
p53-Mediated Radiosensitization of 177Lu-DOTATATE in Neuroblastoma Tumor Spheroids

 , , , ,
, , , ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Statistical Analysis
2.2. Cell Lines
2.3. Drug and Radioconjugate Preparation
2.4. XTT Viability Assay
2.5. Sequencing of the p53 Gene
2.6. Validation of VIP116 Specificity
2.7. Cellular Specificty of 177Lu-DOTATATE and Ligandtracer Experiments
2.8. Multicellular Tumor Spheroids
2.9. Cell Lysate Preparation and Western Blot
2.10. Flow Cytometry
3. Results
3.1. Labeling
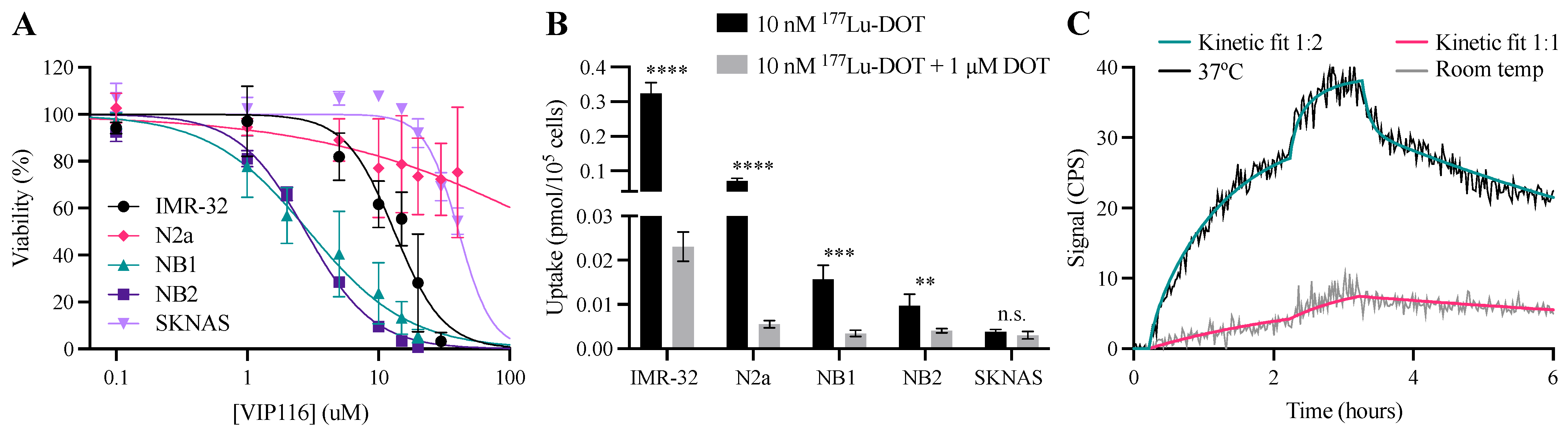
3.2. Characterization of Cell Lines
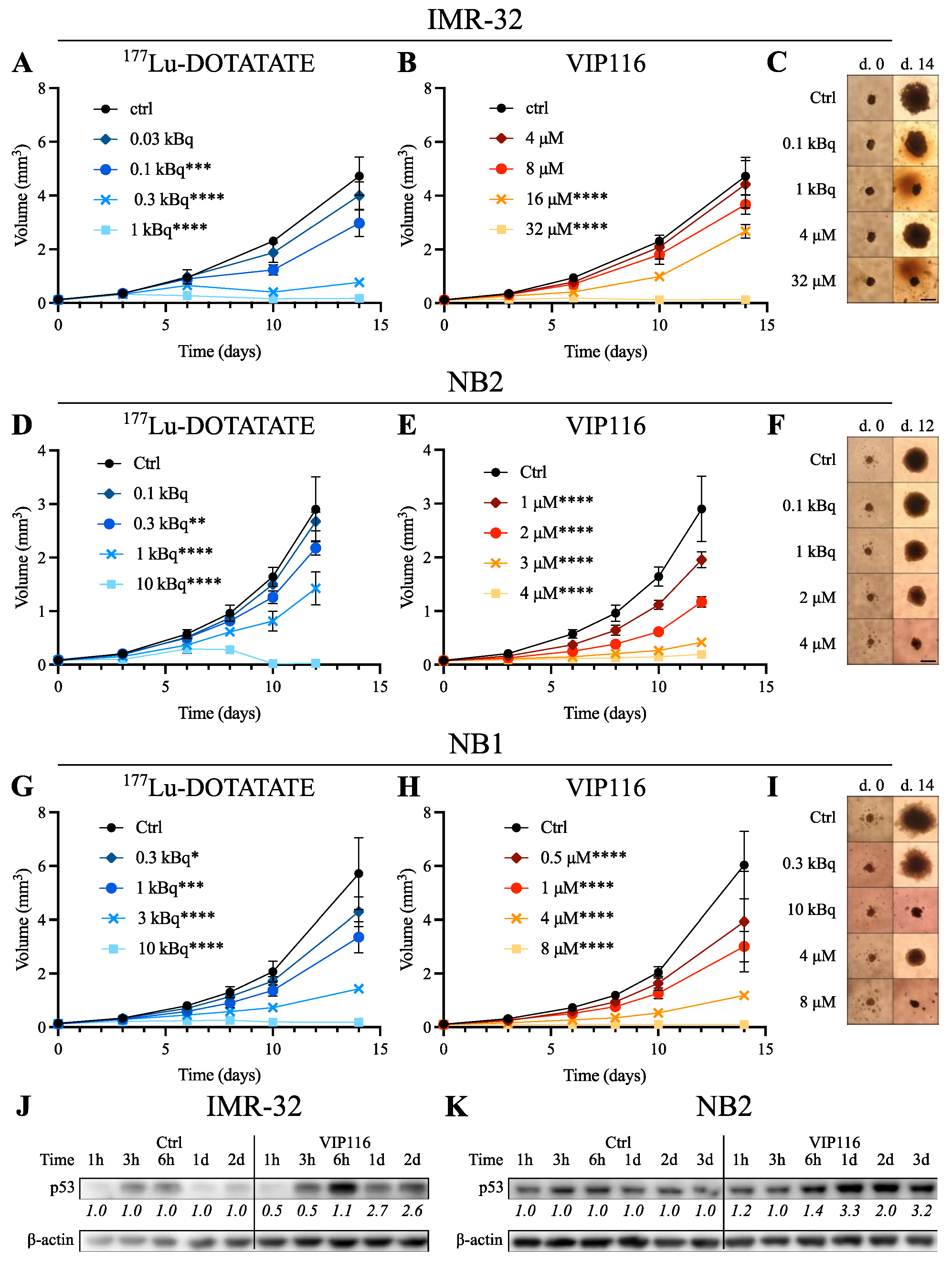
3.3. 177Lu-DOTATATE and VIP116 Monotherapy Inhibit Spheroid Growth
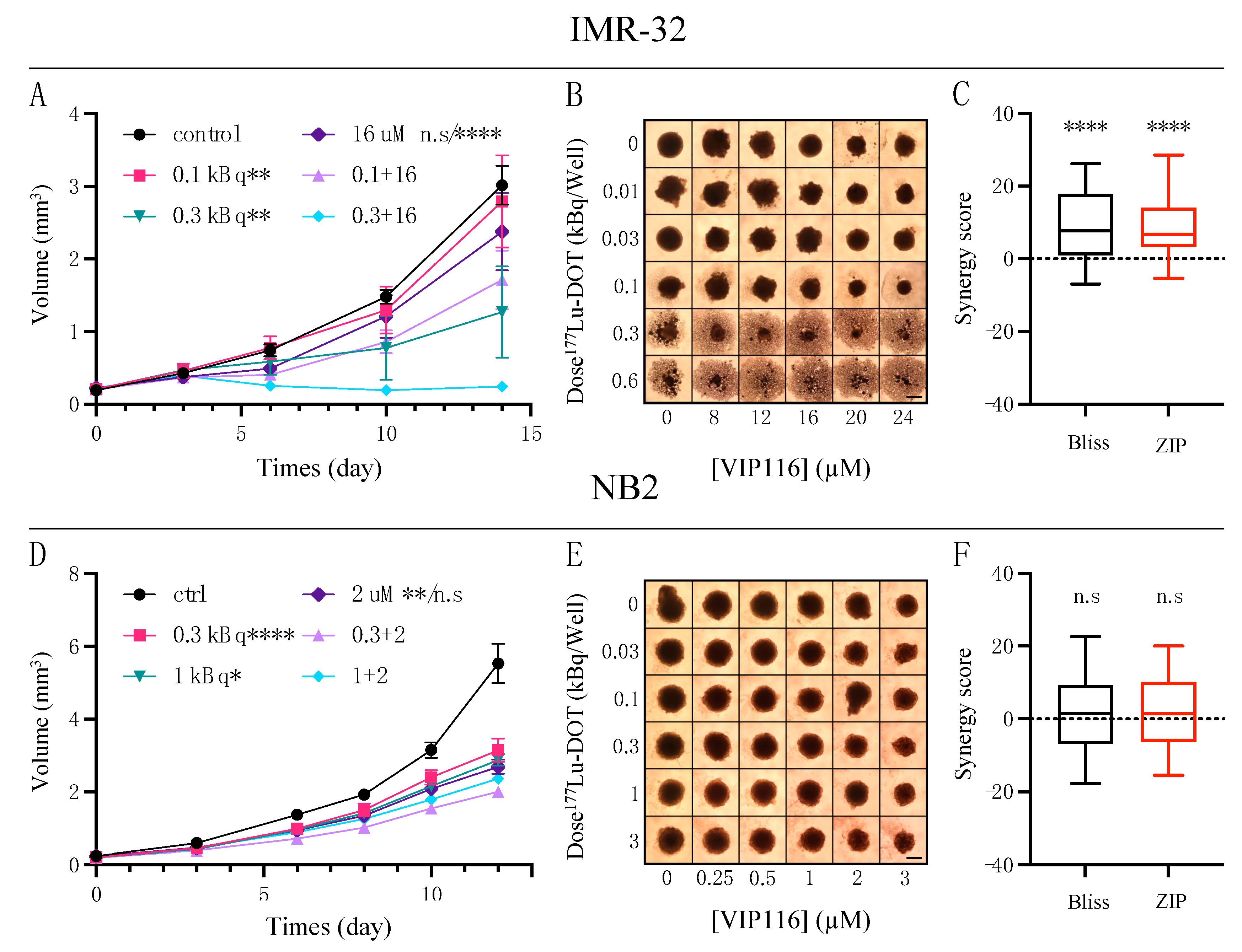
3.4. Combination Therapy Induces Varying Degree of Synergy in IMR-32 and NB2 Spheroids
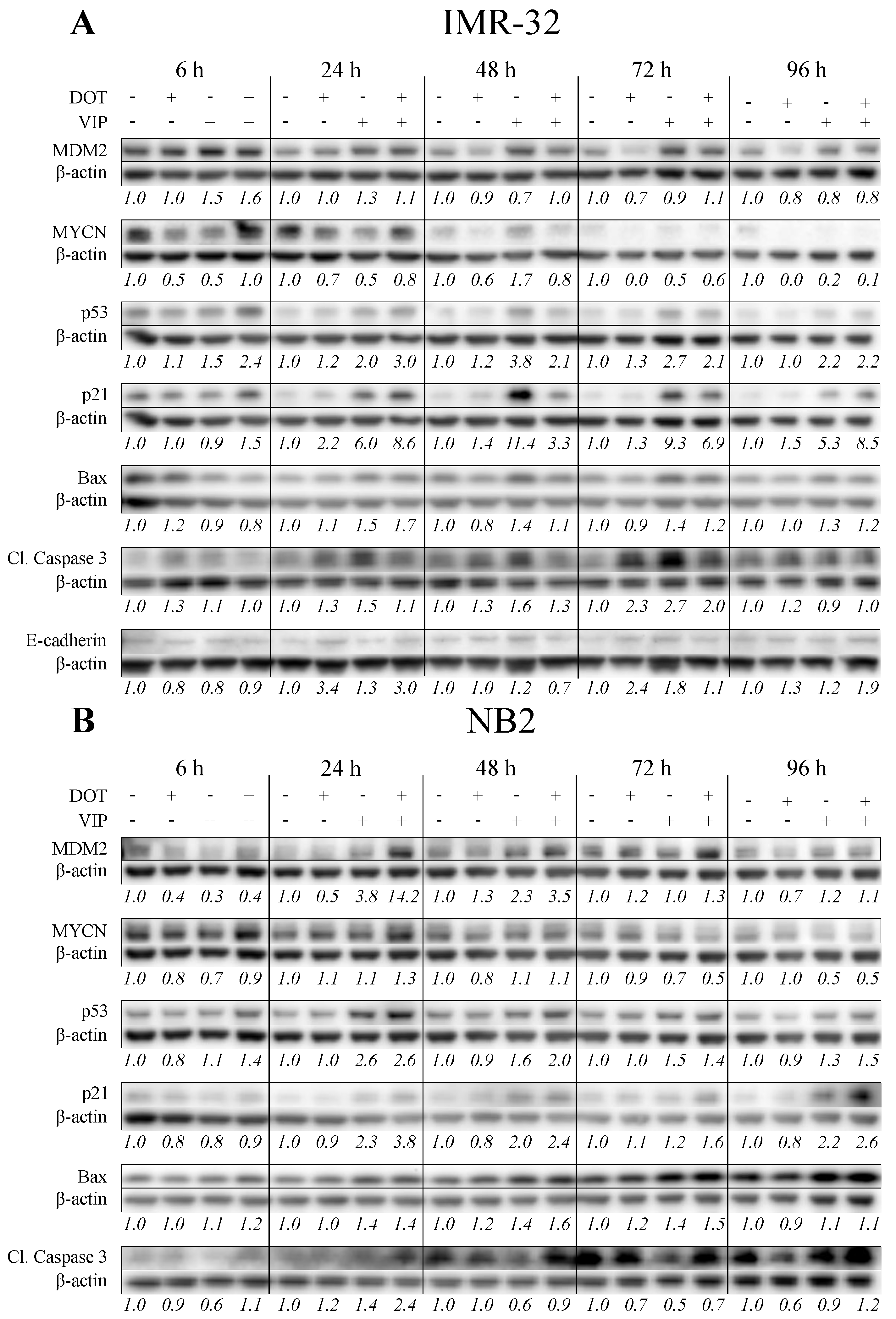
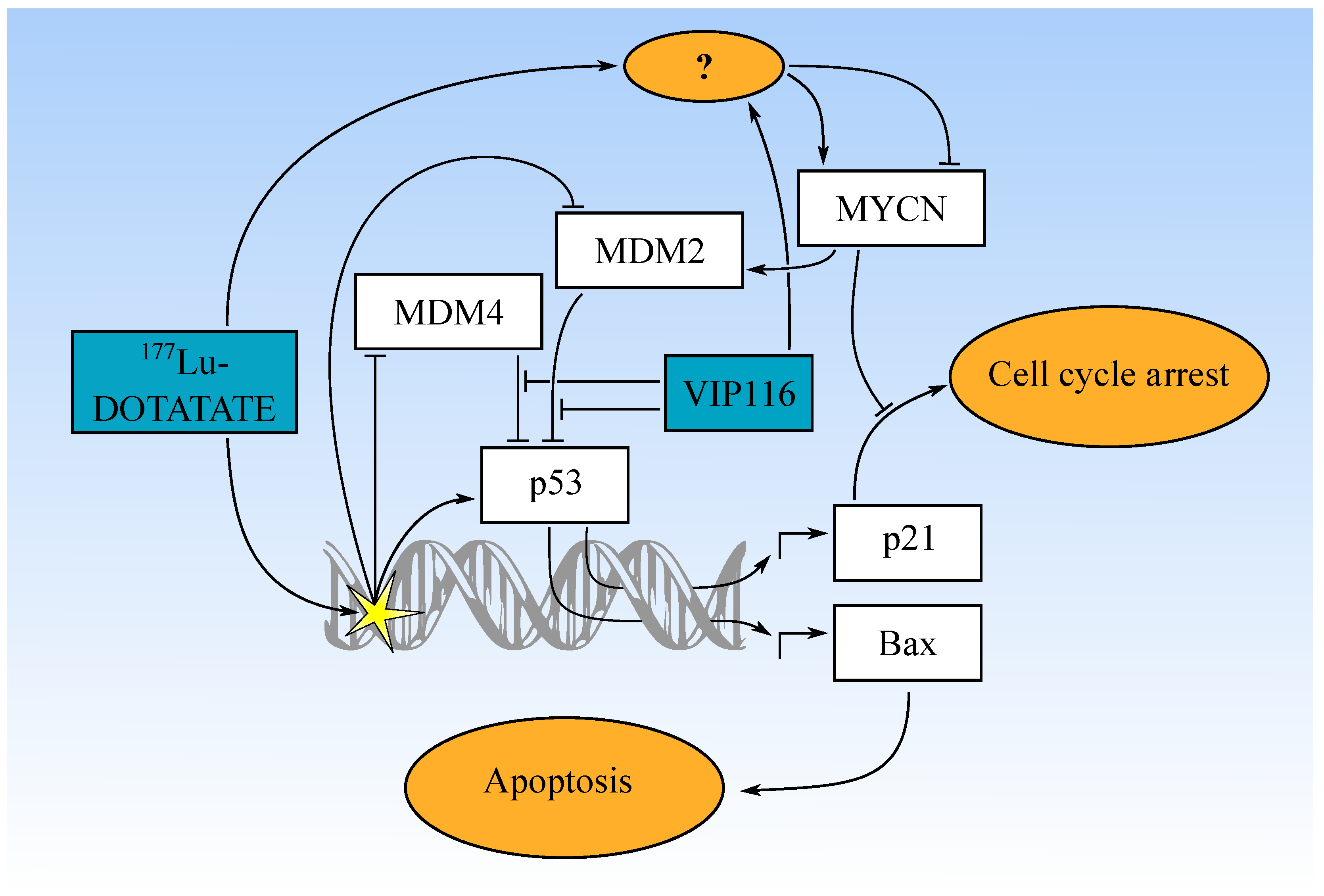
3.5. Combination Treatment Activates p53 Pathway with Differential Effects on MYCN and MDM2
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Vogelstein, B.; Lane, D.; Levine, A.J. Surfing the p53 network. Nature 2000, 408, 307–310. [Google Scholar] [CrossRef]
- Lane, D.P.; Cheok, C.F.; Lain, S. p53-based Cancer Therapy. Cold Spring Harb. Perspect. Biol. 2010, 2. [Google Scholar] [CrossRef] [Green Version]
- Michael, D.; Oren, M. The p53–Mdm2 module and the ubiquitin system. Semin. Cancer Biol. 2003, 13, 49–58. [Google Scholar] [CrossRef]
- Lu, X.; Ma, O.; Nguyen, T.A.; Jones, S.N.; Oren, M.; Donehower, L.A. The Wip1 Phosphatase acts as a gatekeeper in the p53-Mdm2 autoregulatory loop. Cancer Cell 2007, 12, 342–354. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Burgess, A.; Chia, K.M.; Haupt, S.; Thomas, D.; Haupt, Y.; Lim, E. Clinical Overview of MDM2/X-Targeted Therapies. Front. Oncol. 2016, 6, 7. [Google Scholar] [CrossRef] [PubMed]
- ClinicalTrials.gov. Available online: https://clinicaltrials.gov/ct2/home (accessed on 22 August 2021).
- Bernal, F.; Wade, M.; Godes, M.; Davis, T.N.; Whitehead, D.G.; Kung, A.L.; Wahl, G.M.; Walensky, L.D. A stapled p53 helix overcomes HDMX-mediated suppression of p53. Cancer Cell 2010, 18, 411–422. [Google Scholar] [CrossRef] [Green Version]
- Wei, S.J.; Chee, S.; Yurlova, L.; Lane, D.; Verma, C.; Brown, C.; Ghadessy, F. Avoiding drug resistance through extended drug target interfaces: A case for stapled peptides. Oncotarget 2016, 7, 32232–32246. [Google Scholar] [CrossRef] [PubMed]
- Yuen, T.Y.; Brown, C.J.; Xue, Y.; Tan, Y.S.; Ferrer Gago, F.J.; Lee, X.E.; Neo, J.Y.; Thean, D.; Kaan, H.Y.K.; Partridge, A.W.; et al. Stereoisomerism of stapled peptide inhibitors of the p53-Mdm2 interaction: An assessment of synthetic strategies and activity profiles. Chem. Sci. 2019, 10, 6457–6466. [Google Scholar] [CrossRef] [Green Version]
- Lundsten, S.; Hernandez, V.A.; Gedda, L.; Saren, T.; Brown, C.J.; Lane, D.P.; Edwards, K.; Nestor, M. Tumor-Targeted Delivery of the p53-Activating Peptide VIP116 with PEG-Stabilized Lipodisks. Nanomaterials 2020, 10, 783. [Google Scholar] [CrossRef] [Green Version]
- Johansson, P.; Krona, C.; Kundu, S.; Doroszko, M.; Baskaran, S.; Schmidt, L.; Vinel, C.; Almstedt, E.; Elgendy, R.; Elfineh, L.; et al. A Patient-Derived Cell Atlas Informs Precision Targeting of Glioblastoma. Cell Rep. 2020, 32, 107897. [Google Scholar] [CrossRef] [PubMed]
- Spiegelberg, D.; Mortensen, A.C.; Lundsten, S.; Brown, C.J.; Lane, D.P.; Nestor, M. The MDM2/MDMX-p53 antagonist PM2 radiosensitizes wild-type p53 tumors. Cancer Res. 2018. [Google Scholar] [CrossRef] [Green Version]
- Mortensen, A.C.L.; Morin, E.; Brown, C.J.; Lane, D.P.; Nestor, M. Enhancing the therapeutic effects of in vitro targeted radionuclide therapy of 3D multicellular tumor spheroids using the novel stapled MDM2/X-p53 antagonist PM2. EJNMMI Res. 2020, 10, 38. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mortensen, A.C.L.; Spiegelberg, D.; Brown, C.J.; Lane, D.P.; Nestor, M. The Stapled Peptide PM2 Stabilizes p53 Levels and Radiosensitizes Wild-Type p53 Cancer Cells. Front. Oncol. 2019, 9, 923. [Google Scholar] [CrossRef]
- Saleh, M.N.; Patel, M.R.; Bauer, T.M.; Goel, S.; Falchook, G.S.; Shapiro, G.I.; Chung, K.Y.; Infante, J.R.; Conry, R.M.; Rabinowits, G.; et al. Phase 1 Trial of ALRN-6924, a Dual Inhibitor of MDMX and MDM2, in Patients with Solid Tumors and Lymphomas Bearing Wild-Type TP53. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2021. [Google Scholar] [CrossRef] [PubMed]
- Krenning, E.P.; Kooij, P.P.; Bakker, W.H.; Breeman, W.A.; Postema, P.T.; Kwekkeboom, D.J.; Oei, H.Y.; de Jong, M.; Visser, T.J.; Reijs, A.E.; et al. Radiotherapy with a radiolabeled somatostatin analogue, [111In-DTPA-D-Phe1]-octreotide. A case history. Ann. N. Y. Acad. Sci. 1994, 733, 496–506. [Google Scholar] [CrossRef] [Green Version]
- Strosberg, J.; El-Haddad, G.; Wolin, E.; Hendifar, A.; Yao, J.; Chasen, B.; Mittra, E.; Kunz, P.L.; Kulke, M.H.; Jacene, H.; et al. Phase 3 Trial of (177)Lu-Dotatate for Midgut Neuroendocrine Tumors. N. Engl. J. Med. 2017, 376, 125–135. [Google Scholar] [CrossRef]
- Gill, M.R.; Falzone, N.; Du, Y.; Vallis, K.A. Targeted radionuclide therapy in combined-modality regimens. Lancet. Oncol. 2017, 18, e414–e423. [Google Scholar] [CrossRef]
- Brodeur, G.M. Neuroblastoma: Biological insights into a clinical enigma. Nat. Rev. Cancer 2003, 3, 203–216. [Google Scholar] [CrossRef] [PubMed]
- Georgantzi, K.; Tsolakis, A.V.; Stridsberg, M.; Jakobson, A.; Christofferson, R.; Janson, E.T. Differentiated expression of somatostatin receptor subtypes in experimental models and clinical neuroblastoma. Pediatr. Blood Cancer 2011, 56, 584–589. [Google Scholar] [CrossRef]
- Gains, J.E.; Sebire, N.J.; Moroz, V.; Wheatley, K.; Gaze, M.N. Immunohistochemical evaluation of molecular radiotherapy target expression in neuroblastoma tissue. Eur. J. Nucl. Med. Mol. Imaging 2018, 45, 402–411. [Google Scholar] [CrossRef]
- Albers, A.R.; O’Dorisio, M.S.; Balster, D.A.; Caprara, M.; Gosh, P.; Chen, F.; Hoeger, C.; Rivier, J.; Wenger, G.D.; O’Dorisio, T.M.; et al. Somatostatin receptor gene expression in neuroblastoma. Regul. Pept. 2000, 88, 61–73. [Google Scholar] [CrossRef]
- Alexander, N.; Marrano, P.; Thorner, P.; Naranjo, A.; Van Ryn, C.; Martinez, D.; Batra, V.; Zhang, L.; Irwin, M.S.; Baruchel, S. Prevalence and Clinical Correlations of Somatostatin Receptor-2 (SSTR2) Expression in Neuroblastoma. J. Pediatr. Hematol. Oncol. 2019, 41, 222–227. [Google Scholar] [CrossRef] [PubMed]
- Czapiewski, P.; Kunc, M.; Gorczynski, A.; Haybaeck, J.; Okon, K.; Reszec, J.; Lewczuk, A.; Dzierzanowski, J.; Karczewska, J.; Biernat, W.; et al. Frequent expression of somatostatin receptor 2a in olfactory neuroblastomas: A new and distinctive feature. Hum. Pathol. 2018, 79, 144–150. [Google Scholar] [CrossRef] [PubMed]
- Gains, J.E.; Bomanji, J.B.; Fersht, N.L.; Sullivan, T.; D’Souza, D.; Sullivan, K.P.; Aldridge, M.; Waddington, W.; Gaze, M.N. 177Lu-DOTATATE molecular radiotherapy for childhood neuroblastoma. J. Nucl. Med. Off. Publ. Soc. Nucl. Med. 2011, 52, 1041–1047. [Google Scholar] [CrossRef] [Green Version]
- Kong, G.; Hofman, M.S.; Murray, W.K.; Wilson, S.; Wood, P.; Downie, P.; Super, L.; Hogg, A.; Eu, P.; Hicks, R.J. Initial Experience with Gallium-68 DOTA-Octreotate PET/CT and Peptide Receptor Radionuclide Therapy for Pediatric Patients with Refractory Metastatic Neuroblastoma. J. Pediatr. Hematol. Oncol. 2016, 38, 87–96. [Google Scholar] [CrossRef] [Green Version]
- Menda, Y.; O’Dorisio, M.S.; Kao, S.; Khanna, G.; Michael, S.; Connolly, M.; Babich, J.; O’Dorisio, T.; Bushnell, D.; Madsen, M. Phase I trial of 90Y-DOTATOC therapy in children and young adults with refractory solid tumors that express somatostatin receptors. J. Nucl. Med. Off. Publ. Soc. Nucl. Med. 2010, 51, 1524–1531. [Google Scholar] [CrossRef] [Green Version]
- Gains, J.E.; Moroz, V.; Aldridge, M.D.; Wan, S.; Wheatley, K.; Laidler, J.; Peet, C.; Bomanji, J.B.; Gaze, M.N. A phase IIa trial of molecular radiotherapy with 177-lutetium DOTATATE in children with primary refractory or relapsed high-risk neuroblastoma. Eur. J. Nucl. Med. Mol. Imaging 2020, 47, 2348–2357. [Google Scholar] [CrossRef]
- Al-Ghabkari, A.; Narendran, A. In Vitro Characterization of a Potent p53-MDM2 Inhibitor, RG7112 in Neuroblastoma Cancer Cell Lines. Cancer Biother. Radiopharm. 2019, 34, 252–257. [Google Scholar] [CrossRef] [PubMed]
- Arnhold, V.; Schmelz, K.; Proba, J.; Winkler, A.; Wunschel, J.; Toedling, J.; Deubzer, H.E.; Kunkele, A.; Eggert, A.; Schulte, J.H.; et al. Reactivating TP53 signaling by the novel MDM2 inhibitor DS-3032b as a therapeutic option for high-risk neuroblastoma. Oncotarget 2018, 9, 2304–2319. [Google Scholar] [CrossRef]
- Chen, L.; Pastorino, F.; Berry, P.; Bonner, J.; Kirk, C.; Wood, K.M.; Thomas, H.D.; Zhao, Y.; Daga, A.; Veal, G.J.; et al. Preclinical evaluation of the first intravenous small molecule MDM2 antagonist alone and in combination with temozolomide in neuroblastoma. Int. J. Cancer 2019, 144, 3146–3159. [Google Scholar] [CrossRef] [Green Version]
- Chen, L.; Rousseau, R.F.; Middleton, S.A.; Nichols, G.L.; Newell, D.R.; Lunec, J.; Tweddle, D.A. Pre-clinical evaluation of the MDM2-p53 antagonist RG7388 alone and in combination with chemotherapy in neuroblastoma. Oncotarget 2015, 6, 10207–10221. [Google Scholar] [CrossRef] [Green Version]
- Lakoma, A.; Barbieri, E.; Agarwal, S.; Jackson, J.; Chen, Z.; Kim, Y.; McVay, M.; Shohet, J.M.; Kim, E.S. The MDM2 small-molecule inhibitor RG7388 leads to potent tumor inhibition in p53 wild-type neuroblastoma. Cell Death Discov. 2015, 1. [Google Scholar] [CrossRef] [Green Version]
- Lu, J.; Guan, S.; Zhao, Y.; Yu, Y.; Wang, Y.; Shi, Y.; Mao, X.; Yang, K.L.; Sun, W.; Xu, X.; et al. Novel MDM2 inhibitor SAR405838 (MI-773) induces p53-mediated apoptosis in neuroblastoma. Oncotarget 2016, 7, 82757–82769. [Google Scholar] [CrossRef] [Green Version]
- Van Maerken, T.; Rihani, A.; Dreidax, D.; De Clercq, S.; Yigit, N.; Marine, J.C.; Westermann, F.; De Paepe, A.; Vandesompele, J.; Speleman, F. Functional analysis of the p53 pathway in neuroblastoma cells using the small-molecule MDM2 antagonist nutlin-3. Mol. Cancer Ther. 2011, 10, 983–993. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gamble, L.D.; Kees, U.R.; Tweddle, D.A.; Lunec, J. MYCN sensitizes neuroblastoma to the MDM2-p53 antagonists Nutlin-3 and MI-63. Oncogene 2012, 31, 752–763. [Google Scholar] [CrossRef] [Green Version]
- Tesson, M.; Vasan, R.; Hock, A.; Nixon, C.; Rae, C.; Gaze, M.; Mairs, R. An evaluation in vitro of the efficacy of nutlin-3 and topotecan in combination with (177)Lu-DOTATATE for the treatment of neuroblastoma. Oncotarget 2018, 9, 29082–29096. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Persson, C.U.; von Stedingk, K.; Bexell, D.; Merselius, M.; Braekeveldt, N.; Gisselsson, D.; Arsenian-Henriksson, M.; Pahlman, S.; Wigerup, C. Neuroblastoma patient-derived xenograft cells cultured in stem-cell promoting medium retain tumorigenic and metastatic capacities but differentiate in serum. Sci. Rep. 2017, 7, 10274. [Google Scholar] [CrossRef] [PubMed]
- Braekeveldt, N.; Wigerup, C.; Gisselsson, D.; Mohlin, S.; Merselius, M.; Beckman, S.; Jonson, T.; Borjesson, A.; Backman, T.; Tadeo, I.; et al. Neuroblastoma patient-derived orthotopic xenografts retain metastatic patterns and geno-and phenotypes of patient tumours. Int. J. Cancer 2015, 136, E252–E261. [Google Scholar] [CrossRef] [Green Version]
- Lundsten, S.; Spiegelberg, D.; Stenerlow, B.; Nestor, M. The HSP90 inhibitor Onalespib potentiates 177Lu-DOTATATE therapy in neuroendocrine tumor cells. Int. J. Oncol. 2019, 55, 1287–1295. [Google Scholar] [CrossRef]
- Chen, L.; Esfandiari, A.; Reaves, W.; Vu, A.; Hogarty, M.D.; Lunec, J.; Tweddle, D.A. Characterisation of the p53 pathway in cell lines established from TH-MYCN transgenic mouse tumours. Int. J. Oncol. 2018, 52, 967–977. [Google Scholar] [CrossRef] [Green Version]
- Franken, N.A.; Rodermond, H.M.; Stap, J.; Haveman, J.; van Bree, C. Clonogenic assay of cells in vitro. Nat. Protoc. 2006, 1, 2315–2319. [Google Scholar] [CrossRef] [PubMed]
- Leek, R.; Grimes, D.R.; Harris, A.L.; McIntyre, A. Methods: Using Three-Dimensional Culture (Spheroids) as an In Vitro Model of Tumour Hypoxia. Adv. Exp. Med. Biol. 2016, 899, 167–196. [Google Scholar] [CrossRef]
- Friedrich, J.; Seidel, C.; Ebner, R.; Kunz-Schughart, L.A. Spheroid-based drug screen: Considerations and practical approach. Nat. Protoc. 2009, 4, 309–324. [Google Scholar] [CrossRef]
- Schindelin, J.; Arganda-Carreras, I.; Frise, E.; Kaynig, V.; Longair, M.; Pietzsch, T.; Preibisch, S.; Rueden, C.; Saalfeld, S.; Schmid, B.; et al. Fiji: An open-source platform for biological-image analysis. Nat. Methods 2012, 9, 676–682. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ianevski, A.; Giri, A.K.; Aittokallio, T. SynergyFinder 2.0: Visual analytics of multi-drug combination synergies. Nucleic Acids Res. 2020, 48, W488–W493. [Google Scholar] [CrossRef]
- Tweddle, D.A.; Malcolm, A.J.; Cole, M.; Pearson, A.D.; Lunec, J. p53 cellular localization and function in neuroblastoma: Evidence for defective G(1) arrest despite WAF1 induction in MYCN-amplified cells. Am. J. Pathol. 2001, 158, 2067–2077. [Google Scholar] [CrossRef]
- Colicchia, V.; Petroni, M.; Guarguaglini, G.; Sardina, F.; Sahun-Roncero, M.; Carbonari, M.; Ricci, B.; Heil, C.; Capalbo, C.; Belardinilli, F.; et al. PARP inhibitors enhance replication stress and cause mitotic catastrophe in MYCN-dependent neuroblastoma. Oncogene 2017, 36, 4682–4691. [Google Scholar] [CrossRef]
- Amoroso, F.; Capece, M.; Rotondo, A.; Cangelosi, D.; Ferracin, M.; Franceschini, A.; Raffaghello, L.; Pistoia, V.; Varesio, L.; Adinolfi, E. The P2X7 receptor is a key modulator of the PI3K/GSK3beta/VEGF signaling network: Evidence in experimental neuroblastoma. Oncogene 2015, 34, 5240–5251. [Google Scholar] [CrossRef]
- Goldschneider, D.; Horvilleur, E.; Plassa, L.F.; Guillaud-Bataille, M.; Million, K.; Wittmer-Dupret, E.; Danglot, G.; de The, H.; Benard, J.; May, E.; et al. Expression of C-terminal deleted p53 isoforms in neuroblastoma. Nucleic Acids Res. 2006, 34, 5603–5612. [Google Scholar] [CrossRef] [Green Version]
- Taylor, J.S.; Zeki, J.; Ornell, K.; Coburn, J.; Shimada, H.; Ikegaki, N.; Chiu, B. Down-regulation of MYCN protein by CX-5461 leads to neuroblastoma tumor growth suppression. J. Pediatr. Surg. 2019, 54, 1192–1197. [Google Scholar] [CrossRef]
- Tweddle, D.A.; Pearson, A.D.; Haber, M.; Norris, M.D.; Xue, C.; Flemming, C.; Lunec, J. The p53 pathway and its inactivation in neuroblastoma. Cancer Lett. 2003, 197, 93–98. [Google Scholar] [CrossRef]
- Feijtel, D.; Doeswijk, G.N.; Verkaik, N.S.; Haeck, J.C.; Chicco, D.; Angotti, C.; Konijnenberg, M.W.; de Jong, M.; Nonnekens, J. Inter and intra-tumor somatostatin receptor 2 heterogeneity influences peptide receptor radionuclide therapy response. Theranostics 2021, 11, 491–505. [Google Scholar] [CrossRef] [PubMed]
- Mohamed, A.; Blanchard, M.P.; Albertelli, M.; Barbieri, F.; Brue, T.; Niccoli, P.; Delpero, J.R.; Monges, G.; Garcia, S.; Ferone, D.; et al. Pasireotide and octreotide antiproliferative effects and sst2 trafficking in human pancreatic neuroendocrine tumor cultures. Endocr. Relat. Cancer 2014, 21, 691–704. [Google Scholar] [CrossRef] [Green Version]
- De Jong, M.; Bernard, B.F.; De Bruin, E.; Van Gameren, A.; Bakker, W.H.; Visser, T.J.; Macke, H.R.; Krenning, E.P. Internalization of radiolabelled [DTPA0]octreotide and [DOTA0,Tyr3]octreotide: Peptides for somatostatin receptor-targeted scintigraphy and radionuclide therapy. Nucl. Med. Commun. 1998, 19, 283–288. [Google Scholar] [CrossRef] [PubMed]
- O’Neill, E.; Kersemans, V.; Allen, P.D.; Terry, S.Y.A.; Torres, J.B.; Mosley, M.; Smart, S.; Lee, B.Q.; Falzone, N.; Vallis, K.A.; et al. Imaging DNA Damage Repair In Vivo After (177)Lu-DOTATATE Therapy. J. Nucl. Med. Off. Publ. Soc. Nucl. Med. 2020, 61, 743–750. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Karimian, A.; Ahmadi, Y.; Yousefi, B. Multiple functions of p21 in cell cycle, apoptosis and transcriptional regulation after DNA damage. DNA Repair (Amst.) 2016, 42, 63–71. [Google Scholar] [CrossRef] [PubMed]
- Rufini, A.; Tucci, P.; Celardo, I.; Melino, G. Senescence and aging: The critical roles of p53. Oncogene 2013, 32, 5129–5143. [Google Scholar] [CrossRef] [PubMed]
- Schwab, M. MYCN in neuronal tumours. Cancer Lett. 2004, 204, 179–187. [Google Scholar] [CrossRef]
- Slack, A.; Chen, Z.; Tonelli, R.; Pule, M.; Hunt, L.; Pession, A.; Shohet, J.M. The p53 regulatory gene MDM2 is a direct transcriptional target of MYCN in neuroblastoma. Proc. Natl. Acad. Sci. USA 2005, 102, 731–736. [Google Scholar] [CrossRef] [Green Version]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cell Line | p53 Status | MYCN Status | XTT VIP116 (μM) | Sph. VIP116 (μM) | Sph. 177Lu-DOT (kBq/well) | Ref. |
|---|---|---|---|---|---|---|
| IMR-32 | wt | amp | 13.2 (11.1–15.4) | 15.3 (12.7–18.1) | 0.12 (0.10–0.14) | [35,47,48] |
| NB1 | wt | amp | 2.9 (2.3–3.7) | 0.9 (0.8–1.0) | 1.1 (0.8–1.5) | [39] |
| NB2 | wt | amp | 2.8 (2.6–3.0) | 1.4 (1.2–1.6) | 0.88 (0.67–1.21) | [38] |
| N2a | mut 1 | amp | 238.2 (60.5–105) 3 | n.e. | n.e. | [49] |
| SKNAS | del 2 | n.amp | 40.8 (27.8–45.4) 3 | n.e. | n.e. | [35,50,51] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lundsten, S.; Berglund, H.; Jha, P.; Krona, C.; Hariri, M.; Nelander, S.; Lane, D.P.; Nestor, M. p53-Mediated Radiosensitization of 177Lu-DOTATATE in Neuroblastoma Tumor Spheroids. Biomolecules 2021, 11, 1695. https://doi.org/10.3390/biom11111695
Lundsten S, Berglund H, Jha P, Krona C, Hariri M, Nelander S, Lane DP, Nestor M. p53-Mediated Radiosensitization of 177Lu-DOTATATE in Neuroblastoma Tumor Spheroids. Biomolecules. 2021; 11(11):1695. https://doi.org/10.3390/biom11111695
Chicago/Turabian StyleLundsten, Sara, Hanna Berglund, Preeti Jha, Cecilia Krona, Mehran Hariri, Sven Nelander, David P. Lane, and Marika Nestor. 2021. "p53-Mediated Radiosensitization of 177Lu-DOTATATE in Neuroblastoma Tumor Spheroids" Biomolecules 11, no. 11: 1695. https://doi.org/10.3390/biom11111695