
Systemic Biomarkers and Unique Pathways in Different Phenotypes of Heart Failure with Preserved Ejection Fraction
 , ,
, ,  and
and
Abstract
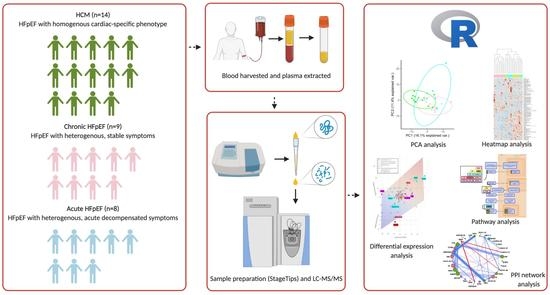
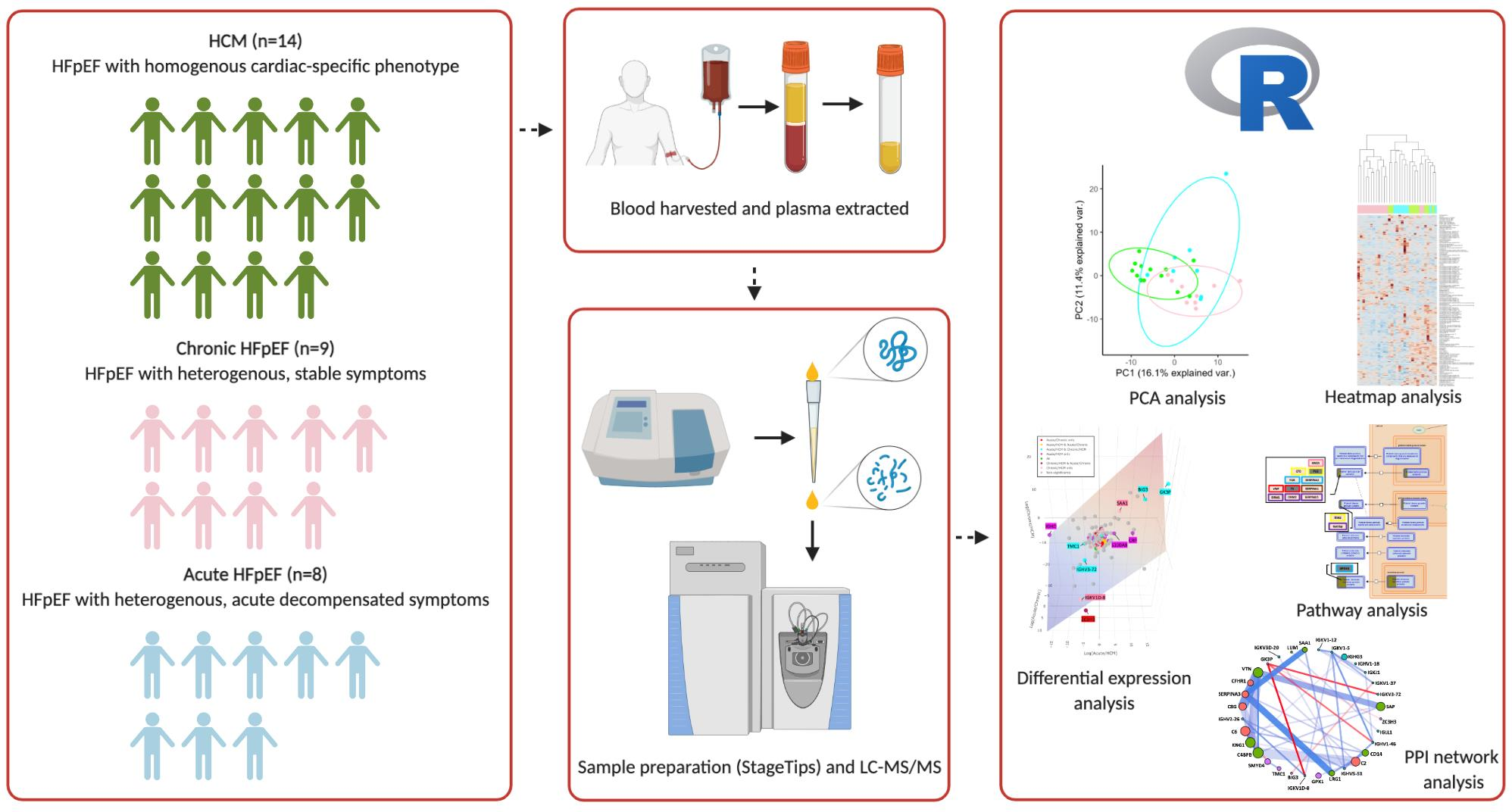
:
1. Introduction
2. Material and Methods
2.1. Collection of Plasma
2.2. Proteomics
2.3. Statistics
2.4. Data Availability
3. Results
3.1. Patient Characteristics
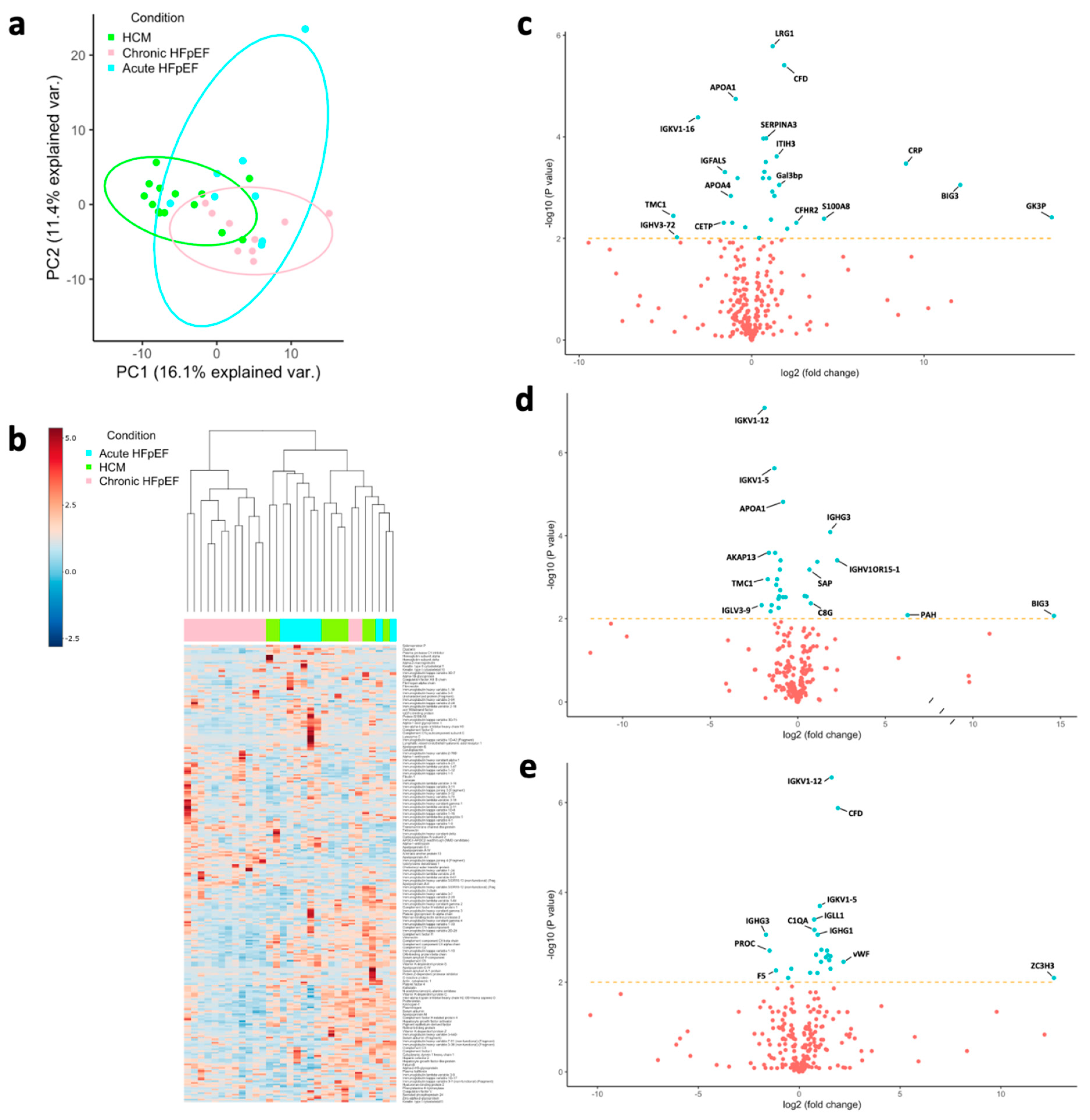
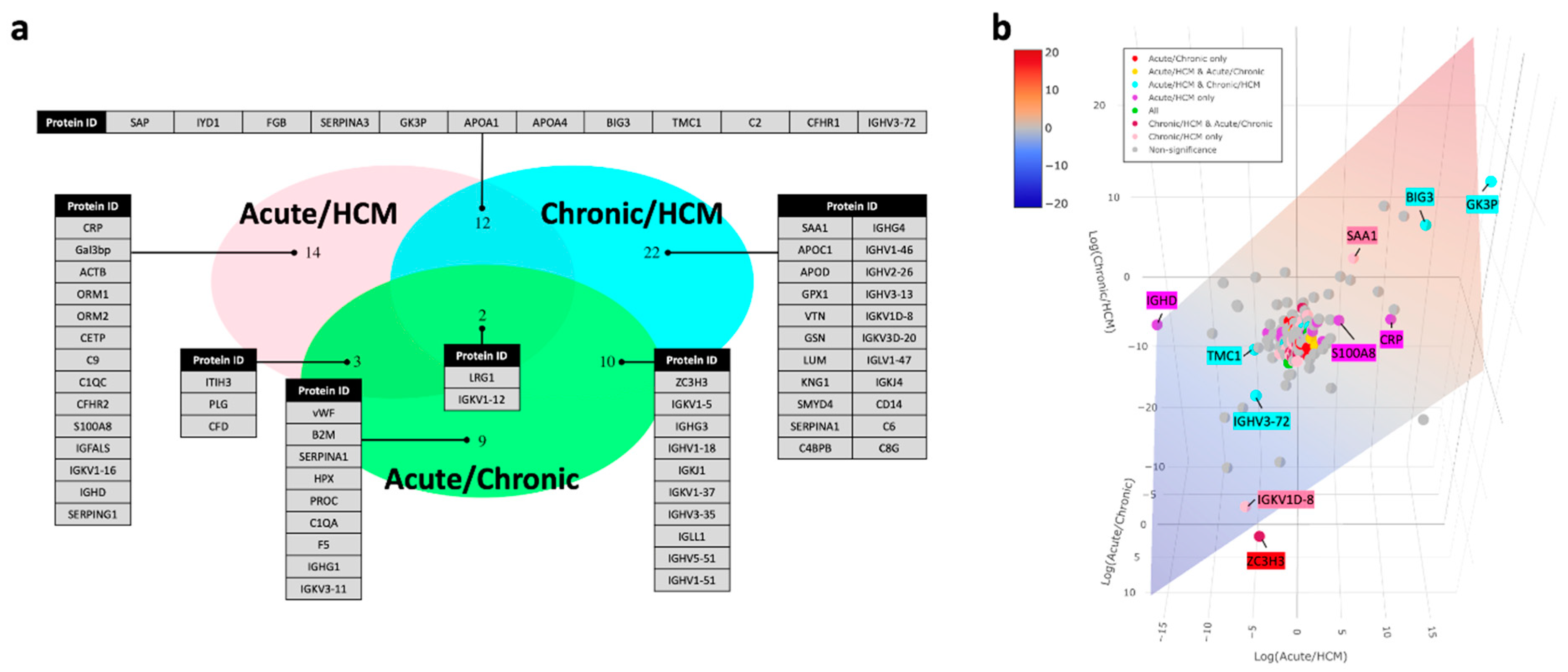
3.2. Protein Markers
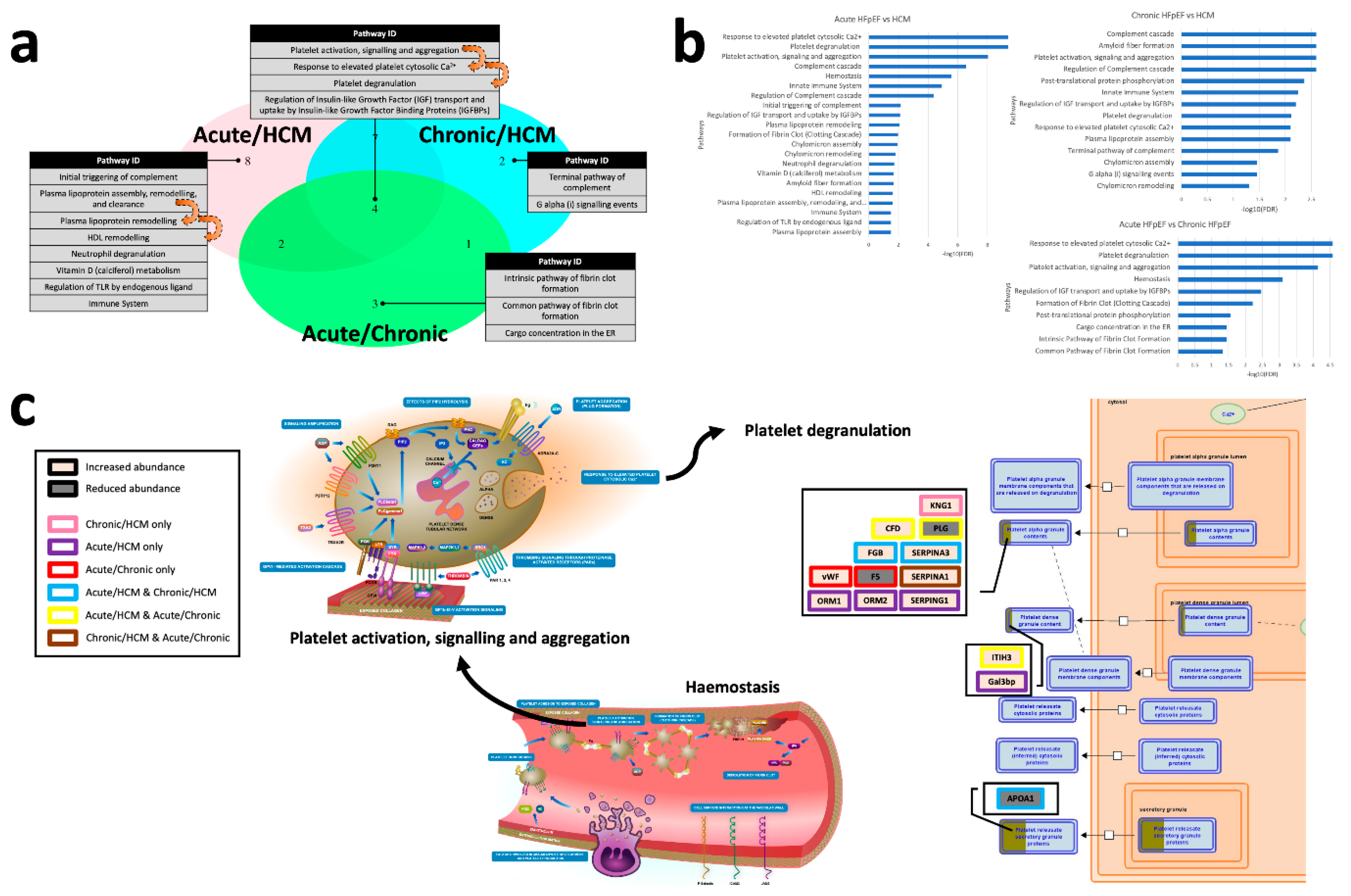
3.3. Pathway Analyses
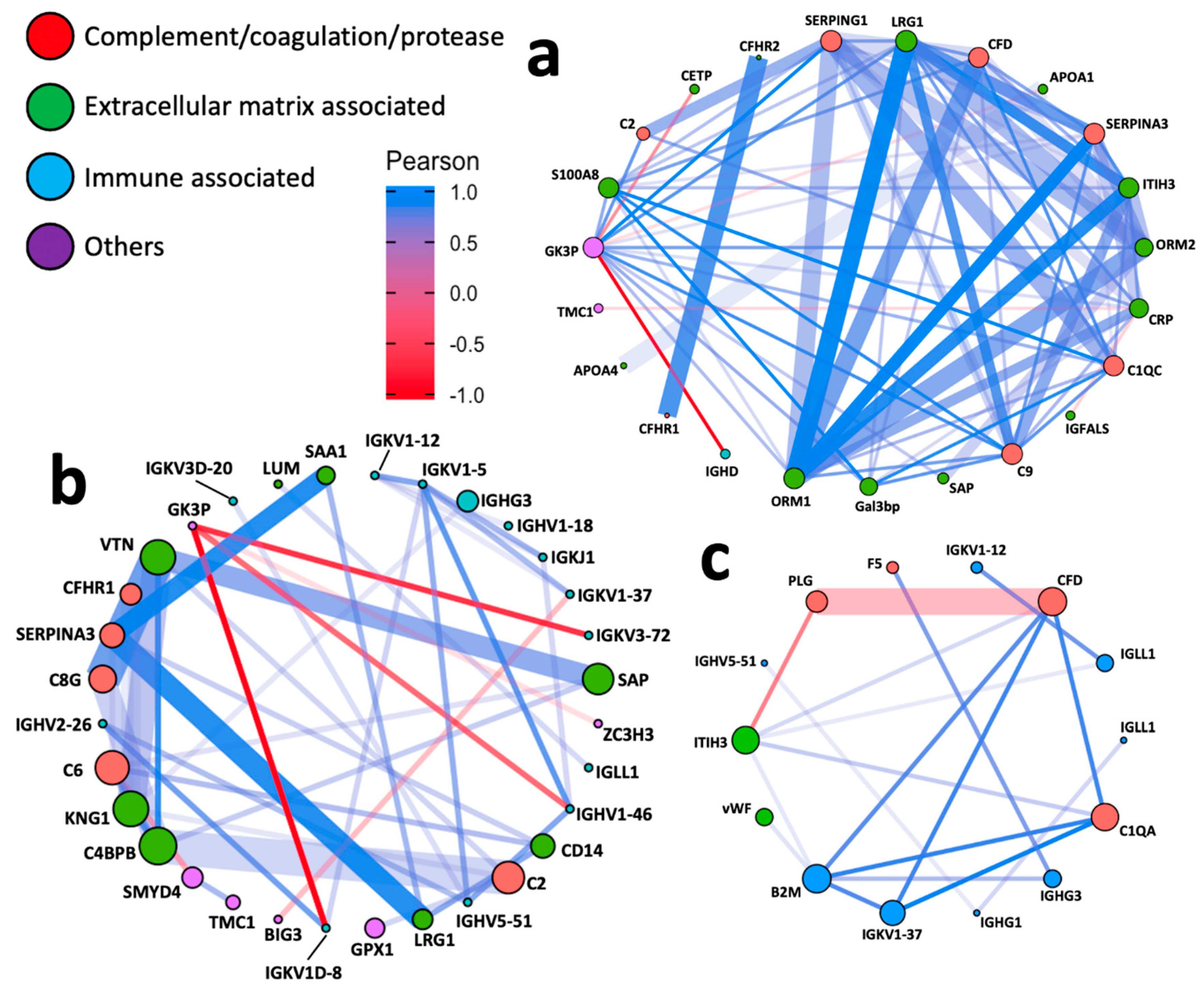
3.4. Network Analyses
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Bhatia, R.S.; Tu, J.V.; Lee, D.S.; Austin, P.C.; Fang, J.; Haouzi, A.; Gong, Y.; Liu, P.P. Outcome of heart failure with preserved ejection fraction in a population-based study. N. Engl. J. Med. 2006, 355, 260–269. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Benjamin, E.J.; Levy, D.; Anderson, K.M.; Wolf, P.A.; Plehn, J.F.; Evans, J.C.; Comai, K.; Fuller, D.L.; St. John Sutton, M. Determinants of Doppler indexes of left ventricular diastolic function in normal subjects (the Framingham Heart Study). Am. J. Cardiol. 1992, 70, 508–515. [Google Scholar] [CrossRef]
- Solomon, S.D.; McMurray, J.J.V.; Anand, I.S.; Ge, J.; Lam, C.S.P.; Maggioni, A.P.; Martinez, F.; Packer, M.; Pfeffer, M.A.; Pieske, B.; et al. Angiotensin-Neprilysin inhibition in heart failure with preserved ejection fraction. N. Engl. J. Med. 2019, 381, 1609–1620. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Neeland, I.J.; Drazner, M.H.; Berry, J.D.; Ayers, C.R.; deFilippi, C.; Seliger, S.L.; Nambi, V.; McGuire, D.K.; Omland, T.; de Lemos, J.A. Biomarkers of chronic cardiac injury and hemodynamic stress identify a malignant phenotype of left ventricular hypertrophy in the general population. J. Am. Coll. Cardiol. 2013, 61, 187–195. [Google Scholar] [CrossRef] [Green Version]
- Shah, S.J.; Katz, D.H.; Deo, R.C. Phenotypic spectrum of heart failure with preserved ejection fraction. Heart Fail. Clin. 2014, 10, 407–418. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Farmakis, D.; Papingiotis, G.; Parissis, J.; Filippatos, G. Ups and downs in heart failure: The case of proteomics. Eur. J. Heart Fail. 2018, 20, 63–66. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, C.Y.; Caporizzo, M.A.; Bedi, K.; Vite, A.; Bogush, A.I.; Robison, P.; Heffler, J.G.; Salomon, A.K.; Kelly, N.A.; Babu, A.; et al. Suppression of detyrosinated microtubules improves cardiomyocyte function in human heart failure. Nat. Med. 2018, 24, 1225–1233. [Google Scholar] [CrossRef]
- Raphael, R.; Purushotham, D.; Gastonguay, C.; Chesnik, M.A.; Kwok, W.M.; Wu, H.E.; Shah, S.J.; Mirza, S.P.; Strande, J.L. Combining patient proteomics and in vitro cardiomyocyte phenotype testing to identify potential mediators of heart failure with preserved ejection fraction. J. Transl. Med. 2016, 14, 18. [Google Scholar] [CrossRef] [Green Version]
- Ponikowski, P.; Voors, A.A.; Anker, S.D.; Bueno, H.; Cleland, J.G.F.; Coats, A.J.S.; Falk, V.; González-Juanatey, J.R.; Harjola, V.-P.; Jankowska, E.A.; et al. 2016 ESC Guidelines for the diagnosis and treatment of acute and chronic heart failure: The Task Force for the diagnosis and treatment of acute and chronic heart failure of the European Society of Cardiology (ESC). Developed with the special contribution of the Heart Failure Association (HFA) of the ESC. Eur. Heart J. 2016, 37, 2129–2200. [Google Scholar]
- Harney, D.J.; Hutchison, A.T.; Hatchwell, L.; Humphrey, S.J.; James, D.E.; Hocking, S.; Heilbronn, L.K.; Larance, M. Proteomic analysis of human plasma during intermittent fasting. J. Proteome Res. 2019, 18, 2228–2240. [Google Scholar] [CrossRef]
- O’Rourke, M.B.; Sahni, S.; Samra, J.; Mittal, A.; Molloy, M.P. Data independent acquisition of plasma biomarkers of response to neoadjuvant chemotherapy in pancreatic ductal adenocarcinoma. J. Proteom. 2021, 231, 103998. [Google Scholar] [CrossRef] [PubMed]
- Roediger, B.; Lee, Q.; Tikoo, S.; Cobbin, J.C.A.; Henderson, J.M.; Jormakka, M.; O’Rourke, M.B.; Padula, M.P.; Pinello, N.; Henry, M.; et al. An atypical parvovirus driving chronic tubulointerstitial nephropathy and kidney fibrosis. Cell 2018, 175, 530–543. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ritchie, M.E.; Phipson, B.; Wu, D.; Hu, Y.; Law, C.W.; Shi, W.; Smyth, G.K. Limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 2015, 43, e47. [Google Scholar] [CrossRef]
- Croft, D.; O’Kelly, G.; Wu, G.; Haw, R.; Gillespie, M.; Matthews, L.; Caudy, M.; Garapati, P.; Gopinath, G.; Jassal, B.; et al. Reactome: A database of reactions, pathways and biological processes. Nucleic Acids Res. 2011, 39, D691–D697. [Google Scholar] [CrossRef] [Green Version]
- Szklarczyk, D.; Gable, A.L.; Lyon, D.; Junge, A.; Wyder, S.; Huerta-Cepas, J.; Simonovic, M.; Doncheva, N.T.; Morris, J.H.; Bork, P.; et al. STRING v11: Protein-protein association networks with increased coverage, supporting functional discovery in genome-wide experimental datasets. Nucleic Acids Res. 2019, 47, D607–D613. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, D.W.; Sherman, B.T.; Lempicki, R. A Bioinformatics enrichment tools: Paths toward the comprehensive functional analysis of large gene lists. Nucleic Acids Res. 2009, 37, 1–13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, D.W.; Sherman, B.T.; Lempicki, R. A Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat. Protoc. 2009, 4, 44–57. [Google Scholar] [CrossRef]
- Andersen, L.-A.C.; Palstrøm, N.B.; Diederichsen, A.; Lindholt, J.S.; Rasmussen, L.M.; Beck, H.C. Determining Plasma Protein Variation Parameters as a Prerequisite for Biomarker Studies—A TMT-Based LC-MSMS Proteome Investigation. Proteomes 2021, 9, 47. [Google Scholar] [CrossRef]
- Wang, X.; Abraham, S.; McKenzie, J.A.G.; Jeffs, N.; Swire, M.; Tripathi, V.B.; Luhmann, U.F.O.; Lange, C.A.K.; Zhai, Z.; Arthur, H.M.; et al. LRG1 promotes angiogenesis by modulating endothelial TGF-ß signalling. Nature 2013, 499, 306–311. [Google Scholar] [CrossRef] [Green Version]
- Li, M.; Parker, B.L.; Pearson, E.; Hunter, B.; Cao, J.; Koay, Y.C.; Guneratne, O.; James, D.E.; Yang, J.; Lal, S.; et al. Core function nodes and sex-specific pathways in human ischaemic and dilated cardiomyopathy. Nat. Commun. 2020, 11, 2843. [Google Scholar] [CrossRef]
- Oberbach, A.; Adams, V.; Schlichting, N.; Heinrich, M.; Kullnick, Y.; Lehmann, S.; Lehmann, S.; Feder, S.; Correia, J.C.; Mohr, F.-W.; et al. Proteome profiles of HDL particles of patients with chronic heart failure are associated with immune response and also include bacteria proteins. Clin. Chim. Acta 2016, 453, 114–122. [Google Scholar] [CrossRef] [PubMed]
- Cleland, J.G.F.; Bunting, K.V.; Flather, M.D.; Altman, D.G.; Holmes, J.; Coats, A.J.S.; Manzano, L.; McMurray, J.J.V.; Ruschitzka, F.; van Veldhuisen, D.J.; et al. Beta-blockers for heart failure with reduced, mid-range, and preserved ejection fraction: An individual patient-level analysis of double-blind randomized trials. Eur. Heart J. 2018, 39, 26–35. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Loffredo, F.S.; Steinhauser, M.L.; Jay, S.M.; Gannon, J.; Pancoast, J.R.; Yalamanchi, P.; Sinha, M.; Dall’Osso, C.; Khong, D.; Shadrach, J.L.; et al. Growth differentiation factor 11 is a circulating factor that reverses age-related cardiac hypertrophy. Cell 2013, 153, 828–839. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Petri, B.; Broermann, A.; Li, H.; Khandoga, A.G.; Zarbock, A.; Krombach, F.; Goerge, T.; Schneider, S.W.; Jones, C.; Nieswandt, B.; et al. Von Willebrand factor promotes leukocyte extravasation. Blood 2010, 116, 4712–4719. [Google Scholar] [CrossRef]
- Kattula, S.; Byrnes, J.R.; Wolberg, A.S. Fibrinogen and fibrin in hemostasis and thrombosis. Arterioscler. Thromb. Vasc. Biol. 2017, 37, e13–e21. [Google Scholar] [CrossRef] [Green Version]
- Moreno, J.A.; Ortega-Gomez, A.; Rubio-Navarro, A.; Louedec, L.; Ho-Tin-Noe, B.; Caligiuri, G.; Nicoletti, A.; Levoye, A.; Plantier, L.; Meilhac, O. High-density lipoproteins potentiate α1-antitrypsin therapy in elastase-induced pulmonary emphysema. Am. J. Respir. Cell Mol. Biol. 2014, 51, 536–549. [Google Scholar] [CrossRef]
- Camici, P.G.; Crea, F. Coronary, Microvascular Dysfunction. N. Engl. J. Med. 2007, 356, 830–840. [Google Scholar] [CrossRef] [Green Version]
- Pries, A.R.; Reglin, B. Coronary microcirculatory pathophysiology: Can we afford it to remain a black box? Eur. Heart J. 2017, 38, 478–488. [Google Scholar] [CrossRef] [Green Version]
- Mohammed, S.F.; Borlaug, B.A.; Roger, V.L.; Mirzoyev, S.A.; Rodeheffer, R.J.; Chirinos, J.A.; Redfield, M.M. Comorbidity and ventricular and vascular structure and function in heart failure with preserved ejection fraction: A community-based study. Circ. Heart Fail. 2012, 5, 710–719. [Google Scholar] [CrossRef] [Green Version]
- Hwang, S.J.; Melenovsky, V.; Borlaug, B.A. Implications of coronary artery disease in heart failure with preserved ejection fraction. J. Am. Coll. Cardiol. 2014, 63, 2817–2827. [Google Scholar] [CrossRef] [Green Version]
- Mohammed, S.F.; Hussain, S.; Mirzoyev, S.A.; Edwards, W.D.; Maleszewski, J.J.; Redfield, M.M. Coronary microvascular rarefaction and myocardial fibrosis in heart failure with preserved ejection fraction. Circulation 2015, 131, 550–559. [Google Scholar] [CrossRef] [PubMed]
- Rao, V.N.; Zhao, D.; Allison, M.A.; Guallar, E.; Sharma, K.; Criqui, M.H.; Cushman, M.; Blumenthal, R.S.; Michos, E.D. Adiposity and Incident Heart Failure and its Subtypes: MESA (Multi-Ethnic Study of Atherosclerosis). JACC Heart Fail. 2018, 6, 999–1007. [Google Scholar] [CrossRef] [PubMed]
- Serneri, G.G.N.; Boddi, M.; Cecioni, I.; Vanni, S.; Coppo, M.; Papa, M.L.; Bandinelli, B.; Bertolozzi, L.; Polidori, G.; Toscano, T.; et al. Cardiac angiotensin II formation in the clinical course of heart failure and its relationship with left ventricular function. Circ. Res. 2001, 88, 961–968. [Google Scholar] [CrossRef] [Green Version]
- Touyz, R.M.; Schiffrin, E.L. Effects of angiotensin II and endothelin-1 on platelet aggregation and cytosolic pH and free Ca2+ concentrations in essential hypertension. Hypertension 1993, 22, 853–862. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baker, P.N.; Kilby, M.D.; Pipkin, F.B. The effect of angiotensin II on platelet intracellular free calcium concentration in human pregnancy. J. Hypertens. 1992, 10, 55–60. [Google Scholar] [CrossRef]
- Bach, L.A. Endothelial cells and the IGF system. J. Mol. Endocrinol. 2015, 54, R1–R13. [Google Scholar] [CrossRef]
- Zeng, H.; Chen, J.X. Microvascular Rarefaction and Heart Failure with Preserved Ejection Fraction. Front. Cardiovasc. Med. 2019, 6, 15. [Google Scholar] [CrossRef] [Green Version]
- Watson, C.J.; Ledwidge, M.T.; Phelan, D.; Collier, P.; Byrne, J.; Dunn, M.J.; McDonald, K.M.; Baugh, J.A. Proteomic analysis of coronary sinus serum reveals leucine-rich α2-glycoprotein as a novel biomarker of ventricular dysfunction and heart failure. Circ. Heart Fail. 2011, 4, 188–197. [Google Scholar] [CrossRef] [Green Version]
- Gonzalez-Lopez, E.; Gallego-Delgado, M.; Guzzo-Merello, G.; de Haro-Del Moral, F.J.; Cobo-Marcos, M.; Robles, C.; Bornstein, B.; Salas, C.; Lara-Pezzi, E.; Alonso-Pulpon, L.; et al. Wild-type transthyretin amyloidosis as a cause of heart failure with preserved ejection fraction. Eur. Heart J. 2015, 36, 2585–2594. [Google Scholar] [CrossRef] [Green Version]
- Mohammed, S.F.; Mirzoyev, S.A.; Edwards, W.D.; Dogan, A.; Grogan, D.R.; Dunlay, S.M.; Roger, V.L.; Gertz, M.A.; Dispenzieri, A.; Zeldenrust, S.R.; et al. Left ventricular amyloid deposition in patients with heart failure and preserved ejection fraction. JACC Heart Fail. 2014, 2, 113–122. [Google Scholar] [CrossRef]
- Wilson, P.G.; Thompson, J.C.; Shridas, P.; McNamara, P.J.; de Beer, M.C.; de Beer, F.C.; Webb, N.R.; Tannock, L.R. Serum amyloid A is an exchangeable apolipoprotein. Arterioscler. Thromb. Vasc. Biol. 2018, 38, 1890–1900. [Google Scholar] [CrossRef] [PubMed]
- Tennent, G.A.; Lovat, L.B.; Pepys, M.B. Serum amyloid P component prevents proteolysis of the amyloid fibrils of Alzheimer disease and systemic amyloidosis. Proc. Natl. Acad. Sci. USA 1995, 92, 4299–4303. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Geyer, P.E.; Voytik, E.; Treit, P.V.; Doll, S.; Kleinhempel, A.; Niu, L.; Müller, J.B.; Buchholtz, M.-L.; Bader, J.M.; Teupser, D.; et al. Plasma Proteome Profiling to detect and avoid sample-related biases in biomarker studies. EMBO Mol. Med. 2019, 11, e10427. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Characteristics | Acute HFpEF (n = 8) | Chronic HFpEF (n = 9) | HCM (n = 14) |
|---|---|---|---|
| Age (years) | 73.1 ± 14.2 ¶ | 64.6 ± 10.6 | 51.2 ± 14.0 ¶ |
| Female (no. [%]) | 3 (37.5) | 3 (33.3) | 3 (21.4) |
| BMI (kg/m2) | 31.4 ± 4.8 | 28.0 ± 2.5 | 26.1 ± 4.1 |
| LVEF (%) | 55.4 ± 10.2 ¶ | 57.4 ± 8.5 | 64.4 ± 4.1 ¶ |
| NYHA class | II/III | I/II | I/II |
| Diabetes (no. %) | 4 (50.0) | 2 (22.2) | 0 (0) |
| NT-proBNP (pg/mL) | 15,417 ± 21,680 ¶ | 2266 ± 3032 | 3155 ± 3037 ¶ |
| Echocardiography measurement | |||
| LVEDD (mm) | 56.5 ± 11.5 | 52.8 ± 6.9 | 47.8 ± 5.6 |
| LVESD (mm) | 38.3 ± 10.4 ¶ | 35.3 ± 8.3 | 29.1 ± 4.3 ¶ |
| LAD (mm) | 48.9 ± 5.9 ¶& | 41.1 ± 3.6 & | 42.2 ± 6.6 ¶ |
| Medications | |||
| Statin (no. [%]) | 3 (37.5) | 6 (66.7) | 1 (7.1) |
| Beta-blocker (no. [%]) | 6 (75.0) | 9 (100.0) | 13 (92.9) |
| Calcium channel blocker (no. [%]) | 2 (25.0) | 3 (33.3) | 1 (7.1) |
| ACEi/ARB (no. [%]) | 5 (62.5) | 7 (77.8) | 4 (28.6) |
| Diuretic (no. [%]) | 4 (50.0) | 4 (44.4) | 4 (28.6) |
| Warfarin (no. [%]) | 1 (12.5) | 1 (11.1) | 0 (0) |
| Acetylsalicylic acid (no. [%]) | 4 (50.0) | 7 (77.8) | 1 (7.1) |
| Amiodarone (no. [%]) | 0 (0) | 0 (0) | 1 (7.1) |
| Isosorbide mononitrate (no. [%]) | 1 (12.5) | 3 (33.3) | 0 (0) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chen, H.; Tesic, M.; Nikolic, V.N.; Pavlovic, M.; Vucic, R.M.; Spasic, A.; Jovanovic, H.; Jovanovic, I.; Town, S.E.L.; Padula, M.P.; et al. Systemic Biomarkers and Unique Pathways in Different Phenotypes of Heart Failure with Preserved Ejection Fraction. Biomolecules 2022, 12, 1419. https://doi.org/10.3390/biom12101419
Chen H, Tesic M, Nikolic VN, Pavlovic M, Vucic RM, Spasic A, Jovanovic H, Jovanovic I, Town SEL, Padula MP, et al. Systemic Biomarkers and Unique Pathways in Different Phenotypes of Heart Failure with Preserved Ejection Fraction. Biomolecules. 2022; 12(10):1419. https://doi.org/10.3390/biom12101419
Chicago/Turabian StyleChen, Hao, Milorad Tesic, Valentina N. Nikolic, Milan Pavlovic, Rada M. Vucic, Ana Spasic, Hristina Jovanovic, Ivana Jovanovic, Stephanie E. L. Town, Matthew P. Padula, and et al. 2022. "Systemic Biomarkers and Unique Pathways in Different Phenotypes of Heart Failure with Preserved Ejection Fraction" Biomolecules 12, no. 10: 1419. https://doi.org/10.3390/biom12101419