
Inhibition of CNOT2 Induces Apoptosis via MID1IP1 in Colorectal Cancer Cells by Activating p53



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cell Culture
2.2. RNA Interference and Plasmid
2.3. Cell Viability
2.4. Western Blot Analysis
2.5. p53 Stability Assay Using Cycloheximide
2.6. Statistical Analysis
3. Results
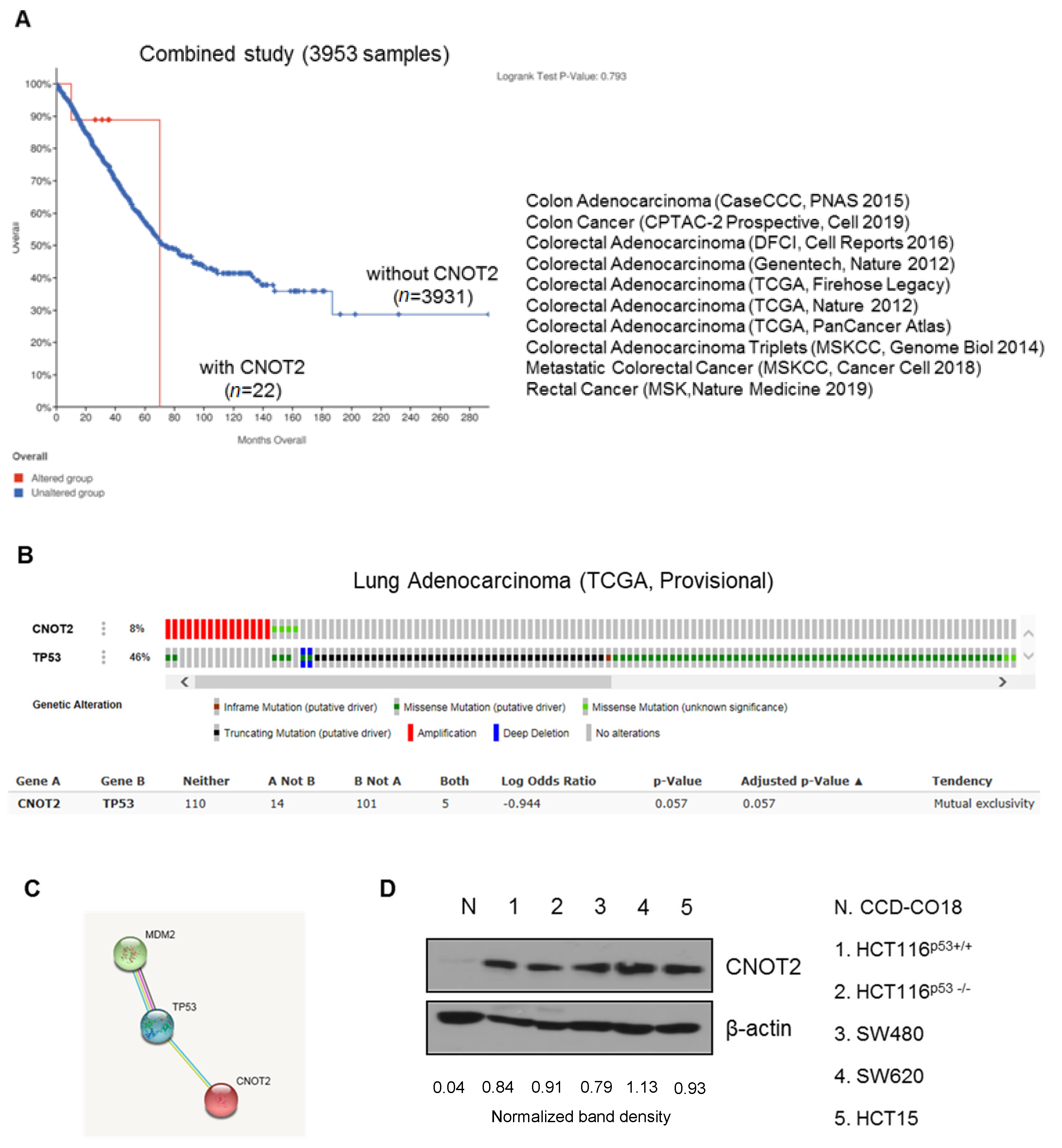
3.1. CNOT2 Is Overexpressed in Colon Cancer Patients and Cancer Cells
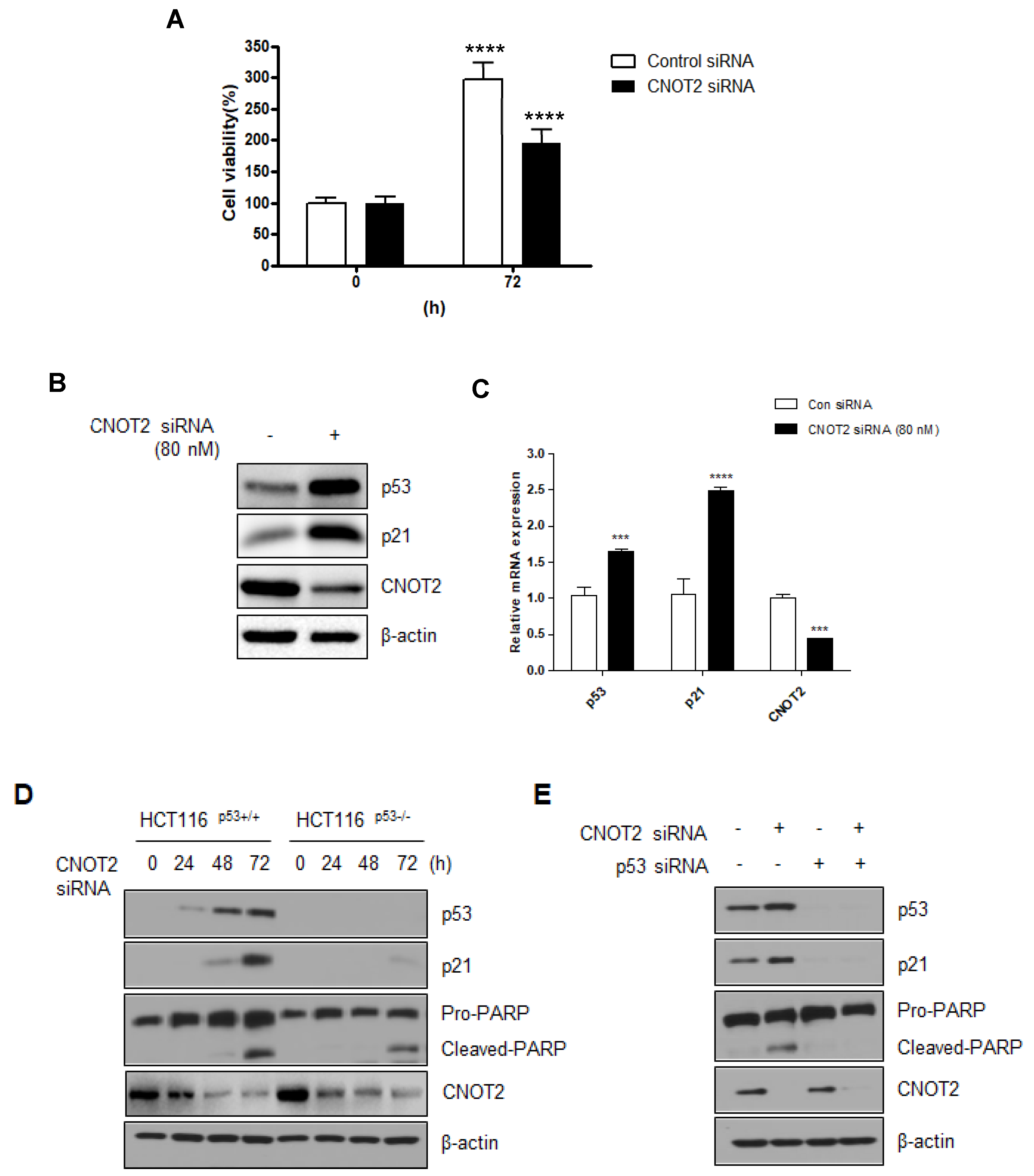
3.2. Knockdown of CNOT2 Induces Apoptosis in Cancer Cells by Activating p53
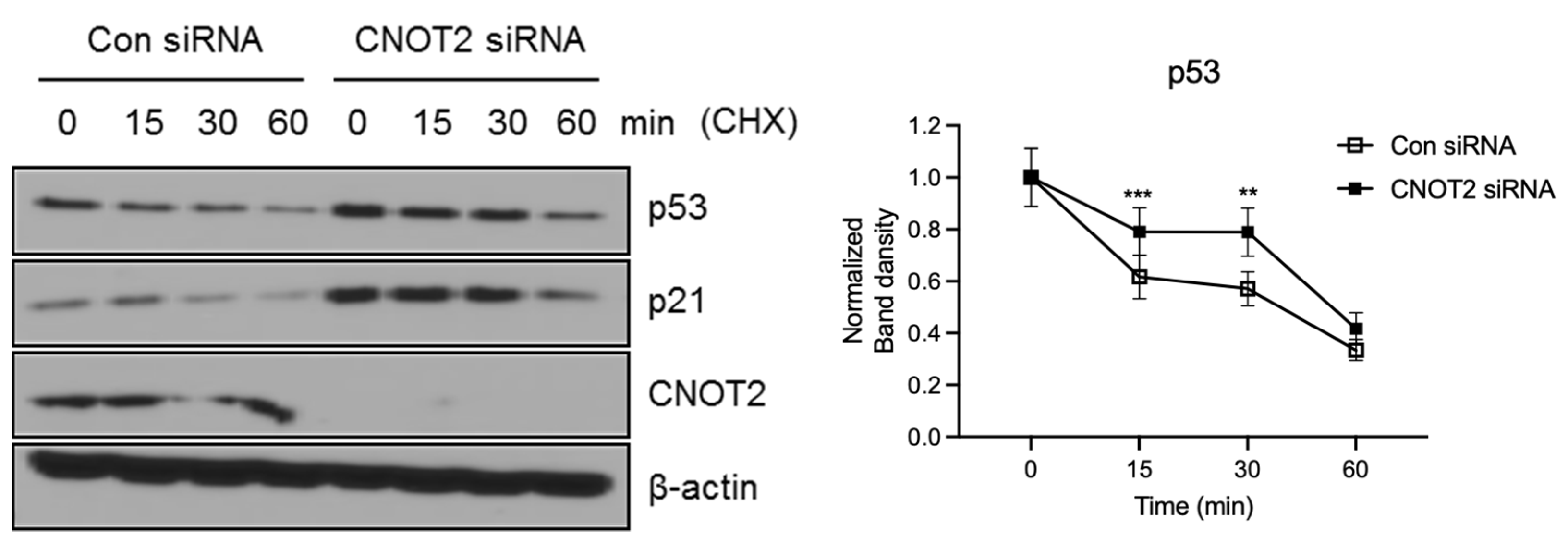
3.3. Knockdown of CNOT2 Induces p53 Protein Stability
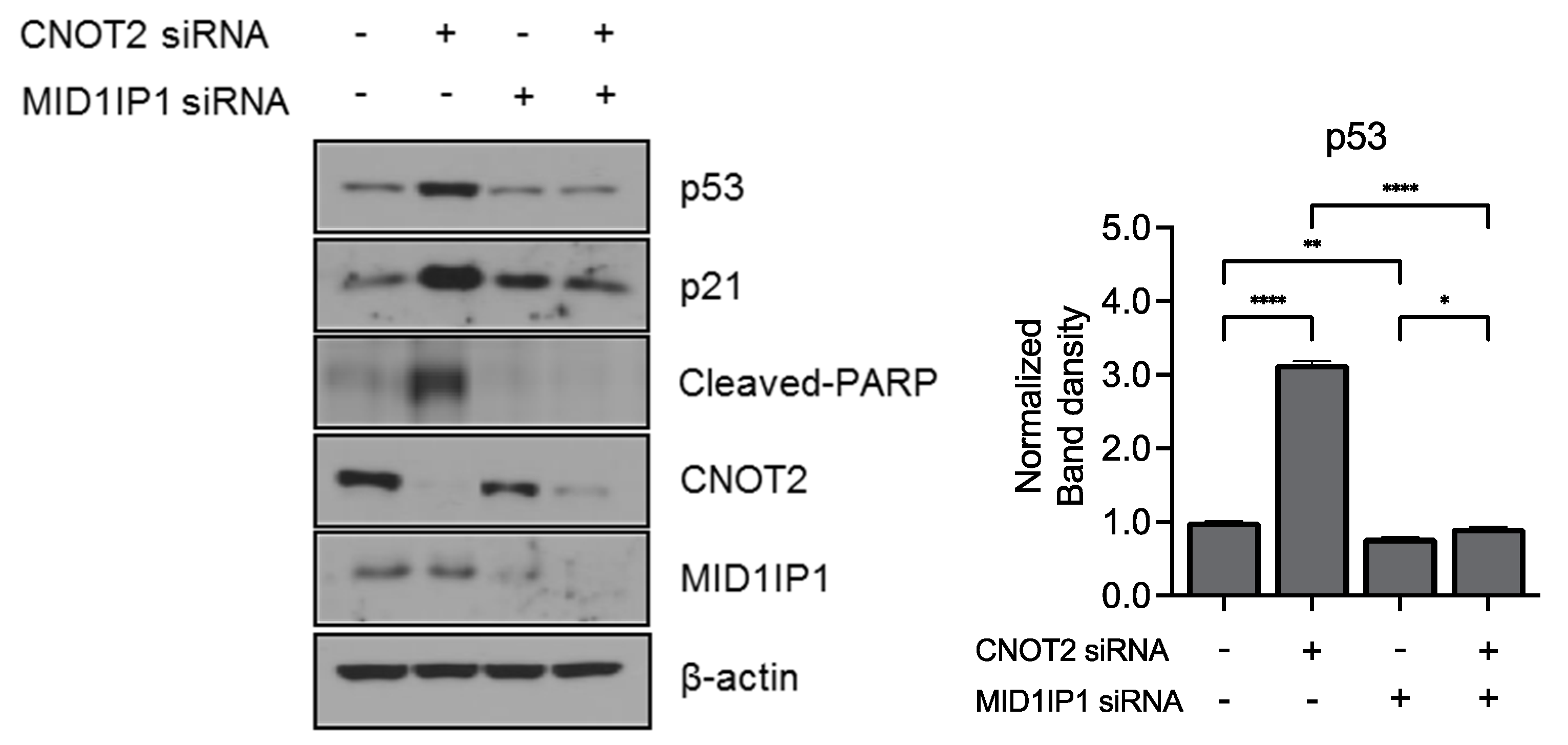
3.4. Knockdown of CNOT2 Enhances Anticancer Effect with MID1IP1
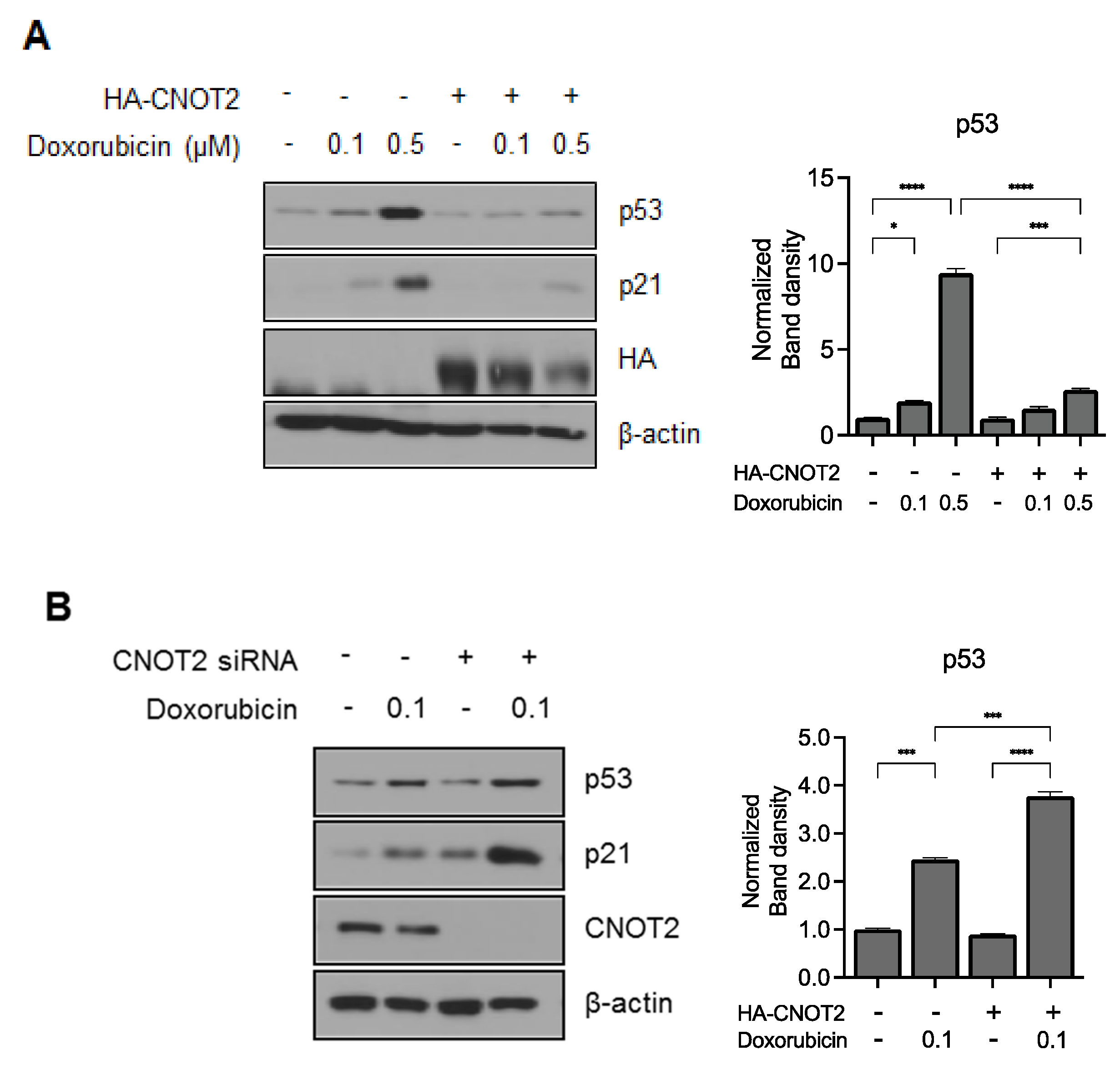
3.5. Knockdown of CNOT2 Potentiated p53 Expression by Doxorubicin in HCT116p53+/+ Cells
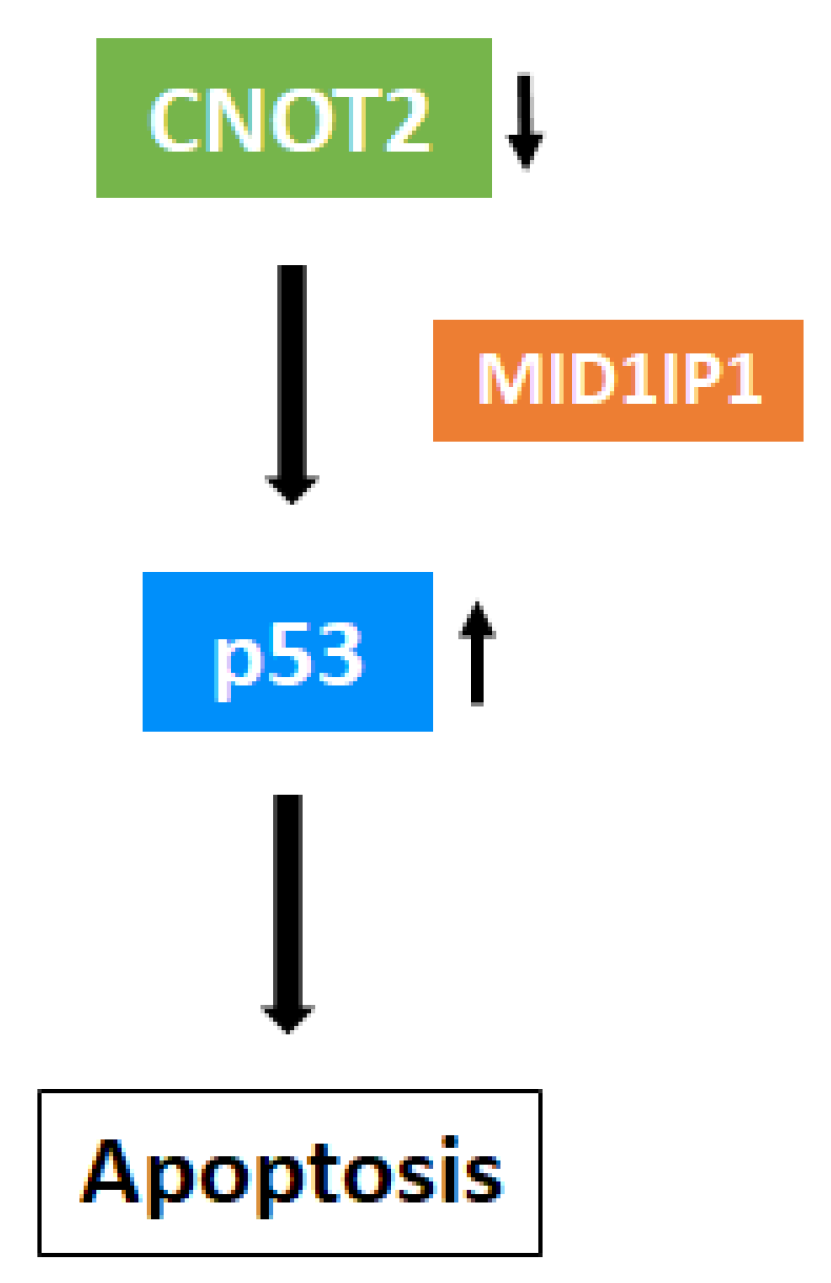
4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Ito, K.; Inoue, T.; Yokoyama, K.; Morita, M.; Suzuki, T.; Yamamoto, T. CNOT2 depletion disrupts and inhibits the CCR4-NOT deadenylase complex and induces apoptotic cell death. Genes Cells 2011, 16, 368–379. [Google Scholar] [CrossRef] [PubMed]
- Zwartjes, C.G.; Jayne, S.; van den Berg, D.L.; Timmers, H.T. Repression of promoter activity by CNOT2, a subunit of the transcription regulatory Ccr4-not complex. J. Biol. Chem. 2004, 279, 10848–10854. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chekulaeva, M.; Mathys, H.; Zipprich, J.T.; Attig, J.; Colic, M.; Parker, R.; Filipowicz, W. miRNA repression involves GW182-mediated recruitment of CCR4-NOT through conserved W-containing motifs. Nat. Struct. Mol. Biol. 2011, 18, 1218–1226. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rodriguez-Gil, A.; Ritter, O.; Saul, V.V.; Wilhelm, J.; Yang, C.Y.; Grosschedl, R.; Imai, Y.; Kuba, K.; Kracht, M.; Schmitz, M.L. The CCR4—NOT complex contributes to repression of Major Histocompatibility Complex class II transcription. Sci. Rep. 2017, 7, 3547. [Google Scholar] [CrossRef] [PubMed]
- Collart, M.A.; Panasenko, O.O. The Ccr4—Not complex. Gene 2012, 492, 42–53. [Google Scholar] [CrossRef] [PubMed]
- Jung, J.H.; Lee, H.J.; Kim, J.H.; Sim, D.Y.; Im, E.; Kim, S.; Chang, S.; Kim, S.H. Colocalization of MID1IP1 and c-Myc is Critically Involved in Liver Cancer Growth via Regulation of Ribosomal Protein L5 and L11 and CNOT2. Cells 2020, 9, 985. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, J.; Jung, J.H.; Hwang, J.; Park, J.E.; Kim, J.H.; Park, W.Y.; Suh, J.Y.; Kim, S.H. CNOT2 Is Critically Involved in Atorvastatin Induced Apoptotic and Autophagic Cell Death in Non-Small Cell Lung Cancers. Cancers 2019, 11, 1470. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sohn, E.J.; Jung, D.B.; Lee, H.; Han, I.; Lee, J.; Lee, H.; Kim, S.H. CNOT2 promotes proliferation and angiogenesis via VEGF signaling in MDA-MB-231 breast cancer cells. Cancer Lett. 2018, 412, 88–98. [Google Scholar] [CrossRef] [PubMed]
- Oren, M. Decision making by p53: Life, death and cancer. Cell Death Differ. 2003, 10, 431–442. [Google Scholar] [CrossRef] [PubMed]
- Fisher, D.E. The p53 tumor suppressor: Critical regulator of life & death in cancer. Apoptosis 2001, 6, 7–15. [Google Scholar] [CrossRef] [PubMed]
- Hao, Q.; Chen, Y.; Zhou, X. The Janus Face of p53-Targeting Ubiquitin Ligases. Cells 2020, 9, 1656. [Google Scholar] [CrossRef] [PubMed]
- Dai, M.S.; Shi, D.; Jin, Y.; Sun, X.X.; Zhang, Y.; Grossman, S.R.; Lu, H. Regulation of the MDM2-p53 pathway by ribosomal protein L11 involves a post-ubiquitination mechanism. J. Biol. Chem. 2006, 281, 24304–24313. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jung, J.H.; Lee, H.; Kim, J.H.; Sim, D.Y.; Ahn, H.; Kim, B.; Chang, S.; Kim, S.H. p53-Dependent Apoptotic Effect of Puromycin via Binding of Ribosomal Protein L5 and L11 to MDM2 and its Combination Effect with RITA or Doxorubicin. Cancers 2019, 11, 582. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dai, M.S.; Lu, H. Inhibition of MDM2-mediated p53 ubiquitination and degradation by ribosomal protein L5. J. Biol. Chem. 2004, 279, 44475–44482. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jung, J.H.; Lee, H.; Cao, B.; Liao, P.; Zeng, S.X.; Lu, H. RNA-binding motif protein 10 induces apoptosis and suppresses proliferation by activating p53. Oncogene 2020, 39, 1031–1040. [Google Scholar] [CrossRef] [PubMed]
- Xiong, S.; Xiao, G.W. Reverting doxorubicin resistance in colon cancer by targeting a key signaling protein, steroid receptor coactivator. Exp. Med. 2018, 15, 3751–3758. [Google Scholar] [CrossRef] [PubMed]
- Sonowal, H.; Pal, P.B.; Wen, J.J.; Awasthi, S.; Ramana, K.V.; Srivastava, S.K. Aldose reductase inhibitor increases doxorubicin-sensitivity of colon cancer cells and decreases cardiotoxicity. Sci. Rep. 2017, 7, 3182. [Google Scholar] [CrossRef] [PubMed]
- Chao, T.; Zhou, X.; Cao, B.; Liao, P.; Liu, H.; Chen, Y.; Park, H.W.; Zeng, S.X.; Lu, H. Pleckstrin homology domain-containing protein PHLDB3 supports cancer growth via a negative feedback loop involving p53. Nat. Commun. 2016, 7, 13755. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, X.; Hao, Q.; Liao, P.; Luo, S.; Zhang, M.; Hu, G.; Liu, H.; Zhang, Y.; Cao, B.; Baddoo, M.; et al. Nerve growth factor receptor negates the tumor suppressor p53 as a feedback regulator. Elife 2016, 5, e15099. [Google Scholar] [CrossRef] [PubMed] [Green Version]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jung, J.H.; Lee, D.; Ko, H.M.; Jang, H.-J. Inhibition of CNOT2 Induces Apoptosis via MID1IP1 in Colorectal Cancer Cells by Activating p53. Biomolecules 2021, 11, 1492. https://doi.org/10.3390/biom11101492
Jung JH, Lee D, Ko HM, Jang H-J. Inhibition of CNOT2 Induces Apoptosis via MID1IP1 in Colorectal Cancer Cells by Activating p53. Biomolecules. 2021; 11(10):1492. https://doi.org/10.3390/biom11101492
Chicago/Turabian StyleJung, Ji Hoon, Duckgue Lee, Hyun Min Ko, and Hyeung-Jin Jang. 2021. "Inhibition of CNOT2 Induces Apoptosis via MID1IP1 in Colorectal Cancer Cells by Activating p53" Biomolecules 11, no. 10: 1492. https://doi.org/10.3390/biom11101492