifying Organelles and Metabolic Hubs in Multiple Organs of Obesity, Insulin Resistance, Metabolic Syndrome, and Type 2 Diabetes: An Observational Ultrastructure Study. Int. J. Mol. Sci. 2022, 23, 4820. [Google Scholar] [CrossRef] [PubMed]
- Mendelsohn, D.H.; Schnabel, K.; Mamilos, A.; Sossalla, S.; Pabel, S.; Duerr, G.D.; Keller, K.; Schmitt, V.H.; Barsch, F.; Walter, N.; et al. Structural Analysis of Mitochondrial Dynamics—From Cardiomyocytes to Osteoblasts: A Critical Review. Int. J. Mol. Sci. 2022, 23, 4571. [Google Scholar] [CrossRef] [PubMed]
- Mishra, E.; Thakur, M.K. Alterations in hippocampal mitochondrial dynamics are associated with neurodegeneration and recognition memory decline in old male mice. Biogerontology 2022, 23, 251–271. [Google Scholar] [CrossRef] [PubMed]
- Ranjbarvaziri, S.; Kooiker, K.B.; Ellenberger, M.; Fajardo, G.; Zhao, M.; Roest, A.S.V.; Woldeyes, R.A.; Koyano, T.T.; Fong, R.; Ma, N.; et al. Altered Cardiac Energetics and Mitochondrial Dysfunction in Hypertrophic Cardiomyopathy. Circulation 2021, 144, 1714–1731. [Google Scholar] [CrossRef] [PubMed]
- Méndez-López, I.; Sancho-Bielsa, F.; Engel, T.; García, A.; Padín, J. Progressive Mitochondrial SOD1G93A Accumulation Causes Severe Structural, Metabolic and Functional Aberrations through OPA1 Down-Regulation in a Mouse Model of Amyotrophic Lateral Sclerosis. Int. J. Mol. Sci. 2021, 22, 8194. [Google Scholar] [CrossRef]
- Knapp-Wilson, A.; Pereira, G.C.; Buzzard, E.; Ford, H.C.; Richardson, A.; Corey, R.A.; Neal, C.; Verkade, P.; Halestrap, A.P.; Gold, V.A.M.; et al. Maintenance of complex I and its supercomplexes by NDUF-11 is essential for mitochondrial structure, function and health. J. Cell Sci. 2021, 134, 258399. [Google Scholar] [CrossRef]
- Mukherjee, I.; Ghosh, M.; Meinecke, M. MICOS and the mitochondrial inner membrane morphology—When things get out of shape. FEBS Lett. 2021, 595, 1159–1183. [Google Scholar] [CrossRef]
- Raaflaub, J. [Swelling of isolated mitochondria of the liver and their susceptibility to physicochemical influences]. Helvetica Physiol. et Pharmacol. Acta 1953, 11, 142–156. [Google Scholar]
- Hunter, E.F.; Ford, L. Inactivation of oxidative and phosphorylative systems in mitochondria by preincubation with phosphate and other ions. J. Biol. Chem. 1955, 216, 357–369. [Google Scholar] [CrossRef]
- Tapley, D.F. The effect of thyroxine and other substances on the swelling of isolated rat liver mitochondria. J. Biol. Chem. 1956, 222, 325–339. [Google Scholar] [CrossRef]
- Humphrey, G.F. The distribution and properties of transhydrogenase from animal tissues. Biochem. J. 1957, 65, 546–550. [Google Scholar] [CrossRef] [Green Version]
- Lehninger, A.L. Reversal of various types of mitochondrial swelling by adenosine triphosphate. J. Biol. Chem. 1959, 234, 2465–2471. [Google Scholar] [CrossRef]
- Lehninger, A.L.; Remmert, L.F. An endogenous uncoupling and swelling agent in liver mitochondria and its enzymic formation. J. Biol. Chem. 1959, 234, 2459–2464. [Google Scholar] [CrossRef]
- Lehninger, A.L.; Schneider, M. Mitochondrial Swelling Induced by Glutathione. J. Cell Biol. 1959, 5, 109–116. [Google Scholar] [CrossRef] [Green Version]
- Wojtczak, L.; Lehninger, A.L. Formation and disappearance of an endogenous uncoupling factor during swelling and contraction of mitochondria. Biochim. et Biophys. Acta 1961, 51, 442–456. [Google Scholar] [CrossRef]
- Azzi, A.; Azzone, G.F. Swelling and shrinkage phenomena in liver mitochondria I. Large amplitude swelling induced by inorganic phosphate and by ATP. Biochim. et Biophys. Acta (BBA)-Enzym. Biol. Oxid. 1965, 105, 253–264. [Google Scholar] [CrossRef]
- Azzone, G.F.; Azzi, A. Volume changes in liver mitochondria. Proc. Natl. Acad. Sci. USA 1965, 53, 1084–1089. [Google Scholar] [CrossRef] [Green Version]
- Chappell, J.B.; Crofts, A.R. Calcium Ion Accumulation and Volume Changes of Isolated Liver Mitochondria. Calcium Ion-Induced Swelling. Biochem. J. 1965, 95, 378–386. [Google Scholar]
- Chappell, J.B.; Crofts, A.R. Calcium ion accumulation and Volume Changes of Isolated Liver Mitochondria. Reversal of Calcium Ion-Induced Swelling. Biochem. J. 1965, 95, 387–392. [Google Scholar]
- Hackenbrock, C.R. Energy-Linked Condensed-Orthodox Ultrastructural Transformations in Mitochondria. Chemotherapy 1981, 27, 21–26. [Google Scholar] [CrossRef] [PubMed]
- Duong, Q.V.; Levitsky, Y.; Dessinger, M.J.; O Strubbe-Rivera, J.; Bazil, J.N. Identifying Site-Specific Superoxide and Hydrogen Peroxide Production Rates From the Mitochondrial Electron Transport System Using a Computational Strategy. Function 2021, 2, 50. [Google Scholar] [CrossRef]
- Hackenbrock, C.R.; Caplan, A. Ion-induced ultrastructural transformations in isolated mitochondria. J. Cell Biol. 1969, 42, 221–234. [Google Scholar] [CrossRef] [PubMed]
- Giorgi, C.; Baldassari, F.; Bononi, A.; Bonora, M.; De Marchi, E.; Marchi, S.; Missiroli, S.; Patergnani, S.; Rimessi, A.; Suski, J.M.; et al. Mitochondrial Ca2+ and apoptosis. Cell Calcium 2012, 52, 36–43. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fink, B.D.; Bai, F.; Yu, L.; Sivitz, W.I. Regulation of ATP production: Dependence on calcium concentration and respiratory state. Am. J. Physiol. Physiol. 2017, 313, C146–C153. [Google Scholar] [CrossRef] [Green Version]
- Wolf, S.G.; Mutsafi, Y.; Dadosh, T.; Ilani, T.; Lansky, Z.; Horowitz, B.; Rubin, S.; Elbaum, M.; Fass, D. 3D visualization of mitochondrial solid-phase calcium stores in whole cells. eLife 2017, 6, e29929. [Google Scholar] [CrossRef]
- Kloner, R.A.; Rude, R.E.; Carlson, N.; Maroko, P.R.; DeBoer, L.W.; Braunwald, E. Ultrastructural evidence of microvascular damage and myocardial cell injury after coronary artery occlusion: Which comes first? Circulation 1980, 62, 945–952. [Google Scholar] [CrossRef] [Green Version]
- Ghosh, S.; Guglielmi, G.; Orfanidis, I.; Spill, F.; Hickey, A.; Hanssen, E.; Rajagopal, V. Effects of altered cellular ultrastructure on energy metabolism in diabetic cardiomyopathy: An in silico study. Philos. Trans. R. Soc. B: Biol. Sci. 2022, 377, 323. [Google Scholar] [CrossRef]
- Afzal, N.; Lederer, W.J.; Jafri, M.S.; Mannella, C.A. Effect of crista morphology on mitochondrial ATP output: A computational study. Curr. Res. Physiol. 2021, 4, 163–176. [Google Scholar] [CrossRef]
- Ghosh, S.; Tran, K.; Delbridge, L.M.D.; Hickey, A.J.R.; Hanssen, E.; Crampin, E.J.; Rajagopal, V. Insights on the impact of mitochondrial organisation on bioenergetics in high-resolution computational models of cardiac cell architecture. PLoS Comput. Biol. 2018, 14, e1006640. [Google Scholar] [CrossRef]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Walkon, L.L.; Strubbe-Rivera, J.O.; Bazil, J.N. Calcium Overload and Mitochondrial Metabolism. Biomolecules 2022, 12, 1891. https://doi.org/10.3390/biom12121891
Walkon LL, Strubbe-Rivera JO, Bazil JN. Calcium Overload and Mitochondrial Metabolism. Biomolecules. 2022; 12(12):1891. https://doi.org/10.3390/biom12121891
Chicago/Turabian StyleWalkon, Lauren L., Jasiel O. Strubbe-Rivera, and Jason N. Bazil. 2022. "Calcium Overload and Mitochondrial Metabolism" Biomolecules 12, no. 12: 1891. https://doi.org/10.3390/biom12121891