
p53-R273H Sustains ROS, Pro-Inflammatory Cytokine Release and mTOR Activation While Reducing Autophagy, Mitophagy and UCP2 Expression, Effects Prevented by wtp53

 , and
, and
Abstract
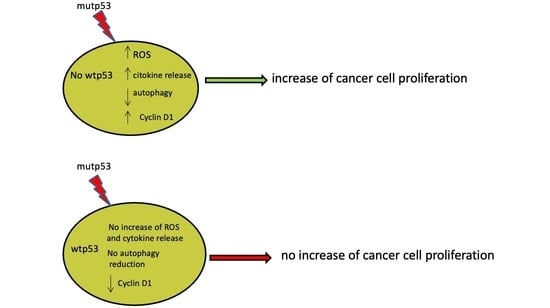
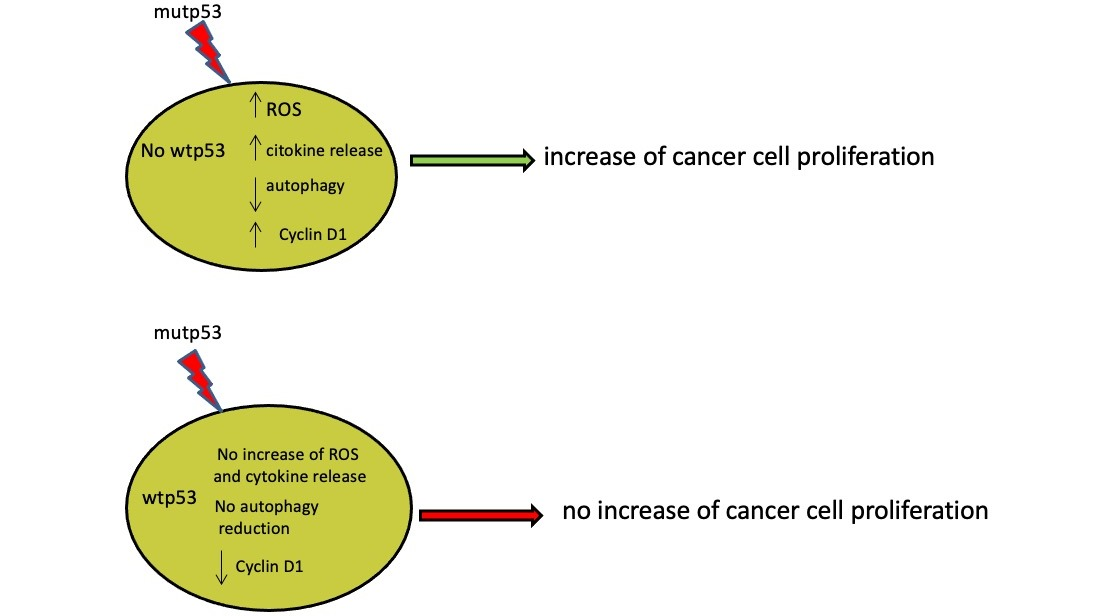
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Material and Methods
2.1. Cell Culture and Treatments
2.2. p53 Silencing, mutp53 and wtp53 Transfection
2.3. Cell Viability
2.4. Measurement of Intracellular Reactive Oxygen Species (ROS) Production
2.5. Western Blot Analysis
2.6. Antibodies
2.7. Chemiluminescent Immunometric Assay
2.8. Indirect Immunofluorescence Assay (IFA)
2.9. Statistical Analysis
3. Results
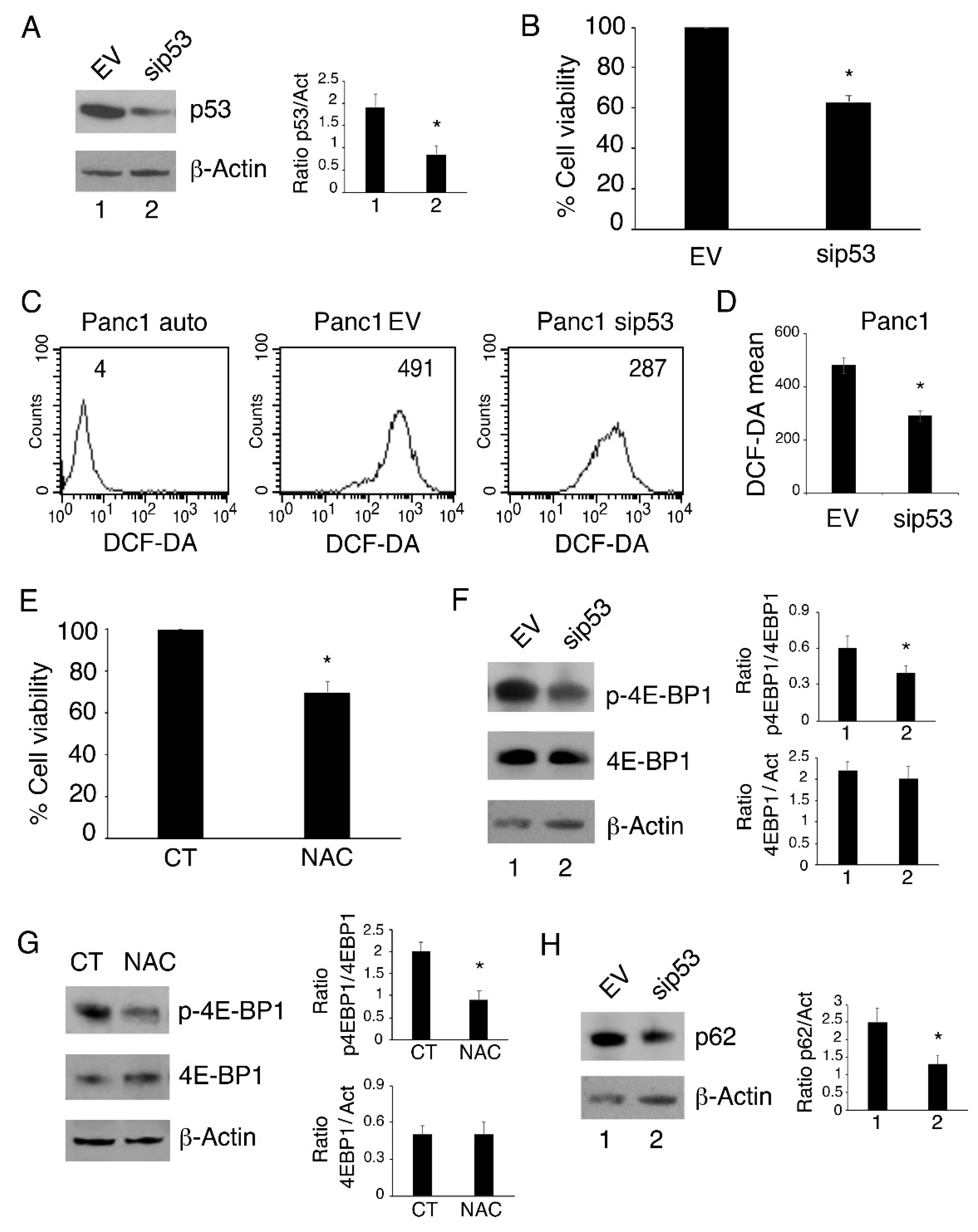
3.1. p53-R273H Silencing Reduces Cell Survival, Intracellular ROS, Cytokine Release and mTOR Activation While Inducing Autophagy in Panc1 Cells
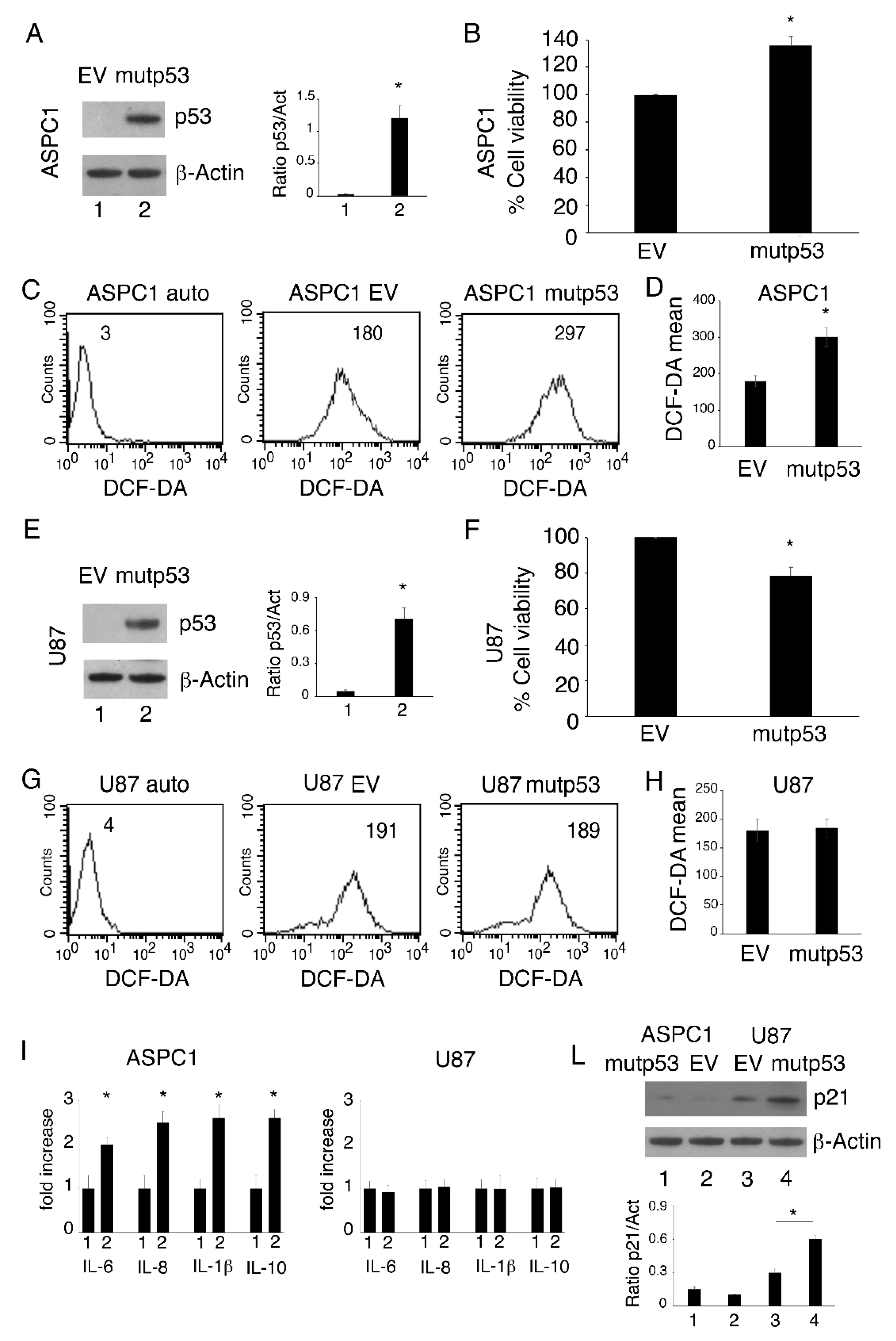
3.2. mutp53-R273H Overexpression Increases Cell Survival, ROS and Inflammatory Cytokine Release in p53 Null Cells; Effects Were Not Observed in wtp53 Cells
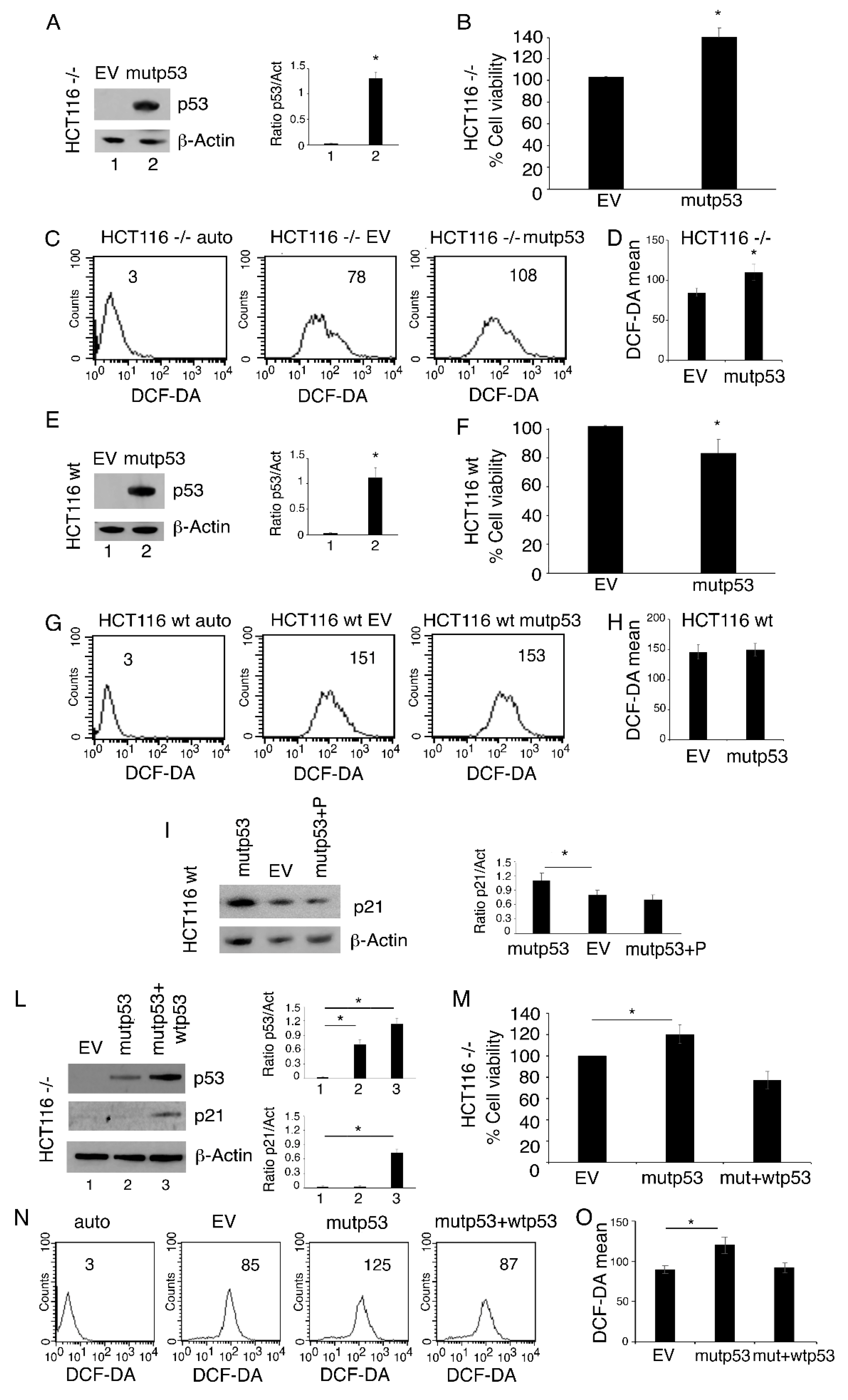
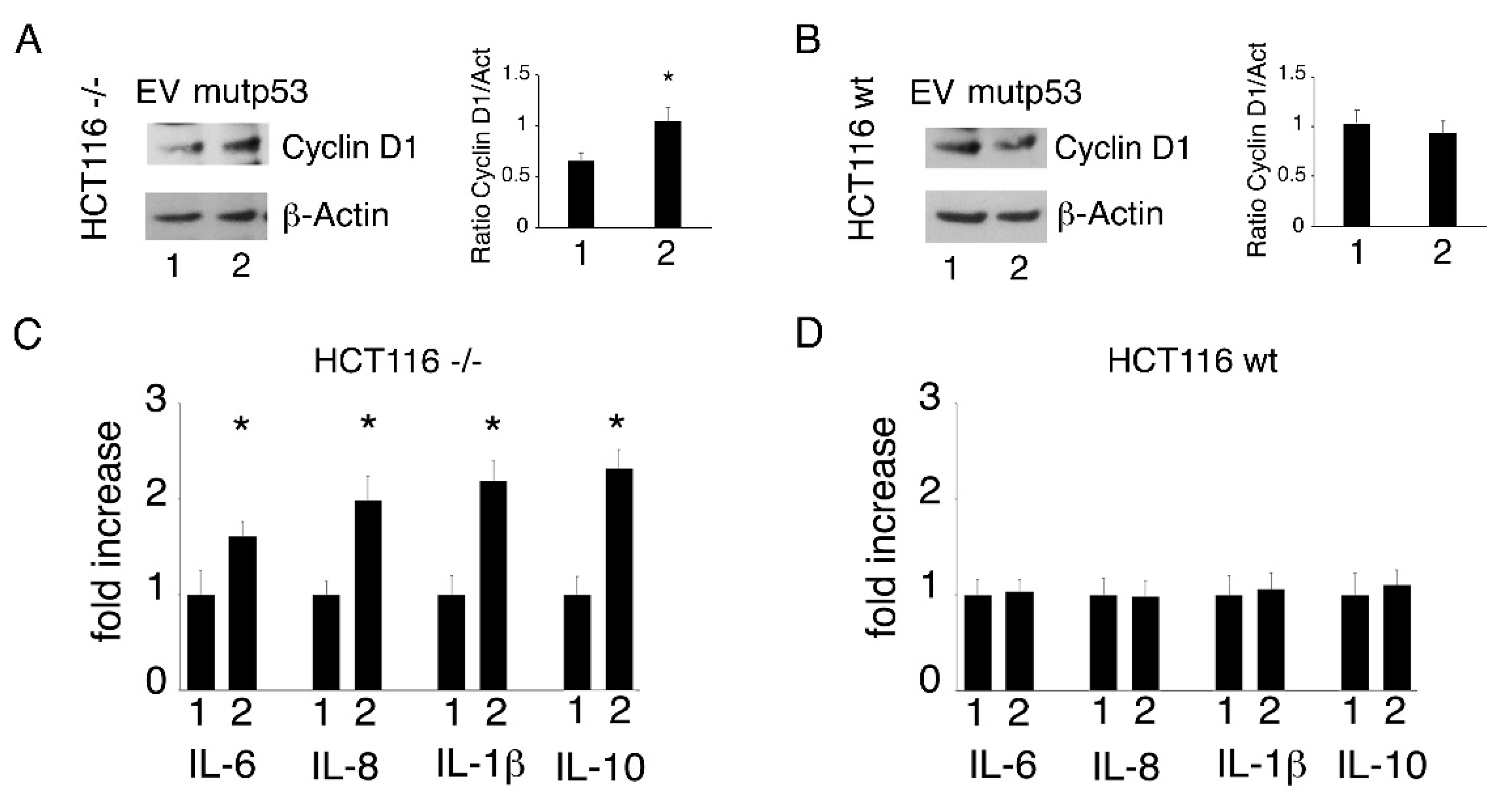
3.3. p53-R273H Overexpression Increases Cell Survival, ROS and Inflammatory Cytokine Release in HCT116 p53-/- But Not in HCT116 wtp53 Cells
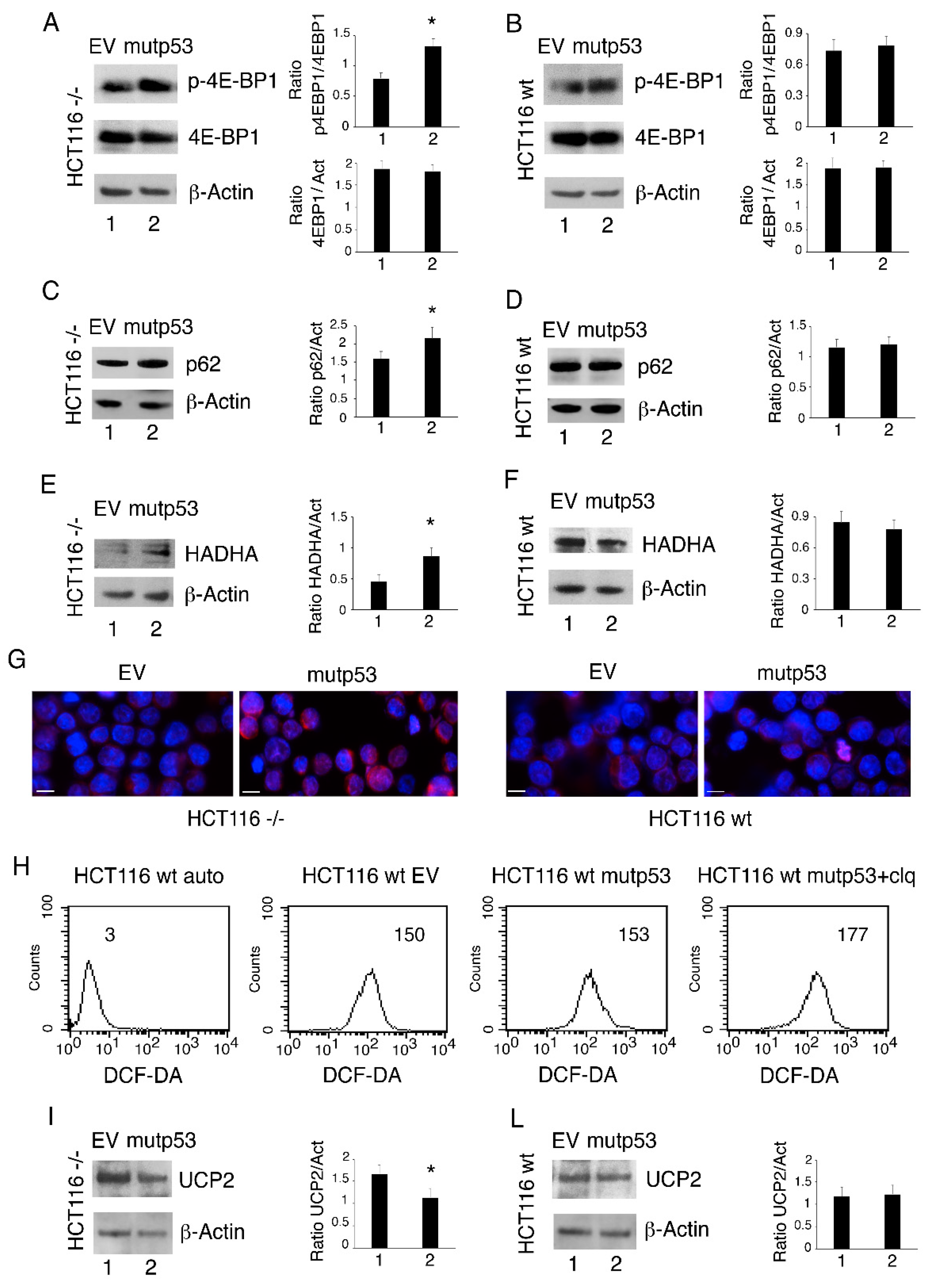
3.4. p53-R273H Overexpression in HCT116 p53-/- Cells Induces mTOR Activation, Reduces Autophagy and Mitophagy and Downregulates UCP2 Expression
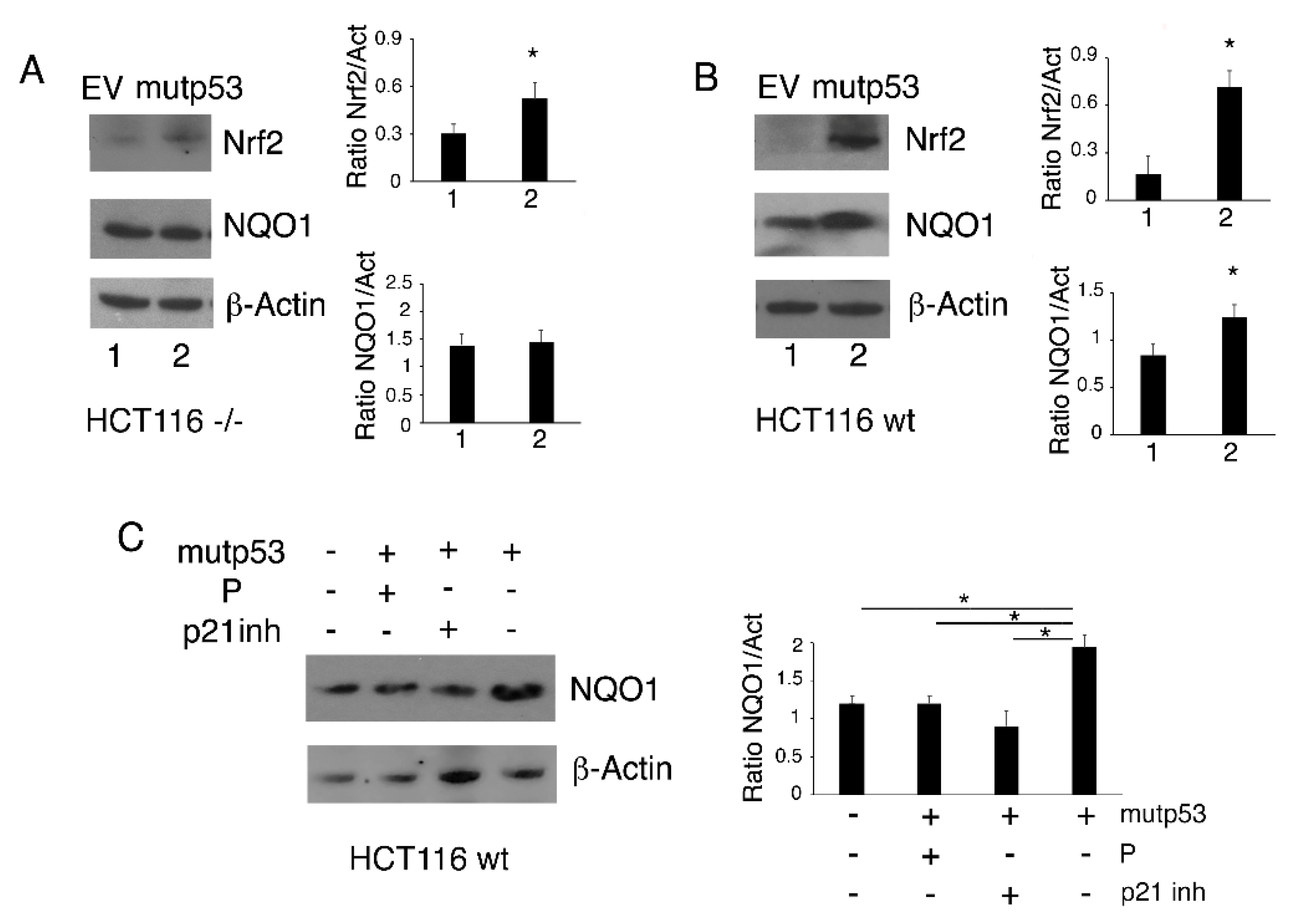
3.5. The Nrf2 Antioxidant Program is Activated in HCT116 p53-/- But Not in HCT116 wtp53 Following p53-R273H Transfection
4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- D’Orazi, G.; Cirone, M. Mutant p53 and Cellular Stress Pathways: A Criminal Alliance That Promotes Cancer Progression. Cancers 2019, 11, 614. [Google Scholar] [CrossRef] [Green Version]
- Dando, I.; Cordani, M.; Donadelli, M. Mutant p53 and mTOR/PKM2 regulation in cancer cells. IUBMB Life 2016, 68, 722–726. [Google Scholar] [CrossRef] [Green Version]
- Romeo, M.A.; Gilardini Montani, M.S.; Benedetti, R.; Santarelli, R.; D’Orazi, G.; Cirone, M. STAT3 and mutp53 Engage a Positive Feedback Loop Involving HSP90 and the Mevalonate Pathway. Front. Oncol. 2020, 10, 1102. [Google Scholar] [CrossRef] [PubMed]
- Parrales, A.; Thoenen, E.; Iwakuma, T. The interplay between mutant p53 and the mevalonate pathway. Cell Death Differ. 2018, 25, 460–470. [Google Scholar] [CrossRef]
- Ingallina, E.; Sorrentino, G.; Bertolio, R.; Lisek, K.; Zannini, A.; Azzolin, L.; Severino, L.U.; Scaini, D.; Mano, M.; Mantovani, F.; et al. Mechanical cues control mutant p53 stability through a mevalonate-RhoA axis. Nat. Cell Biol. 2018, 20, 28–35. [Google Scholar] [CrossRef] [PubMed]
- Cordani, M.; Butera, G.; Pacchiana, R.; Masetto, F.; Mullappilly, N.; Riganti, C.; Donadelli, M. Mutant p53-Associated Molecular Mechanisms of ROS Regulation in Cancer Cells. Biomolecules 2020, 10, 361. [Google Scholar] [CrossRef] [Green Version]
- Ma, L.; Fu, Q.; Xu, B.; Zhou, H.; Gao, J.; Shao, X.; Xiong, J.; Gu, Q.; Wen, S.; Li, F.; et al. Breast cancer-associated mitochondrial DNA haplogroup promotes neoplastic growth via ROS-mediated AKT activation. Int. J. Cancer 2018, 142, 1786–1796. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Granato, M.; Rizzello, C.; Gilardini Montani, M.S.; Cuomo, L.; Vitillo, M.; Santarelli, R.; Gonnella, R.; D’Orazi, G.; Faggioni, A.; Cirone, M. Quercetin induces apoptosis and autophagy in primary effusion lymphoma cells by inhibiting PI3K/AKT/mTOR and STAT3 signaling pathways. J. Nutr. Biochem. 2017, 41, 124–136. [Google Scholar] [CrossRef]
- Bhatt, A.P.; Bhende, P.M.; Sin, S.H.; Roy, D.; Dittmer, D.P.; Damania, B. Dual inhibition of PI3K and mTOR inhibits autocrine and paracrine proliferative loops in PI3K/Akt/mTOR-addicted lymphomas. Blood 2010, 115, 4455–4463. [Google Scholar] [CrossRef] [PubMed]
- D’Orazi, G.; Cordani, M.; Cirone, M. Oncogenic pathways activated by pro-inflammatory cytokines promote mutant p53 stability: Clue for novel anticancer therapies. Cell. Mol. Life Sci. 2020. [Google Scholar] [CrossRef]
- Moon, S.H.; Huang, C.H.; Houlihan, S.L.; Regunath, K.; Freed-Pastor, W.A.; Morris, J.P.t.; Tschaharganeh, D.F.; Kastenhuber, E.R.; Barsotti, A.M.; Culp-Hill, R.; et al. p53 Represses the Mevalonate Pathway to Mediate Tumor Suppression. Cell 2019, 176, 564–580.e519. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sorrentino, G.; Mantovani, F.; Del Sal, G. The stiff RhoAd from mevalonate to mutant p53. Cell Death Differ. 2018, 25, 645–647. [Google Scholar] [CrossRef] [PubMed]
- Granato, M.; Gilardini Montani, M.S.; Santarelli, R.; D’Orazi, G.; Faggioni, A.; Cirone, M. Apigenin, by activating p53 and inhibiting STAT3, modulates the balance between pro-apoptotic and pro-survival pathways to induce PEL cell death. J. Exp. Clin. Cancer Res. 2017, 36, 167. [Google Scholar] [CrossRef] [Green Version]
- Feng, Z. p53 regulation of the IGF-1/AKT/mTOR pathways and the endosomal compartment. Cold Spring Harb. Perspect. Biol. 2010, 2, a001057. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- He, Z.; Simon, H.U. A novel link between p53 and ROS. Cell Cycle 2013, 12, 201–202. [Google Scholar] [CrossRef]
- Boudreau, H.E.; Ma, W.F.; Korzeniowska, A.; Park, J.J.; Bhagwat, M.A.; Leto, T.L. Histone modifications affect differential regulation of TGFbeta- induced NADPH oxidase 4 (NOX4) by wild-type and mutant p53. Oncotarget 2017, 8, 44379–44397. [Google Scholar] [CrossRef] [Green Version]
- Desaint, S.; Luriau, S.; Aude, J.C.; Rousselet, G.; Toledano, M.B. Mammalian antioxidant defenses are not inducible by H2O2. J. Biol. Chem. 2004, 279, 31157–31163. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rotblat, B.; Melino, G.; Knight, R.A. NRF2 and p53: Januses in cancer? Oncotarget 2012, 3, 1272–1283. [Google Scholar] [CrossRef] [Green Version]
- D’Orazi, G.; Garufi, A.; Cirone, M. Nuclear factor erythroid 2 (NF-E2) p45-related factor 2 interferes with homeodomain-interacting protein kinase 2/p53 activity to impair solid tumors chemosensitivity. IUBMB Life 2020, 72, 1634–1639. [Google Scholar] [CrossRef]
- Tasdemir, E.; Chiara Maiuri, M.; Morselli, E.; Criollo, A.; D’Amelio, M.; Djavaheri-Mergny, M.; Cecconi, F.; Tavernarakis, N.; Kroemer, G. A dual role of p53 in the control of autophagy. Autophagy 2008, 4, 810–814. [Google Scholar] [CrossRef] [Green Version]
- Cecchinelli, B.; Lavra, L.; Rinaldo, C.; Iacovelli, S.; Gurtner, A.; Gasbarri, A.; Ulivieri, A.; Del Prete, F.; Trovato, M.; Piaggio, G.; et al. Repression of the antiapoptotic molecule galectin-3 by homeodomain-interacting protein kinase 2-activated p53 is required for p53-induced apoptosis. Mol. Cell. Biol. 2006, 26, 4746–4757. [Google Scholar] [CrossRef] [Green Version]
- Cordani, M.; Oppici, E.; Dando, I.; Butturini, E.; Dalla Pozza, E.; Nadal-Serrano, M.; Oliver, J.; Roca, P.; Mariotto, S.; Cellini, B.; et al. Mutant p53 proteins counteract autophagic mechanism sensitizing cancer cells to mTOR inhibition. Mol. Oncol. 2016, 10, 1008–1029. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pitolli, C.; Wang, Y.; Mancini, M.; Shi, Y.; Melino, G.; Amelio, I. Do Mutations Turn p53 into an Oncogene? Int. J. Mol. Sci. 2019, 20, 6241. [Google Scholar] [CrossRef] [Green Version]
- Rocha, S.; Campbell, K.J.; Perkins, N.D. p53- and Mdm2-independent repression of NF-kappa B transactivation by the ARF tumor suppressor. Mol. Cell 2003, 12, 15–25. [Google Scholar] [CrossRef]
- Santarelli, R.; Arteni, A.M.B.; Gilardini Montani, M.S.; Romeo, M.A.; Gaeta, A.; Gonnella, R.; Faggioni, A.; Cirone, M. KSHV dysregulates bulk macroautophagy, mitophagy and UPR to promote endothelial to mesenchymal transition and CCL2 release, key events in viral-driven sarcomagenesis. Int. J. Cancer 2020, 147, 3500–3510. [Google Scholar] [CrossRef] [PubMed]
- Cordani, M.; Butera, G.; Dando, I.; Torrens-Mas, M.; Butturini, E.; Pacchiana, R.; Oppici, E.; Cavallini, C.; Gasperini, S.; Tamassia, N.; et al. Mutant p53 blocks SESN1/AMPK/PGC-1alpha/UCP2 axis increasing mitochondrial O2-. production in cancer cells. Br. J. Cancer 2018, 119, 994–1008. [Google Scholar] [CrossRef]
- Dando, I.; Pacchiana, R.; Pozza, E.D.; Cataldo, I.; Bruno, S.; Conti, P.; Cordani, M.; Grimaldi, A.; Butera, G.; Caraglia, M.; et al. UCP2 inhibition induces ROS/Akt/mTOR axis: Role of GAPDH nuclear translocation in genipin/everolimus anticancer synergism. Free Radic. Biol. Med. 2017, 113, 176–189. [Google Scholar] [CrossRef]
- Lisek, K.; Campaner, E.; Ciani, Y.; Walerych, D.; Del Sal, G. Mutant p53 tunes the NRF2-dependent antioxidant response to support survival of cancer cells. Oncotarget 2018, 9, 20508–20523. [Google Scholar] [CrossRef] [Green Version]
- Kalo, E.; Kogan-Sakin, I.; Solomon, H.; Bar-Nathan, E.; Shay, M.; Shetzer, Y.; Dekel, E.; Goldfinger, N.; Buganim, Y.; Stambolsky, P.; et al. Mutant p53R273H attenuates the expression of phase 2 detoxifying enzymes and promotes the survival of cells with high levels of reactive oxygen species. J. Cell Sci. 2012, 125, 5578–5586. [Google Scholar] [CrossRef] [Green Version]
- Williams, A.B.; Schumacher, B. p53 in the DNA-Damage-Repair Process. Cold Spring Harb. Perspect. Med. 2016, 6. [Google Scholar] [CrossRef] [Green Version]
- Garlid, K.D.; Jaburek, M.; Jezek, P.; Varecha, M. How do uncoupling proteins uncouple? Biochim. Biophys. Acta 2000, 1459, 383–389. [Google Scholar] [CrossRef] [Green Version]
- Qin, N.; Cai, T.; Ke, Q.; Yuan, Q.; Luo, J.; Mao, X.; Jiang, L.; Cao, H.; Wen, P.; Zen, K.; et al. UCP2-dependent improvement of mitochondrial dynamics protects against acute kidney injury. J. Pathol. 2019, 247, 392–405. [Google Scholar] [CrossRef]
- Cirone, M.; Gilardini Montani, M.S.; Granato, M.; Garufi, A.; Faggioni, A.; D’Orazi, G. Autophagy manipulation as a strategy for efficient anticancer therapies: Possible consequences. J. Exp. Clin. Cancer Res. 2019, 38, 262. [Google Scholar] [CrossRef]
- Gilardini Montani, M.S.; Cecere, N.; Granato, M.; Romeo, M.A.; Falcinelli, L.; Ciciarelli, U.; D’Orazi, G.; Faggioni, A.; Cirone, M. Mutant p53, Stabilized by Its Interplay with HSP90, Activates a Positive Feed-Back Loop Between NRF2 and p62 that Induces Chemo-Resistance to Apigenin in Pancreatic Cancer Cells. Cancers 2019, 11, 703. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, W.; Sun, Z.; Wang, X.J.; Jiang, T.; Huang, Z.; Fang, D.; Zhang, D.D. Direct interaction between Nrf2 and p21(Cip1/WAF1) upregulates the Nrf2-mediated antioxidant response. Mol. Cell 2009, 34, 663–673. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kumari, S.; Badana, A.K.; Malla, R. Reactive Oxygen Species: A Key Constituent in Cancer Survival. Biomark. Insights 2018, 13, 1177271918755391. [Google Scholar] [CrossRef] [Green Version]
- Naik, E.; O’Reilly, L.A.; Asselin-Labat, M.L.; Merino, D.; Lin, A.; Cook, M.; Coultas, L.; Bouillet, P.; Adams, J.M.; Strasser, A. Destruction of tumor vasculature and abated tumor growth upon VEGF blockade is driven by proapoptotic protein Bim in endothelial cells. J. Exp. Med. 2011, 208, 1351–1358. [Google Scholar] [CrossRef] [Green Version]
- Yang, D.; Elner, S.G.; Bian, Z.M.; Till, G.O.; Petty, H.R.; Elner, V.M. Pro-inflammatory cytokines increase reactive oxygen species through mitochondria and NADPH oxidase in cultured RPE cells. Exp. Eye Res. 2007, 85, 462–472. [Google Scholar] [CrossRef] [Green Version]
- Gruosso, T.; Mieulet, V.; Cardon, M.; Bourachot, B.; Kieffer, Y.; Devun, F.; Dubois, T.; Dutreix, M.; Vincent-Salomon, A.; Miller, K.M.; et al. Chronic oxidative stress promotes H2AX protein degradation and enhances chemosensitivity in breast cancer patients. EMBO Mol. Med. 2016, 8, 527–549. [Google Scholar] [CrossRef] [PubMed]
- Song, H.; Hollstein, M.; Xu, Y. p53 gain-of-function cancer mutants induce genetic instability by inactivating ATM. Nat. Cell Biol. 2007, 9, 573–580. [Google Scholar] [CrossRef]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Romeo, M.A.; Gilardini Montani, M.S.; Benedetti, R.; Arena, A.; D’Orazi, G.; Cirone, M. p53-R273H Sustains ROS, Pro-Inflammatory Cytokine Release and mTOR Activation While Reducing Autophagy, Mitophagy and UCP2 Expression, Effects Prevented by wtp53. Biomolecules 2021, 11, 344. https://doi.org/10.3390/biom11030344
Romeo MA, Gilardini Montani MS, Benedetti R, Arena A, D’Orazi G, Cirone M. p53-R273H Sustains ROS, Pro-Inflammatory Cytokine Release and mTOR Activation While Reducing Autophagy, Mitophagy and UCP2 Expression, Effects Prevented by wtp53. Biomolecules. 2021; 11(3):344. https://doi.org/10.3390/biom11030344
Chicago/Turabian StyleRomeo, Maria Anele, Maria Saveria Gilardini Montani, Rossella Benedetti, Andrea Arena, Gabriella D’Orazi, and Mara Cirone. 2021. "p53-R273H Sustains ROS, Pro-Inflammatory Cytokine Release and mTOR Activation While Reducing Autophagy, Mitophagy and UCP2 Expression, Effects Prevented by wtp53" Biomolecules 11, no. 3: 344. https://doi.org/10.3390/biom11030344