
Metformin and Cancer Glucose Metabolism: At the Bench or at the Bedside?

 , ,
, , 
{kind=link}
{kind=link}
Abstract
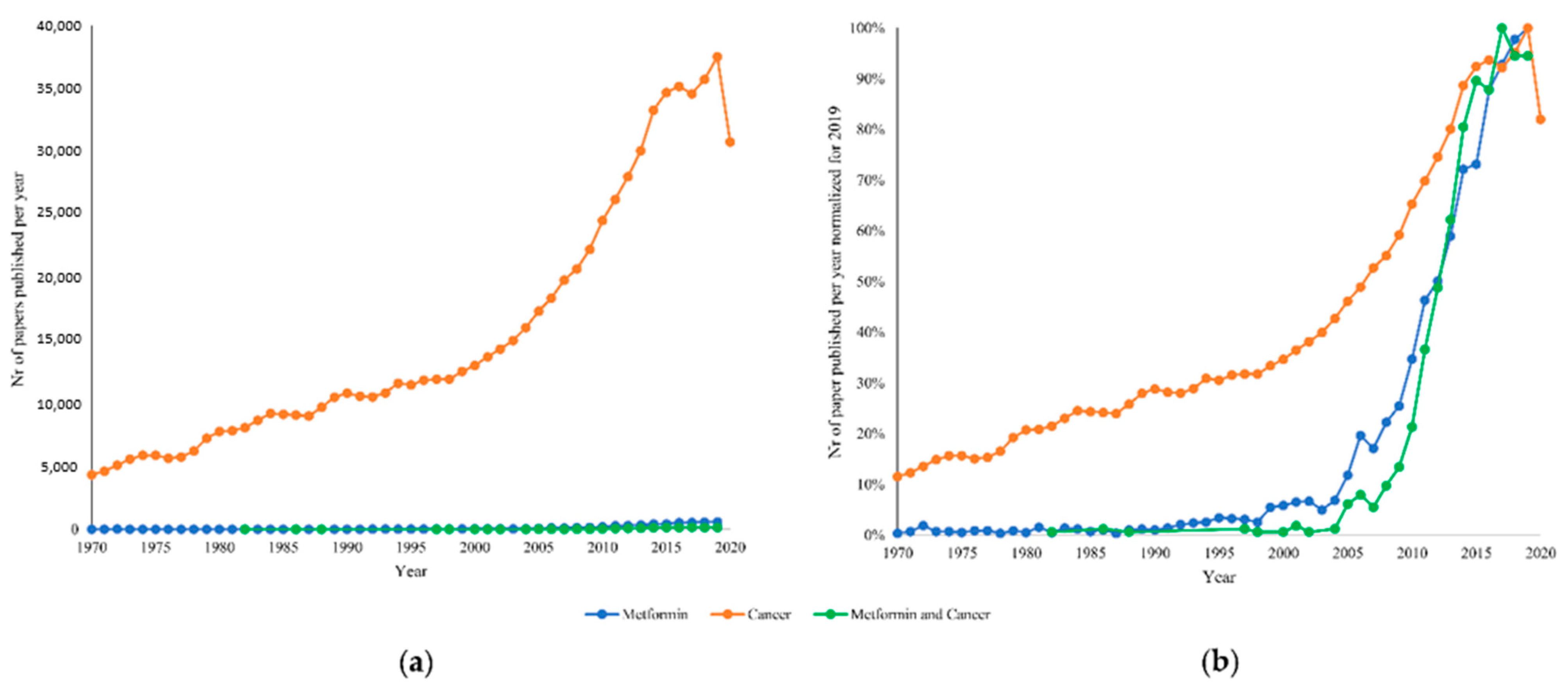
:1. Introduction
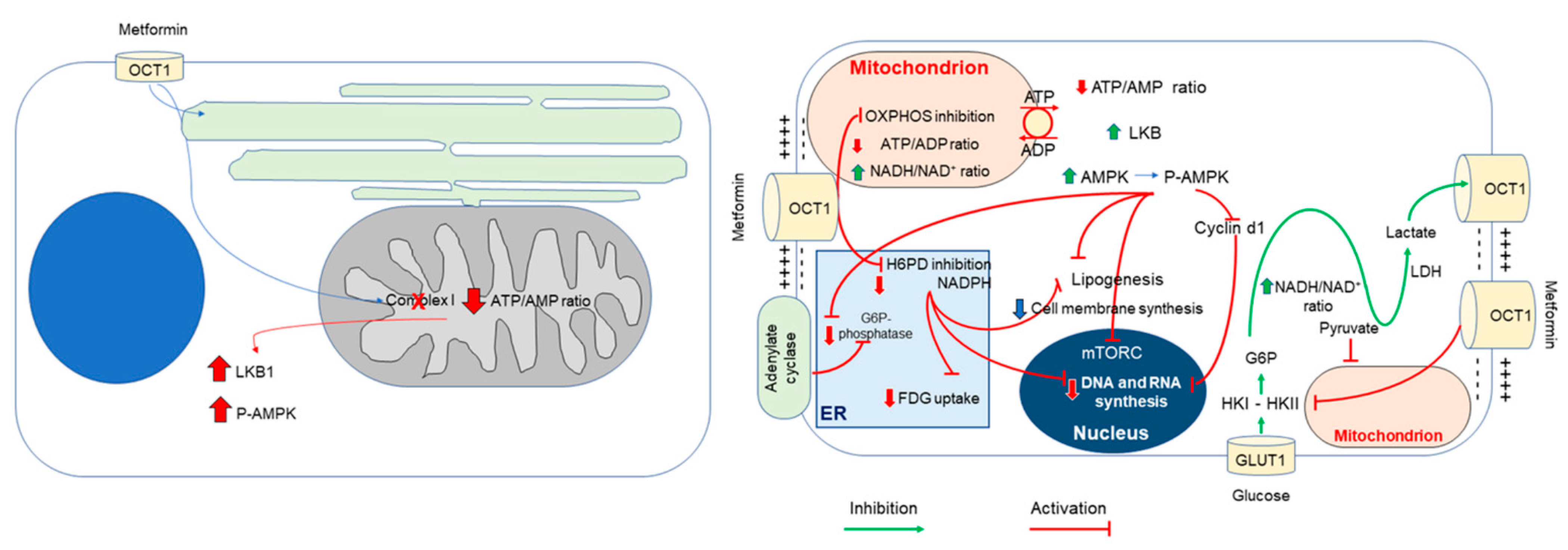
2. Effect of Metformin on Glucose Metabolism in Cancer
3. Metformin and Cancer Glucose Metabolism by Direct Inhibition of Hexokinase-II
4. Moving from the Bench to the Bedside: What Is Anticancer Potential of Metformin?
5. Moving from the Bench to the Bedside: Does Metformin Hamper the Clinical Accuracy of FDG Imaging?
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
References
- Foretz, M.; Hébrard, S.; Leclerc, J.; Zarrinpashneh, E.; Soty, M.; Mithieux, G.; Sakamoto, K.; Andreelli, F.; Viollet, B. Metformin inhibits hepatic gluconeogenesis in mice independently of the LKB1/AMPK pathway via a decrease in hepatic energy state. J. Clin. Investig. 2010, 120, 2355–2369. [Google Scholar] [CrossRef] [Green Version]
- Kim, Y.D.; Park, K.G.; Lee, Y.S.; Park, Y.Y.; Kim, D.K.; Nedumaran, B.; Jang, W.G.; Cho, W.J.; Ha, J.; Lee, I.K.; et al. Metformin inhibits hepatic gluconeogenesis through AMP-activated protein kinase-dependent regulation of the orphan nuclear receptor SHP. Diabetes 2008, 57, 306–314. [Google Scholar] [CrossRef] [Green Version]
- McIntyre, H.D.; Ma, A.; Bird, D.M.; Paterson, C.A.; Ravenscroft, P.J.; Cameron, D.P. Metformin increases insulin sensitivity and basal glucose clearance in type 2 (non-insulin dependent) diabetes mellitus. Aust. N. Z. J. Med. 1991, 21, 714–719. [Google Scholar] [CrossRef]
- Rena, G.; Hardie, D.G.; Pearson, E.R. The mechanisms of action of metformin. Diabetologia 2017, 60, 1577–1585. [Google Scholar] [CrossRef] [Green Version]
- Foretz, M.; Guigas, B.; Bertrand, L.; Pollak, M.; Viollet, B. Metformin: From mechanisms of action to therapies. Cell Metab. 2014, 20, 953–966. [Google Scholar] [CrossRef] [Green Version]
- Schulten, H.J. Pleiotropic Effects of Metformin on Cancer. Int. J. Mol. Sci. 2018, 19, 2850. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bridgeman, S.C.; Ellison, G.C.; Melton, P.E.; Newsholme, P.; Mamotte, C.D.S. Epigenetic effects of metformin: From molecular mechanisms to clinical implications. Diabetes Obes. Metab. 2018, 20, 1553–1562. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saraei, P.; Asadi, I.; Kakar, M.A.; Moradi-Kor, N. The beneficial effects of metformin on cancer prevention and therapy: A comprehensive review of recent advances. Cancer Manag. Res. 2019, 11, 3295–3313. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gandini, S.; Puntoni, M.; Heckman-Stoddard, B.M.; Dunn, B.K.; Ford, L.; DeCensi, A.; Szabo, E. Metformin and cancer risk and mortality: A systematic review and meta-analysis taking into account biases and confounders. Cancer Prev. Res. 2014, 7, 867–885. [Google Scholar] [CrossRef] [Green Version]
- Fontaine, E. Metformin-Induced Mitochondrial Complex I Inhibition: Facts, Uncertainties, and Consequences. Front. Endocrinol. 2018, 9, 753. [Google Scholar] [CrossRef]
- Bridges, H.R.; Jones, A.J.; Pollak, M.N.; Hirst, J. Effects of metformin and other biguanides on oxidative phosphorylation in mitochondria. Biochem. J. 2014, 462, 475–487. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- El-Mir, M.Y.; Nogueira, V.; Fontaine, E.; Avéret, N.; Rigoulet, M.; Leverve, X. Dimethylbiguanide inhibits cell respiration via an indirect effect targeted on the respiratory chain complex I. J. Biol. Chem. 2000, 275, 223–228. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Owen, M.R.; Doran, E.; Halestrap, A.P. Evidence that metformin exerts its anti-diabetic effects through inhibition of complex 1 of the mitochondrial respiratory chain. Biochem. J. 2000, 348, 607–614. [Google Scholar] [CrossRef]
- Muller, S.; Denet, S.; Candiloros, H.; Barrois, R.; Wiernsperger, N.; Donner, M.; Drouin, P. Action of metformin on erythrocyte membrane fluidity in vitro and in vivo. Eur. J. Pharmacol. 1997, 337, 103–110. [Google Scholar] [CrossRef]
- Shaw, R.J.; Lamia, K.A.; Vasquez, D.; Koo, S.H.; Bardeesy, N.; Depinho, R.A.; Montminy, M.; Cantley, L.C. The kinase LKB1 mediates glucose homeostasis in liver and therapeutic effects of metformin. Science 2005, 310, 1642–1646. [Google Scholar] [CrossRef] [Green Version]
- Hadad, S.M.; Hardie, D.G.; Appleyard, V.; Thompson, A.M. Effects of metformin on breast cancer cell proliferation, the AMPK pathway and the cell cycle. Clin. Transl. Oncol. 2014, 16, 746–752. [Google Scholar] [CrossRef]
- Zhou, G.; Myers, R.; Li, Y.; Chen, Y.; Shen, X.; Fenyk-Melody, J.; Wu, M.; Ventre, J.; Doebber, T.; Fujii, N.; et al. Role of AMP-activated protein kinase in mechanism of metformin action. J. Clin. Investig. 2001, 108, 1167–1174. [Google Scholar] [CrossRef]
- Baur, J.A.; Birnbaum, M.J. Control of gluconeogenesis by metformin: Does redox trump energy charge? Cell Metab. 2014, 20, 197–199. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miller, R.A.; Chu, Q.; Xie, J.; Foretz, M.; Viollet, B.; Birnbaum, M.J. Biguanides suppress hepatic glucagon signalling by decreasing pro-duction of cyclic AMP. Nature 2013, 494, 256–260. [Google Scholar] [CrossRef] [Green Version]
- Foretz, M.; Guigas, B.; Viollet, B. Understanding the glucoregulatory mechanisms of metformin in type 2 diabetes mellitus. Nat. Rev. Endocrinol. 2019, 15, 569–589. [Google Scholar] [CrossRef] [Green Version]
- Sengupta, S.; Peterson, T.R.; Sabatini, D.M. Regulation of the mTOR complex 1 pathway by nutrients, growth factors, and stress. Mol. Cell 2010, 40, 310–322. [Google Scholar] [CrossRef] [Green Version]
- Hindupur, S.K.; González, A.; Hall, M.N. The opposing actions of target of rapamycin and AMP-activated protein kinase in cell growth control. Cold Spring Harb. Perspect. Biol. 2015, 7, a019141. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Inoki, K.; Ouyang, H.; Zhu, T.; Lindvall, C.; Wang, Y.; Zhang, X.; Yang, Q.; Bennett, C.; Harada, Y.; Stankunas, K.; et al. TSC2 integrates Wnt and energy signals via a coordinated phosphorylation by AMPK and GSK3 to regulate cell growth. Cell 2006, 126, 955–968. [Google Scholar] [CrossRef] [Green Version]
- Zakikhani, M.; Dowling, R.; Fantus, I.G.; Sonenberg, N.; Pollak, M. Metformin is an AMP kinase-dependent growth inhibitor for breast cancer cells. Cancer Res. 2006, 66, 10269–10273. [Google Scholar] [CrossRef] [Green Version]
- Vakana, E.; Altman, J.K.; Glaser, H.; Donato, N.J.; Platanias, L.C. Antileukemic effects of AMPK activators on BCR-ABL-expressing cells. Blood 2011, 118, 6399–6402. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Faubert, B.; Vincent, E.E.; Poffenberger, M.C.; Jones, R.G. The AMP-activated protein kinase (AMPK) and cancer: Many faces of a metabolic regulator. Cancer Lett. 2015, 356, 165–170. [Google Scholar] [CrossRef] [PubMed]
- Wu, D.; Hu, D.; Chen, H.; Shi, G.; Fetahu, I.S.; Wu, F.; Rabidou, K.; Fang, R.; Tan, L.; Xu, S.; et al. Glucose-regulated phosphorylation of TET2 by AMPK reveals a pathway linking diabetes to cancer. Nature 2018, 559, 637–641. [Google Scholar] [CrossRef] [PubMed]
- Zoncu, R.; Efeyan, A.; Sabatini, D.M. mTOR: From growth signal integration to cancer, diabetes and ageing. Nat. Rev. Mol. Cell Biol. 2011, 12, 21–35. [Google Scholar] [CrossRef] [Green Version]
- Pollak, M.N. Investigating metformin for cancer prevention and treatment: The end of the beginning. Cancer Discov. 2012, 2, 778–790. [Google Scholar] [CrossRef] [Green Version]
- Koppenol, W.H.; Bounds, P.L.; Dang, C.V. Otto Warburg’s contributions to current concepts of cancer metabolism. Nat. Rev. Cancer 2011, 11, 618. [Google Scholar] [CrossRef] [Green Version]
- Vander Heiden, M.G.; Cantley, L.C.; Thompson, C.B. Understanding the Warburg effect: The metabolic requirements of cell proliferation. Science 2009, 324, 1029–1033. [Google Scholar] [CrossRef] [Green Version]
- Wilson, J.E. Isozymes of mammalian hexokinase: Structure, subcellular localization and metabolic function. J. Exp. Biol. 2003, 206, 2049–2057. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pastorino, J.G.; Hoek, J.B. Hexokinase II: The integration of energy metabolism and control of apoptosis. Curr. Med. Chem. 2003, 10, 1535–1551. [Google Scholar] [CrossRef]
- Mathupala, S.P.; Ko, Y.H.; Pedersen, P.L. Hexokinase-2 bound to mitochondria: Cancer’s stygian link to the “Warburg Effect” and a pivotal target for effective therapy. Semin. Cancer Biol. 2009, 19, 17–24. [Google Scholar] [CrossRef] [Green Version]
- Salani, B.; Marini, C.; Rio, A.D.; Ravera, S.; Massollo, M.; Orengo, A.M.; Amaro, A.; Passalacqua, M.; Maffioli, S.; Pfeffer, U.; et al. Metformin impairs glucose consumption and survival in Calu-1 cells by direct inhibition of hexokinase-II. Sci. Rep. 2013, 3, 2070. [Google Scholar] [CrossRef]
- Storozhuk, Y.; Hopmans, S.N.; Sanli, T.; Barron, C.; Tsiani, E.; Cutz, J.C.; Pond, G.; Wright, J.; Singh, G.; Tsakiridis, T. Metformin inhibits growth and enhances radiation response of non-small cell lung cancer (NSCLC) through ATM and AMPK. Br. J. Cancer 2013, 108, 2021–2032. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Coyle, C.; Cafferty, F.H.; Vale, C.; Langley, R.E. Metformin as an adjuvant treatment for cancer: A systematic review and meta-analysis. Ann. Oncol. 2016, 27, 2184–2195. [Google Scholar] [CrossRef]
- Marini, C.; Ravera, S.; Buschiazzo, A.; Bianchi, G.; Orengo, A.M.; Bruno, S.; Bottoni, G.; Emionite, L.; Pastorino, F.; Monteverde, E.; et al. Discovery of a novel glucose metabolism in cancer: The role of endoplasmic reticulum beyond glycolysis and pentose phosphate shunt. Sci. Rep. 2016, 6, 25092. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mashhedi, H.; Blouin, M.J.; Zakikhani, M.; David, S.; Zhao, Y.; Bazile, M.; Birman, E.; Algire, C.; Aliaga, A.; Bedell, B.J.; et al. Metformin abolishes increased tumor (18)F-2-fluoro-2-deoxy-D-glucose uptake associated with a high energy diet. Cell Cycle 2011, 10, 2770–2778. [Google Scholar] [CrossRef] [Green Version]
- Cossu, V.; Marini, C.; Piccioli, P.; Rocchi, A.; Bruno, S.; Orengo, A.M.; Emionite, L.; Bauckneht, M.; Grillo, F.; Capitanio, S.; et al. Obligatory role of endoplasmic reticulum in brain FDG uptake. Eur. J. Nucl. Med. Mol. Imaging 2019, 46, 1184–1196. [Google Scholar] [CrossRef]
- Cossu, V.; Bonanomi, M.; Bauckneht, M.; Ravera, S.; Righi, N.; Miceli, A.; Morbelli, S.; Orengo, A.M.; Piccioli, P.; Bruno, S.; et al. Two high-rate pentose-phosphate pathways in cancer cells. Sci. Rep. 2020, 10, 22111. [Google Scholar] [CrossRef]
- Gormsen, L.C.; Sundelin, E.I.; Jensen, J.B.; Vendelbo, M.H.; Jakobsen, S.; Munk, O.L.; Hougaard Christensen, M.M.; Brøsen, K.; Frøkiær, J.; Jessen, N. In Vivo Imaging of Human 11C-Metformin in Peripheral Organs: Dosimetry, Biodistribution, and Kinetic Analyses. J. Nucl. Med. Off. Publ. Soc. Nucl. Med. 2016, 57, 1920–1926. [Google Scholar] [CrossRef] [Green Version]
- Graham, G.G.; Punt, J.; Arora, M.; Day, R.O.; Doogue, M.P.; Duong, J.K.; Furlong, T.J.; Greenfield, J.R.; Greenup, L.C.; Kirkpatrick, C.M.; et al. Clinical pharmacokinetics of metformin. Clin. Pharmacokinet. 2011, 50, 81–98. [Google Scholar] [CrossRef] [PubMed]
- Bonora, E.; Cigolini, M.; Bosello, O.; Zancanaro, C.; Capretti, L.; Zavaroni, I.; Coscelli, C.; Butturini, U. Lack of effect of intravenous metformin on plasma concentrations of glucose, insulin, C-peptide, glucagon and growth hormone in non-diabetic subjects. Curr. Med. Res. Opin. 1984, 9, 47–51. [Google Scholar] [CrossRef]
- Ng, C.W.; Jiang, A.A.; Toh, E.; Ng, C.H.; Ong, Z.H.; Peng, S.; Tham, H.Y.; Sundar, R.; Chong, C.S.; Khoo, C.M. Metformin and colorectal cancer: A systematic review, meta-analysis and meta-regression. Int. J. Colorectal Dis. 2020, 35, 1501–1512. [Google Scholar] [CrossRef]
- Jones, V.C.; Dietze, E.C.; Jovanovic-Talisman, T.; McCune, J.S.; Seewaldt, V.L. Metformin and Chemoprevention: Potential for Heart-Healthy Targeting of Biologically Aggressive Breast Cancer. Front. Public Health 2020, 8, 509714. [Google Scholar] [CrossRef] [PubMed]
- Xiao, K.; Liu, F.; Liu, J.; Xu, J.; Wu, Q.; Li, X. The effect of metformin on lung cancer risk and survival in patients with type 2 diabetes mellitus: A meta-analysis. J. Clin. Pharm. Ther. 2020, 45, 783–792. [Google Scholar] [CrossRef] [PubMed]
- Ahn, H.K.; Lee, Y.H.; Koo, K.C. Current Status and Application of Metformin for Prostate Cancer: A Comprehensive Review. Int. J. Mol. Sci. 2020, 21, 8540. [Google Scholar] [CrossRef]
- Buzzai, M.; Jones, R.G.; Amaravadi, R.K.; Lum, J.J.; DeBerardinis, R.J.; Zhao, F.; Viollet, B.; Thompson, C.B. Systemic treatment with the antidiabetic drug metformin selectively impairs p53-deficient tumor cell growth. Cancer Res. 2007, 67, 6745–6752. [Google Scholar] [CrossRef] [Green Version]
- Sokoloff, L.; Reivich, M.; Kennedy, C.; Des Rosiers, M.H.; Patlak, C.S.; Pettigrew, K.D.; Sakurada, O.; Shinohara, M. The [14C]deoxyglucose method for the measurement of local cerebral glucose utilization: Theory, procedure, and normal values in the conscious and anesthetized albino rat. J. Neurochem. 1977, 28, 897–916. [Google Scholar] [CrossRef]
- Phelps, M.E.; Huang, S.C.; Hoffman, E.J.; Selin, C.; Sokoloff, L.; Kuhl, D.E. Tomographic measurement of local cerebral glucose metabolic rate in humans with (F-18)2-fluoro-2-deoxy-D-glucose: Validation of method. Ann. Neurol. 1979, 6, 371–388. [Google Scholar] [CrossRef] [PubMed]
- Bachelard, H.S. Specificity and kinetic properties of monosaccharide uptake into guinea pig cerebral cortex in vitro. J. Neurochem. 1971, 18, 213–222. [Google Scholar] [CrossRef]
- Sols, A.; Crane, R.K. Substrate specificity of brain hexokinase. J. Biol. Chem. 1954, 210, 581–595. [Google Scholar] [CrossRef]
- Fathinul, F.; Nordin, A.J.; Lau, W.F. 18[F]FDG-PET/CT is a useful molecular marker in evaluating tumour aggressiveness: A revised understanding of an in-vivo FDG-PET imaging that alludes the alteration of cancer biology. Cell Biochem. Biophys. 2013, 66, 37–43. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Southworth, R.; Parry, C.R.; Parkes, H.G.; Medina, R.A.; Garlick, P.B. Tissue-specific differences in 2-fluoro-2-deoxyglucose metabolism beyond FDG-6-P: A 19F NMR spectroscopy study in the rat. NMR Biomed. 2003, 16, 494–502. [Google Scholar] [CrossRef] [PubMed]
- Foster, J.D.; Pederson, B.A.; Nordlie, R.C. Glucose-6-phosphatase structure, regulation, and function: An update. Proc. Soc. Exp. Biol. Med. 1997, 215, 314–332. [Google Scholar] [CrossRef] [PubMed]
- Csala, M.; Bánhegyi, G.; Benedetti, A. Endoplasmic reticulum: A metabolic compartment. FEBS Lett. 2006, 580, 2160–2165. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bánhegyi, G.; Benedetti, A.; Fulceri, R.; Senesi, S. Cooperativity between 11beta-hydroxysteroid dehydrogenase type 1 and hexose-6-phosphate dehydrogenase in the lumen of the endoplasmic reticulum. J. Biol. Chem. 2004, 279, 27017–27021. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stincone, A.; Prigione, A.; Cramer, T.; Wamelink, M.M.; Campbell, K.; Cheung, E.; Olin-Sandoval, V.; Grüning, N.M.; Krüger, A.; Tauqeer Alam, M.; et al. The return of metabolism: Biochemistry and physiology of the pentose phosphate pathway. Biol. Rev. Camb. Philos. Soc. 2015, 90, 927–963. [Google Scholar] [CrossRef] [Green Version]
- Jiang, P.; Du, W.; Wu, M. Regulation of the pentose phosphate pathway in cancer. Protein Cell 2014, 5, 592–602. [Google Scholar] [CrossRef] [Green Version]
- Boellaard, R. Standards for PET image acquisition and quantitative data analysis. J. Nucl. Med. 2009, 50, 11–20. [Google Scholar] [CrossRef] [Green Version]
- Cossu, V.; Bauckneht, M.; Bruno, S.; Orengo, A.M.; Emionite, L.; Balza, E.; Castellani, P.; Piccioli, P.; Miceli, A.; Raffa, S.; et al. The Elusive Link Between Cancer FDG Uptake and Glycolytic Flux Explains the Preserved Diagnostic Accuracy of PET/CT in Diabetes. Transl. Oncol. 2020, 13, 100752. [Google Scholar] [CrossRef]
- Zaafar, D.K.; Zaitone, S.A.; Moustafa, Y.M. Role of metformin in suppressing 1,2-dimethylhydrazine-induced colon cancer in diabetic and non-diabetic mice: Effect on tumor angiogenesis and cell proliferation. PLoS ONE 2014, 9, e100562. [Google Scholar] [CrossRef]
- Massollo, M.; Marini, C.; Brignone, M.; Emionite, L.; Salani, B.; Riondato, M.; Capitanio, S.; Fiz, F.; Democrito, A.; Amaro, A.; et al. Metformin temporal and localized effects on gut glucose metabolism assessed using 18F-FDG PET in mice. J. Nucl. Med. 2013, 54, 259–266. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Steenkamp, D.W.; McDonnell, M.E.; Meibom, S. Metformin may be associated with false-negative cancer detection in the gastrointestinal tract on PET/CT. Endocr. Pract. 2014, 20, 1079–1083. [Google Scholar] [CrossRef] [PubMed]
- Hamidizadeh, R.; Eftekhari, A.; Wiley, E.A.; Wilson, D.; Alden, T.; Bénard, F. Metformin Discontinuation prior to FDG PET/CT: A Randomized Controlled Study to Compare 24- and 48-hour Bowel Activity. Radiology 2018, 289, 418–425. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cho, S.Y. To Hold or Not to Hold Metformin for FDG PET Scans: That Is the Question. Radiology 2018, 289, 426–427. [Google Scholar] [CrossRef]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Marini, C.; Cossu, V.; Bauckneht, M.; Lanfranchi, F.; Raffa, S.; Orengo, A.M.; Ravera, S.; Bruno, S.; Sambuceti, G. Metformin and Cancer Glucose Metabolism: At the Bench or at the Bedside? Biomolecules 2021, 11, 1231. https://doi.org/10.3390/biom11081231
Marini C, Cossu V, Bauckneht M, Lanfranchi F, Raffa S, Orengo AM, Ravera S, Bruno S, Sambuceti G. Metformin and Cancer Glucose Metabolism: At the Bench or at the Bedside? Biomolecules. 2021; 11(8):1231. https://doi.org/10.3390/biom11081231
Chicago/Turabian StyleMarini, Cecilia, Vanessa Cossu, Matteo Bauckneht, Francesco Lanfranchi, Stefano Raffa, Anna Maria Orengo, Silvia Ravera, Silvia Bruno, and Gianmario Sambuceti. 2021. "Metformin and Cancer Glucose Metabolism: At the Bench or at the Bedside?" Biomolecules 11, no. 8: 1231. https://doi.org/10.3390/biom11081231