Unstructured Biology of Proteins from Ubiquitin-Proteasome System: Roles in Cancer and Neurodegenerative Diseases

 , ,
, ,  and
and
Abstract
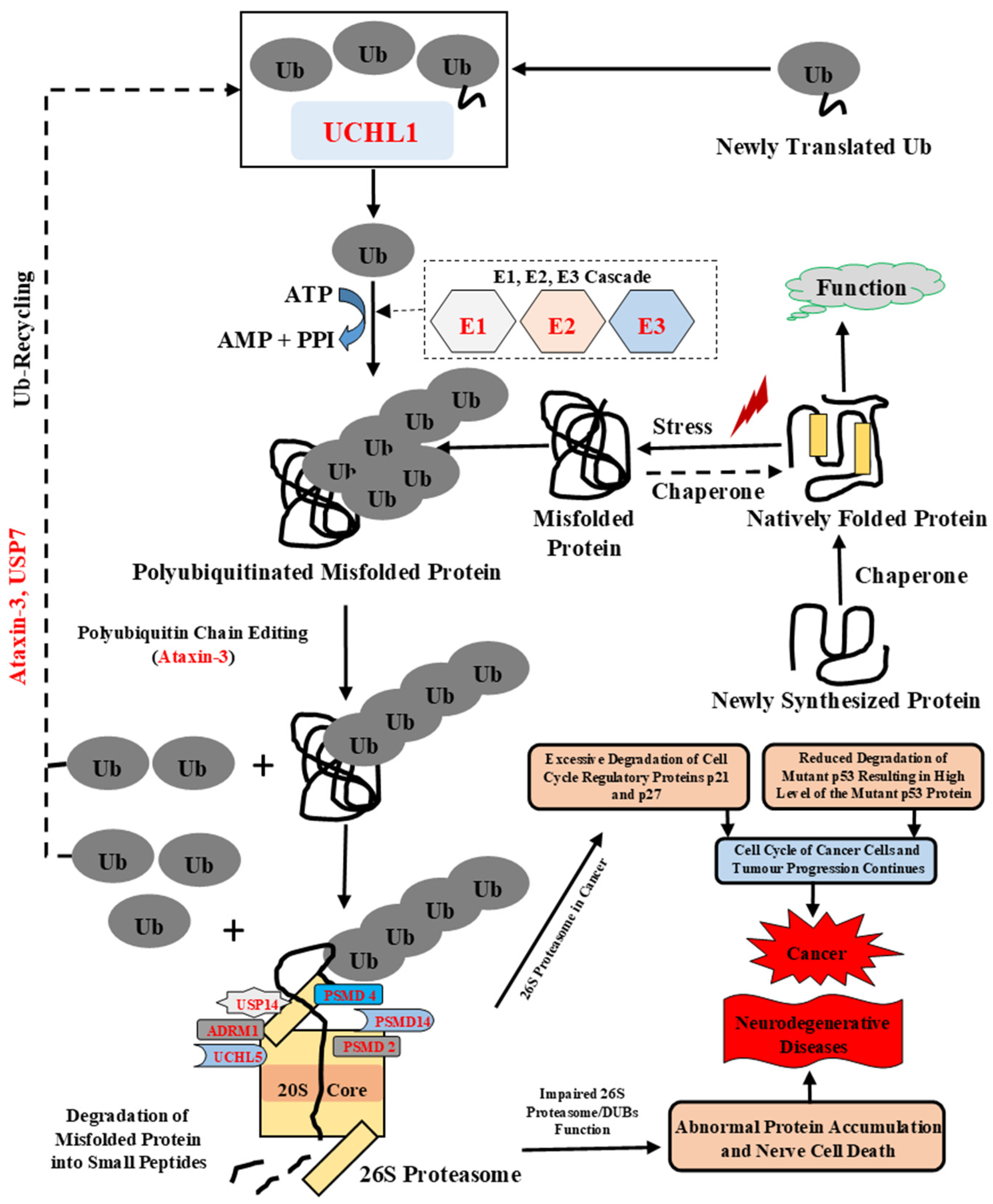
:1. Introduction
2. Materials and Methods
2.1. Retrieval of Sequences and Structures
2.2. Identification of Intrinsically Disordered Protein Regions (IDPRs)
2.3. Molecular Recognition Features (MoRFs) Prediction
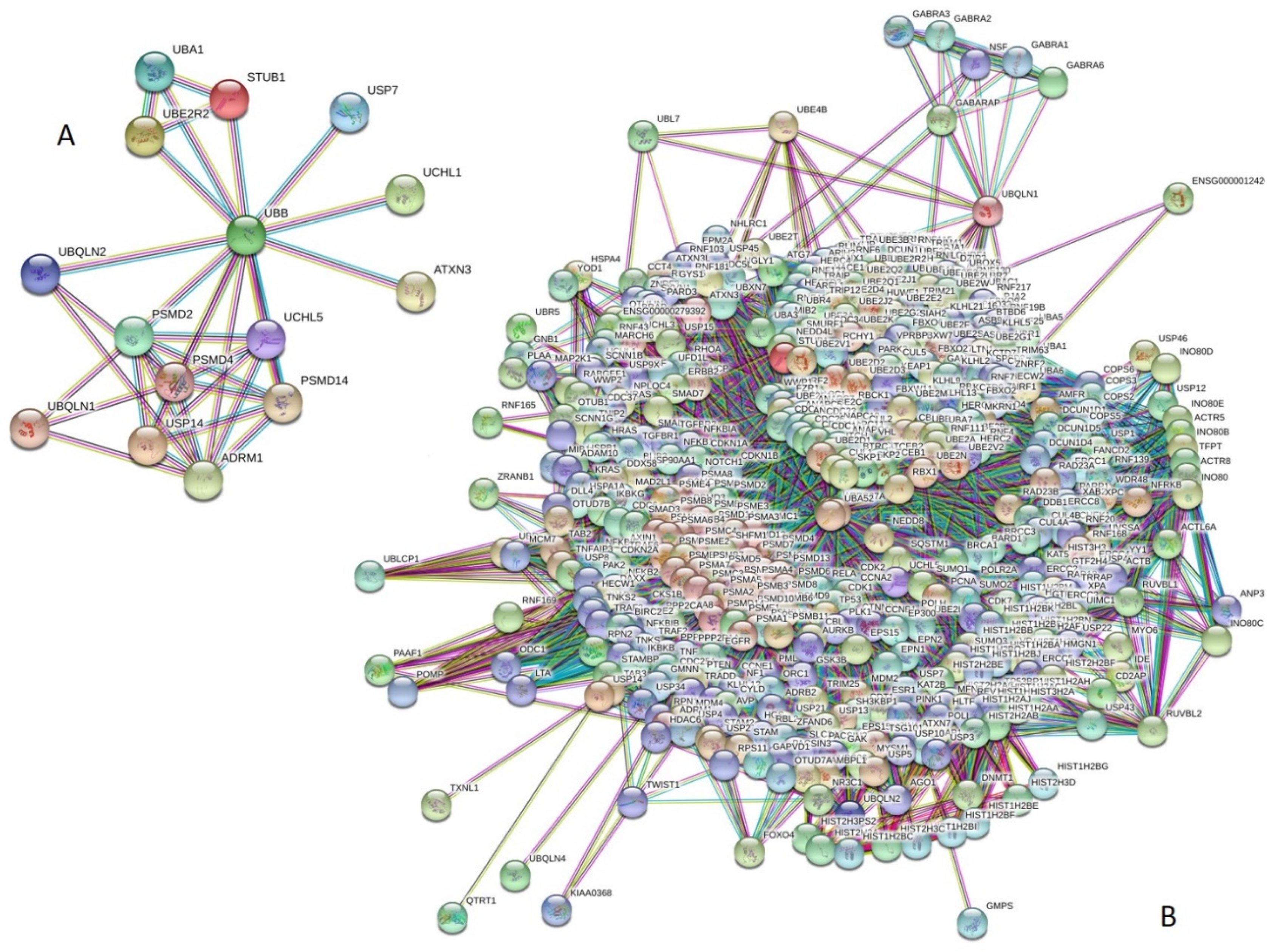
2.4. Protein–Protein Interaction Using STRING
2.5. Representation of IDPs and MoRFs
3. Results and Discussion
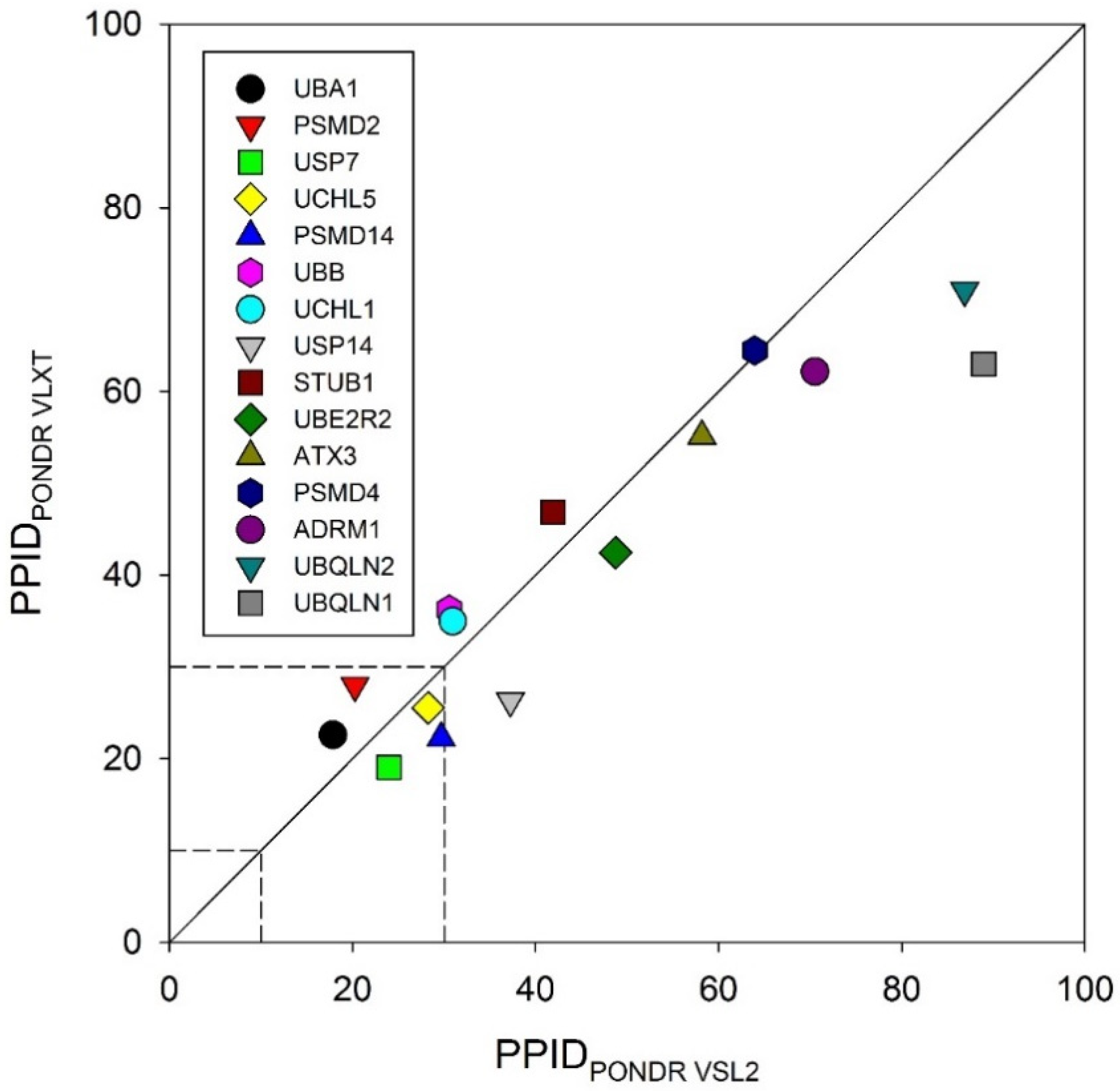
3.1. Intrinsic Disorder in the Proteins of Ubiquitin Proteasomal System
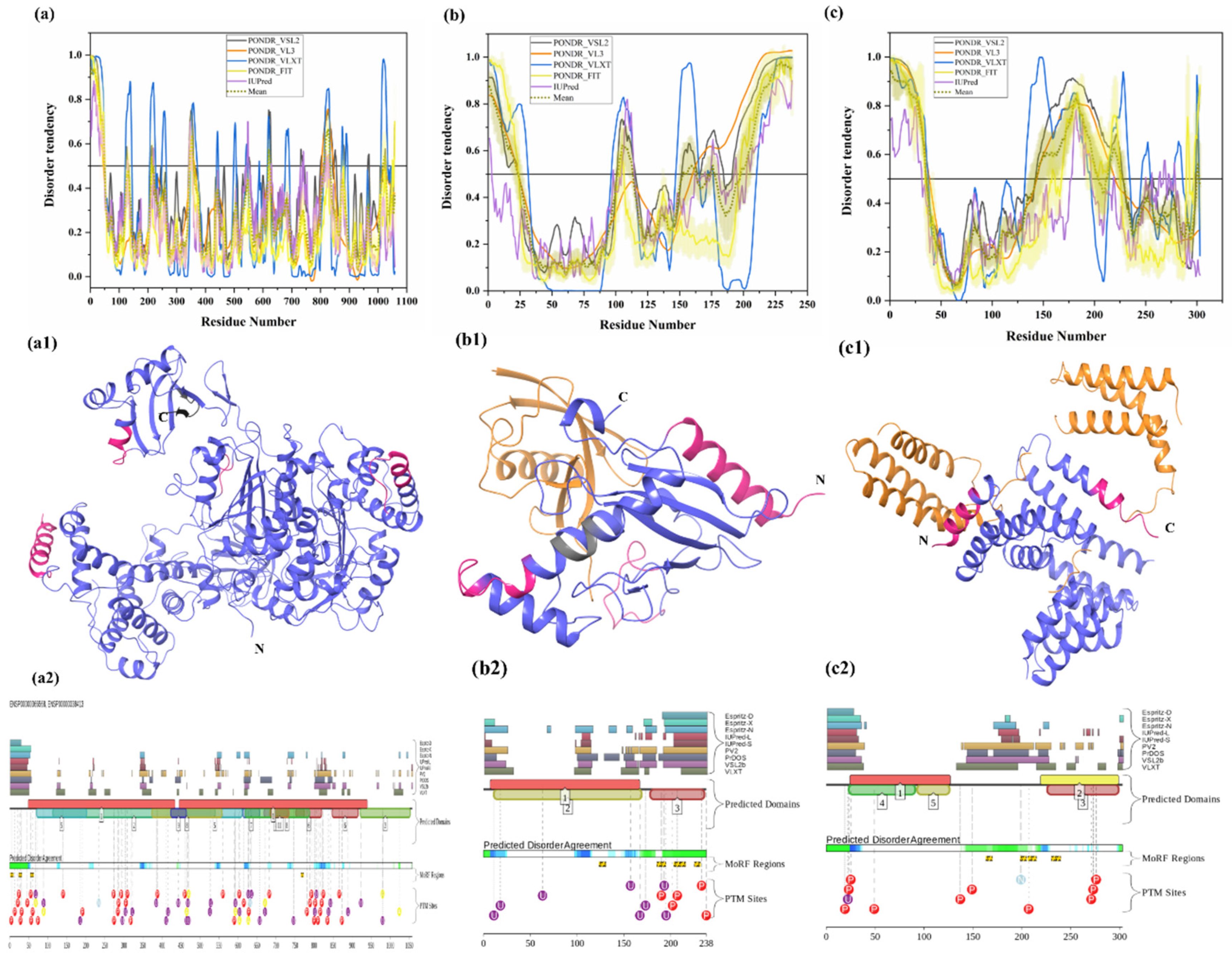
3.1.1. Intrinsic Disorder in Ubiquitin-Activating Enzyme (E1 Enzyme)
3.1.2. Intrinsic Disorder in Ubiquitin-Conjugating Enzyme (E2 Enzyme)
3.1.3. Intrinsic Disorder in Ubiquitin Ligase (E3 Enzyme)
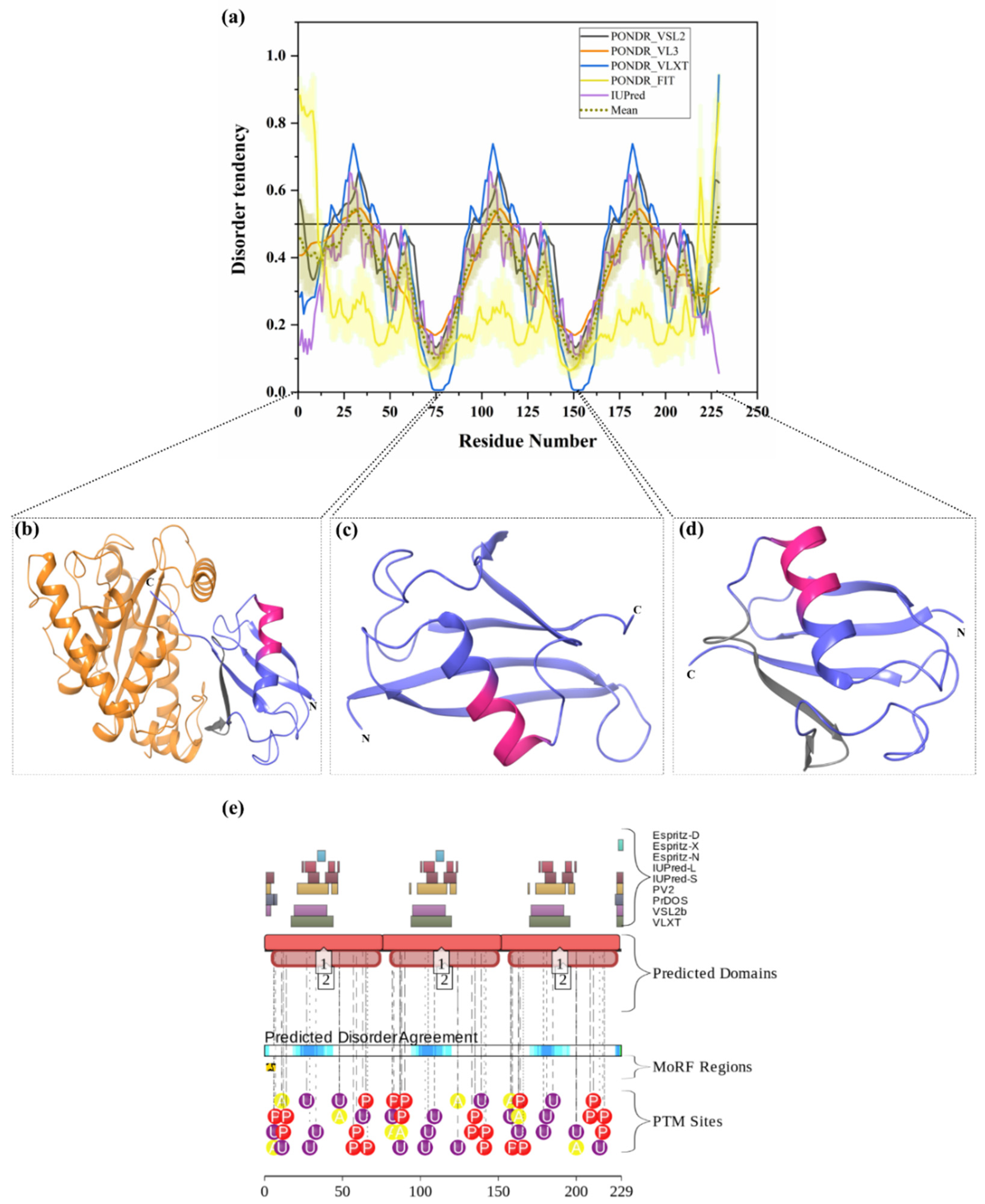
3.1.4. Intrinsic Disorder in Polyubiquitin-B (UBB)
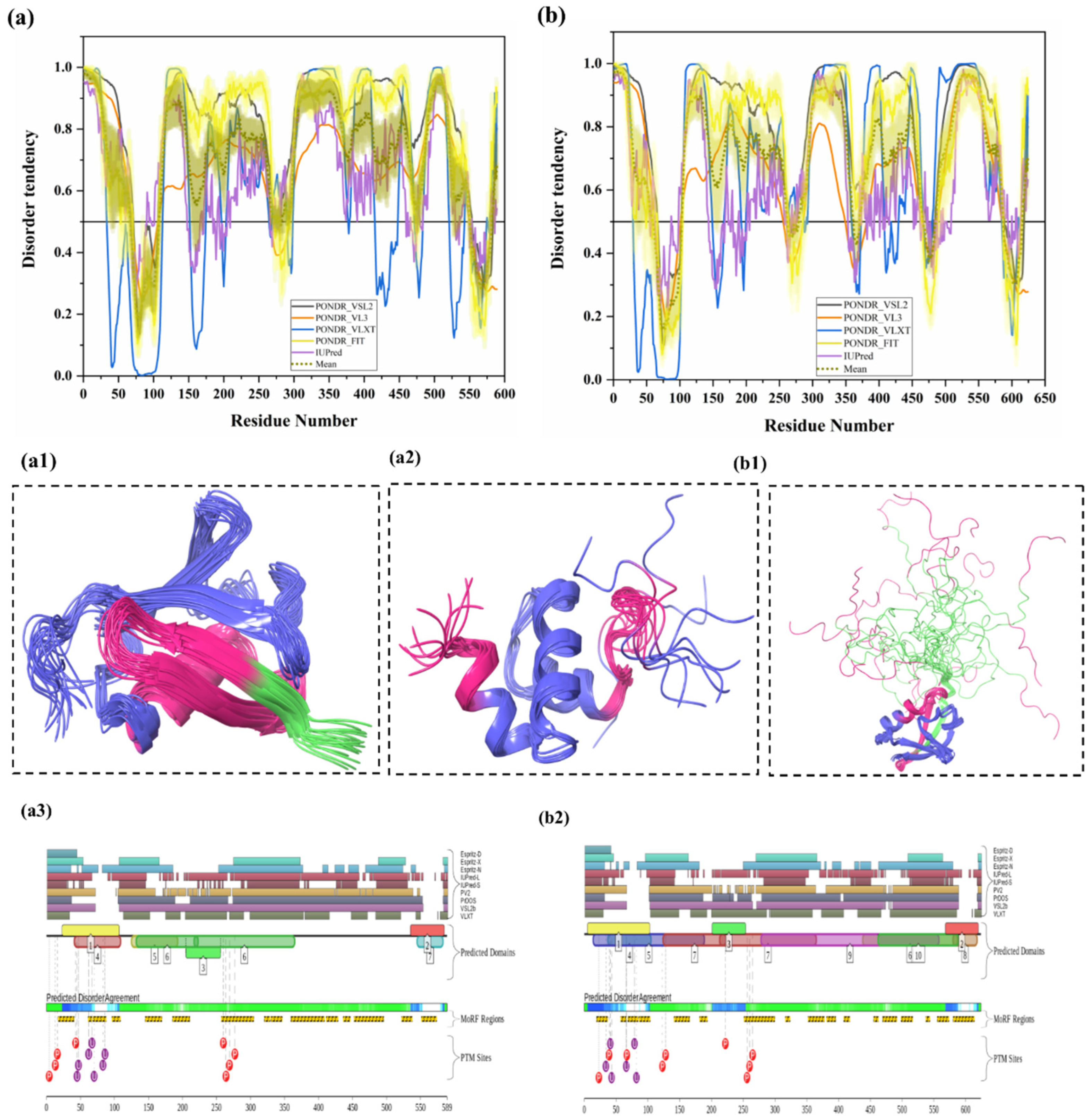
3.1.5. Intrinsic Disorder in Ubiquilin 1 (UBQLN1)
3.1.6. Intrinsic Disorder in Ubiquilin 2 (UBQLN2)
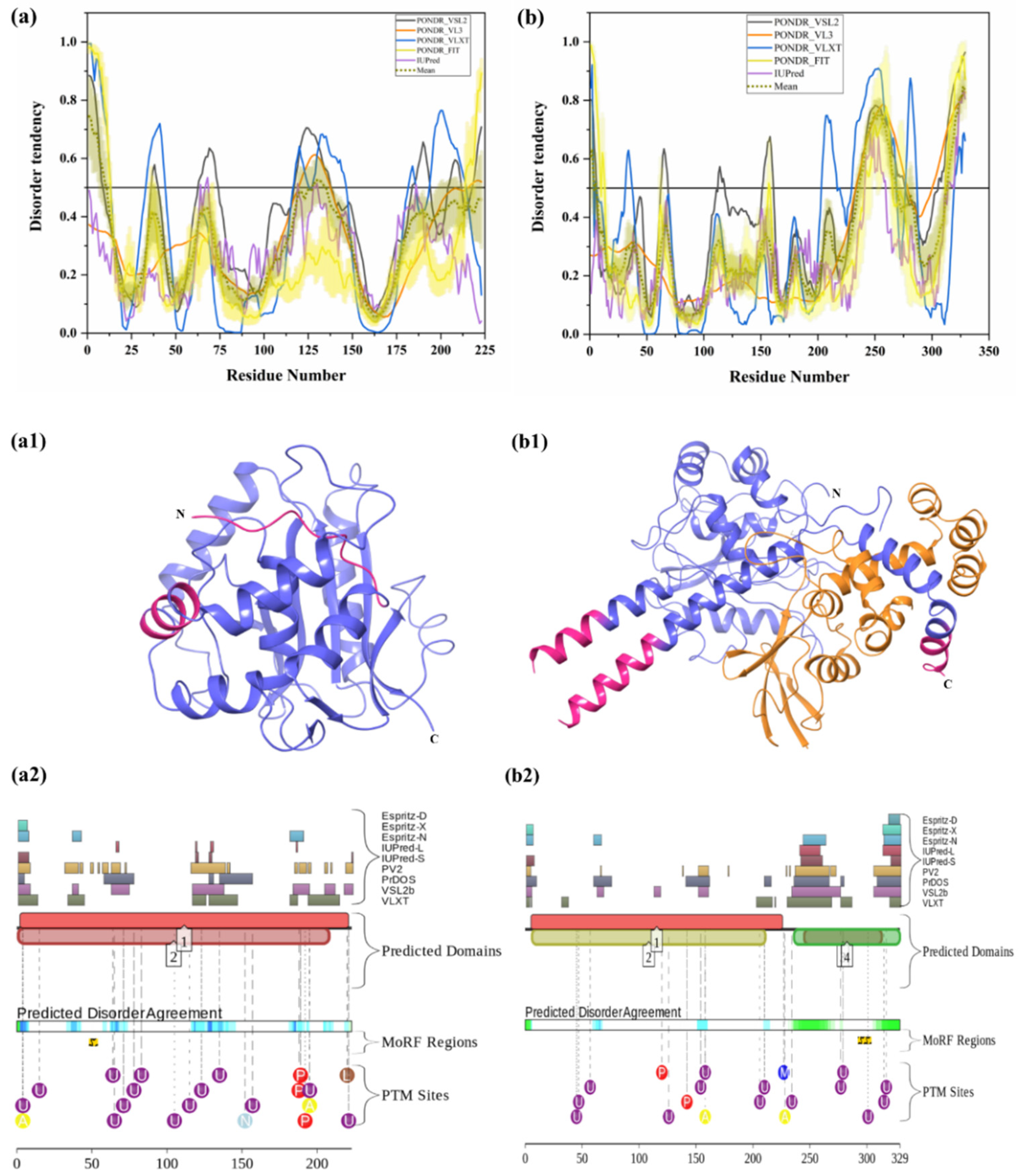
3.1.7. Intrinsic Disorder in Ubiquitin C-Terminal Hydrolase Isozyme L1 (UCHL1)
3.1.8. Intrinsic Disorder in Ubiquitin C-terminal Hydrolase Isozyme L5 (UCHL5)
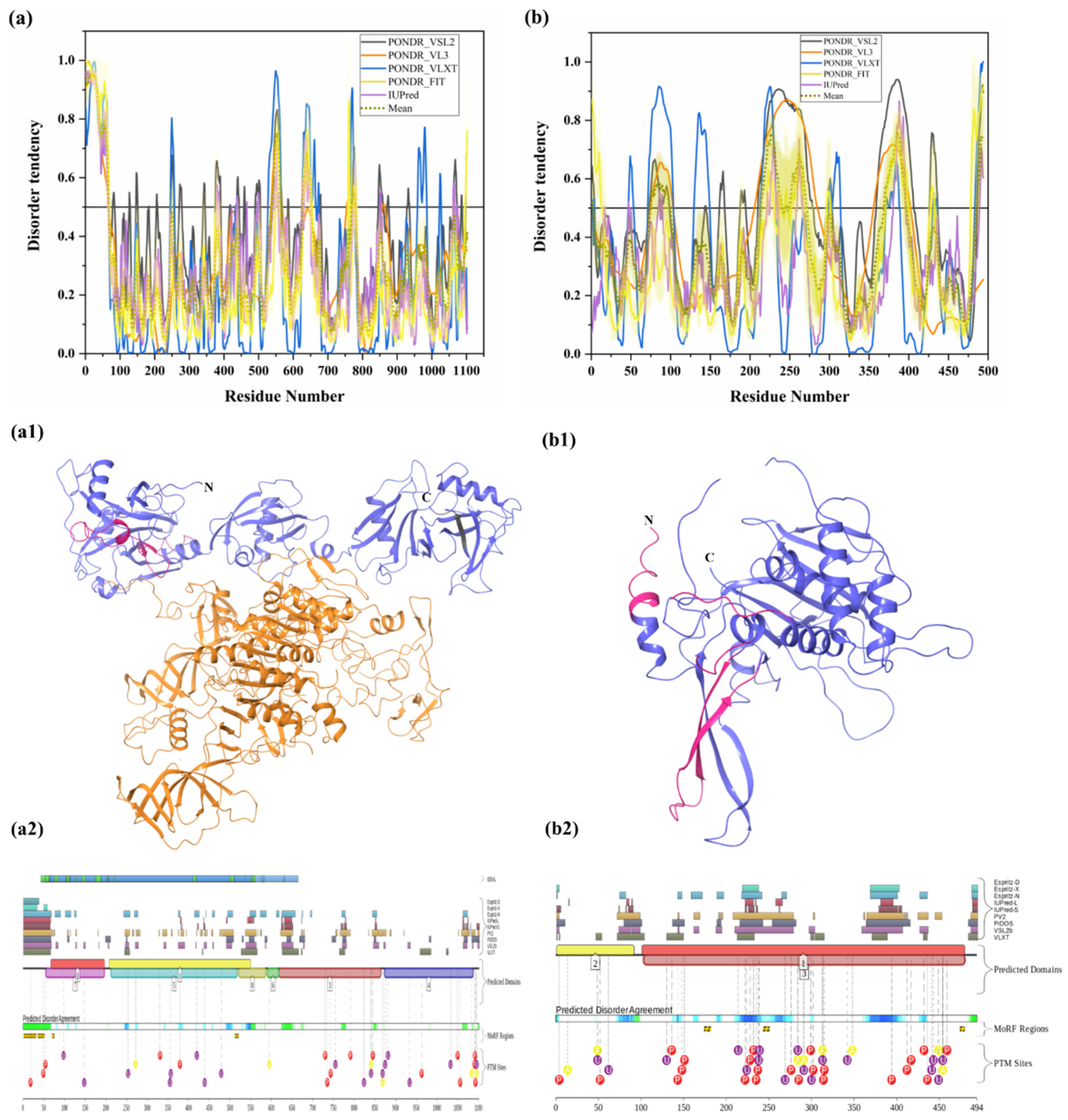
3.1.9. Intrinsic Disorder in Ubiquitin-Specific-Processing Protease 7 (USP7)
3.1.10. Intrinsic Disorder in Ubiquitin Carboxyl-Terminal Hydrolase 14 (USP14)
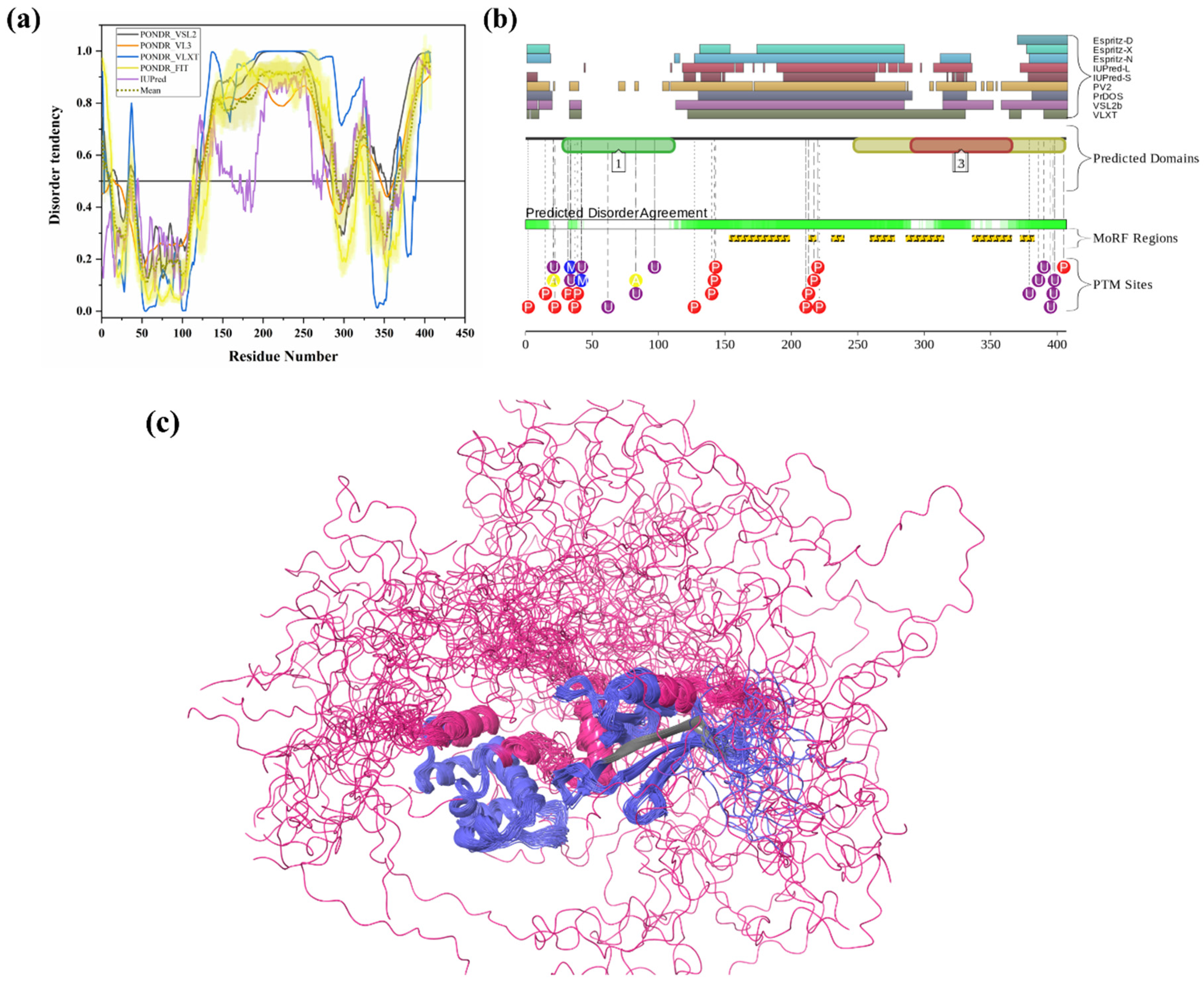
3.1.11. Intrinsic Disorder in Ataxin-3 (ATXN3)
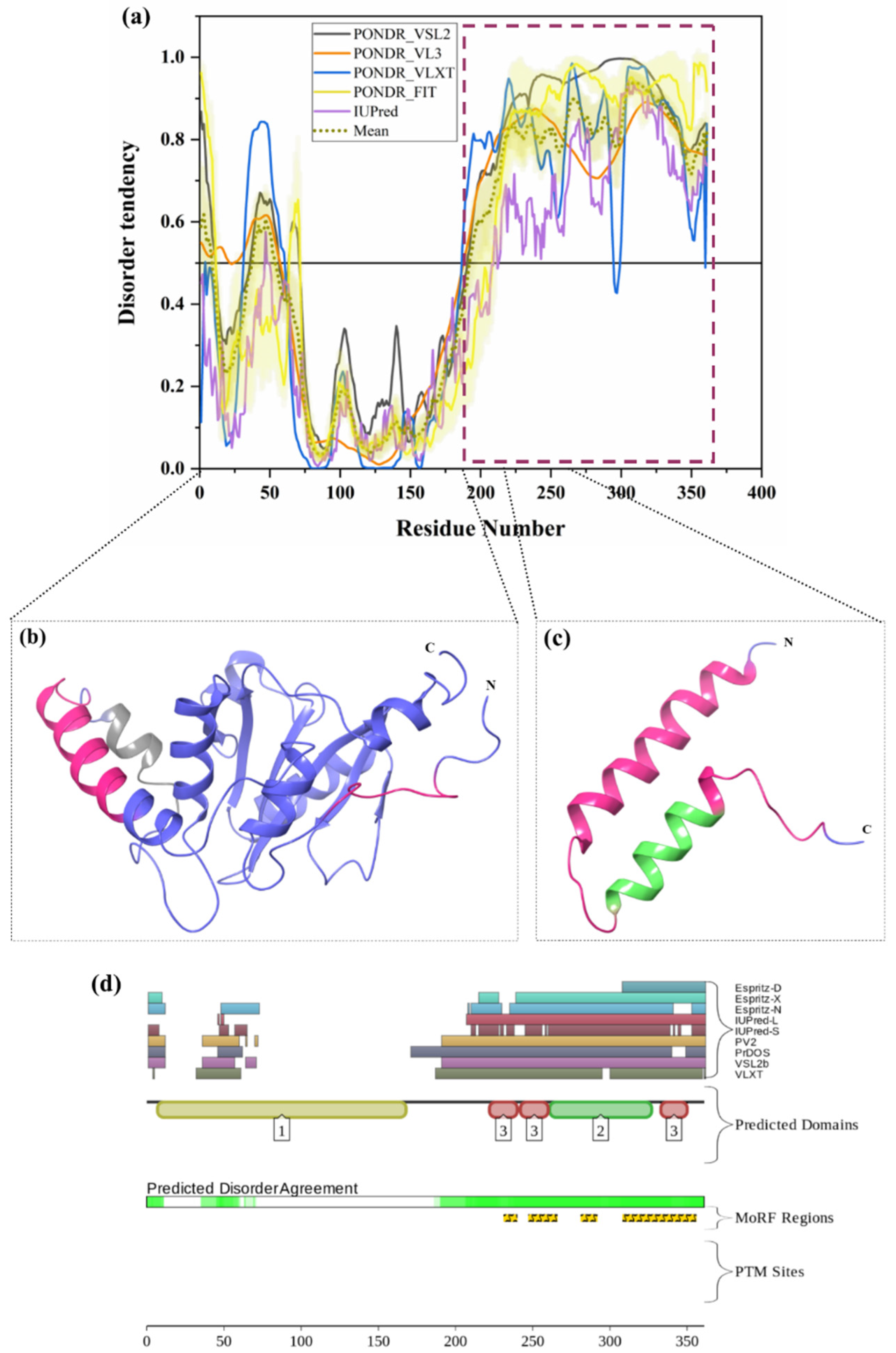
3.1.12. Intrinsic Disorder in Adhesion-Regulating Molecule 1 (ADRM1)
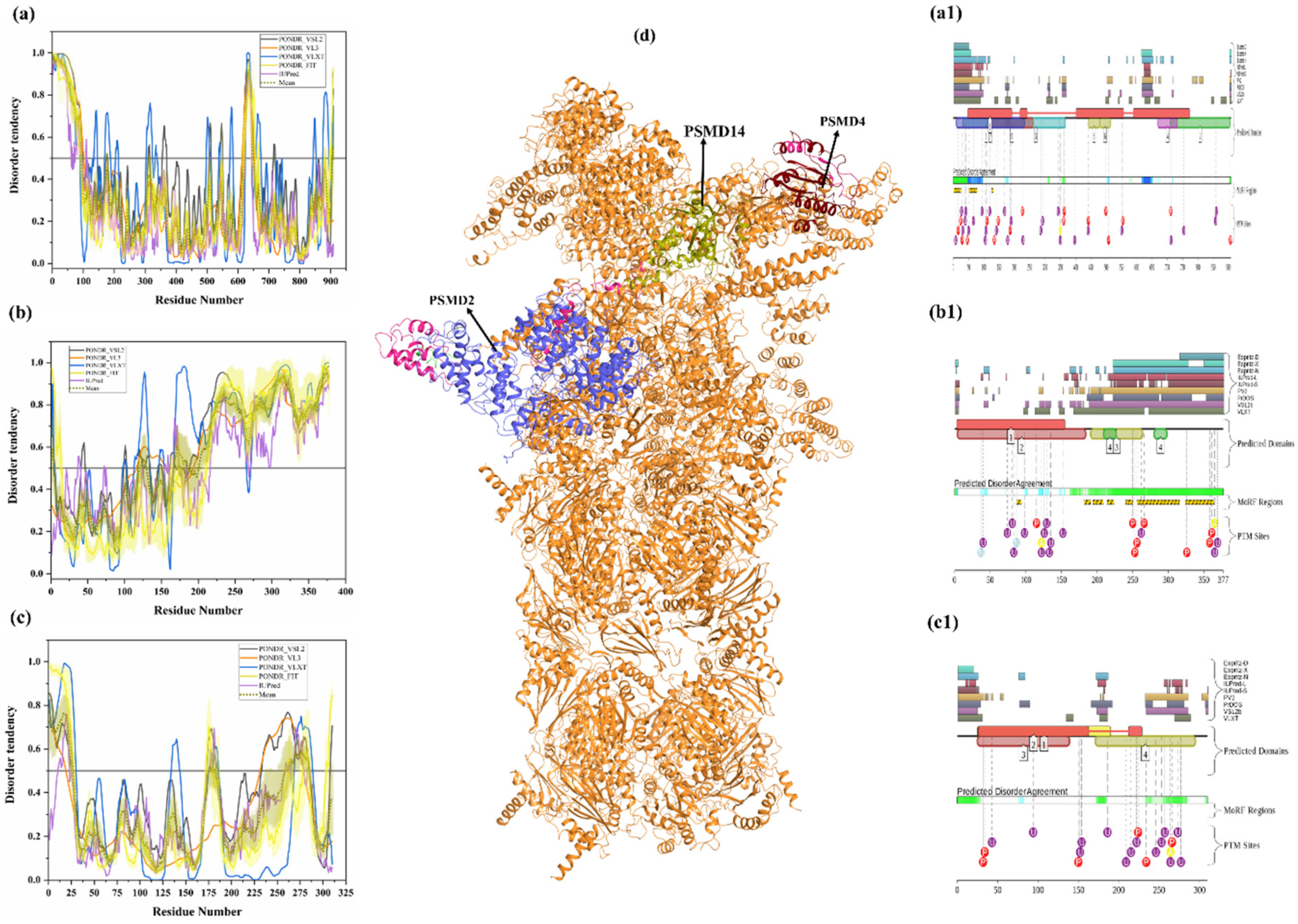
3.1.13. Intrinsic Disorder in 26S Proteasome Non-ATPase Regulatory Subunit 2 (PSMD2)
3.1.14. Intrinsic Disorder in 26S Proteasome Non-ATPase Regulatory Subunit 4 (PSMD4)
3.1.15. Intrinsic Disorder in 26S Proteasome Non-ATPase Regulatory Subunit 14 (PSMD14)
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Etlinger, J.D.; Goldberg, A.L. A soluble ATP dependent proteolytic system responsible for the degradation of abnormal proteins in reticulocytes. Proc. Natl. Acad. Sci. USA 1977, 74, 54–58. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wilkinson, K.D. The discovery of ubiquitin-dependent proteolysis. Proc. Natl. Acad. Sci. USA 2005, 102, 15280–15282. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ciechanover, A.; Heller, H.; Elias, S.; Haas, A.L.; Hershko, A. U2 R ATP dependence1980 Ciechanover. Proc. Natl. Acad. Sci. USA 1980, 77, 1365–1368. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hershko, A.; Ciechanover, A.; Rose, I.A. Resolution of the ATP dependent proteolytic system from reticulocytes: A component that interacts with ATP. Proc. Natl. Acad. Sci. USA 1979, 76, 3107–3110. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goldberg, A.L. Protein degradation and protection against misfolded or damaged proteins. Nature 2003, 426, 895–899. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Saunders, A.J. An emerging role for Ubiquilin 1 in regulating protein quality control system and in disease pathogenesis. Discov. Med. 2009, 8, 18–22. [Google Scholar]
- He, M.; Zhou, Z.; Shah, A.A.; Zou, H.; Tao, J.; Chen, Q.; Wan, Y. The emerging role of deubiquitinating enzymes in genomic integrity, diseases, and therapeutics. Cell Biosci. 2016, 6, 1–15. [Google Scholar] [CrossRef] [Green Version]
- Tu, Y.; Chen, C.; Pan, J.; Xu, J.; Zhou, Z.G.; Wang, C.Y. The ubiquitin proteasome pathway (UPP) in the regulation of cell cycle control and DNA damage repair and its implication in tumorigenesis. Int. J. Clin. Exp. Pathol. 2012, 5, 726–738. [Google Scholar]
- Dwane, L.; Gallagher, W.M.; Chonghaile, T.N.; O’Connor, D.P. The emerging role of nontraditional ubiquitination in oncogenic pathways. J. Biol. Chem. 2017, 292, 3543–3551. [Google Scholar] [CrossRef] [Green Version]
- Bhattacharya, S.; Chakraborty, D.; Basu, M.; Ghosh, M.K. Emerging insights into HAUSP (USP7) in physiology, cancer and other diseases. Signal Transduct. Target. Ther. 2018, 3, 1–12. [Google Scholar] [CrossRef]
- Gadhave, K.; Bolshette, N.; Ahire, A.; Pardeshi, R.; Thakur, K.; Trandafir, C.; Istrate, A.; Ahmed, S.; Lahkar, M.; Muresanu, D.F.; et al. The ubiquitin proteasomal system: A potential target for the management of Alzheimer’s disease. J. Cell. Mol. Med. 2016, 20, 1392–1407. [Google Scholar] [CrossRef] [PubMed]
- Bedford, L.; Paine, S.; Sheppard, P.W.; Mayer, R.J.; Roelofs, J. Assembly, structure, and function of the 26S proteasome. Trends Cell Biol. 2010, 20, 391–401. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kulathu, Y.; Komander, D. Atypical ubiquitylation-the unexplored world of polyubiquitin beyond Lys48 and Lys63 linkages. Nat. Rev. Mol. Cell Biol. 2012, 13, 508–523. [Google Scholar] [CrossRef]
- Li, W.; Ye, Y. Polyubiquitin chains: Functions, structures, and mechanisms. Cell. Mol. Life Sci. 2008, 65, 2397–2406. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tian, Y.; Ding, W.; Wang, Y.; Ji, T.; Sun, S.; Mo, Q.; Chen, P.; Fang, Y.; Liu, J.; Wang, B.; et al. Ubiquitin B in cervical cancer: Critical for the maintenance of cancer stem-like cell characters. PLoS ONE 2013, 8, e84457. [Google Scholar] [CrossRef] [PubMed]
- Dennissen, F.J.A.; Kholod, N.; Hermes, D.J.H.P.; Kemmerling, N.; Steinbusch, H.W.M.; Dantuma, N.P.; van Leeuwen, F.W. Mutant ubiquitin (UBB+1) associated with neurodegenerative disorders is hydrolyzed by ubiquitin C-terminal hydrolase L3 (UCH-L3). FEBS Lett. 2011, 585, 2568–2574. [Google Scholar] [CrossRef] [Green Version]
- Dennissen, F.J.A.; Kholod, N.; Steinbusch, H.W.M.; Van Leeuwen, F.W. Misframed proteins and neurodegeneration: A novel view on Alzheimer’s and Parkinson’s diseases. Neurodegener. Dis. 2010, 7, 76–79. [Google Scholar] [CrossRef]
- Van Leeuwen, F.W.; De Kleijn, D.P.V.; Van Den Hurk, H.H.; Neubauer, A.; Sonnemans, M.A.F.; Sluijs, J.A.; Köycü, S.; Ramdjielal, R.D.J.; Salehi, A.; Martens, G.J.M.; et al. Frameshift mutants of β amyloid precursor protein and ubiquitin-B in Alzheimer’s and Down patients. Science 1998, 279, 242–247. [Google Scholar] [CrossRef]
- Fischer, D.F.; De Vos, R.A.I.; Van Dijk, R.; De Vrij, F.M.S.; Proper, E.A.; Sonnemans, M.A.F.; Verhage, M.C.; Sluijs, J.A.; Hobo, B.; Zouambia, M.; et al. Disease-specific accumulation of mutant ubiquitin as a marker for proteasomal dysfunction in the brain. FASEB J. 2003, 17, 2014–2024. [Google Scholar] [CrossRef] [Green Version]
- Cook, J.C.; Chock, P.B. Isoforms of mammalian ubiquitin-activating enzyme. J. Biol. Chem. 1992, 267, 24315–24321. [Google Scholar]
- Dlamini, N.; Josifova, D.J.; Paine, S.M.L.; Wraige, E.; Pitt, M.; Murphy, A.J.; King, A.; Buk, S.; Smith, F.; Abbs, S.; et al. Clinical and neuropathological features of X-linked spinal muscular atrophy (SMAX2) associated with a novel mutation in the UBA1 gene. Neuromuscul. Disord. 2013, 23, 391–398. [Google Scholar] [CrossRef]
- David, Y.; Ziv, T.; Admon, A.; Navon, A. The E2 ubiquitin-conjugating enzymes direct polyubiquitination to preferred lysines. J. Biol. Chem. 2010, 285, 8595–8604. [Google Scholar] [CrossRef] [Green Version]
- Alpi, A.F.; Chaugule, V.; Walden, H. Mechanism and disease association of E2-conjugating enzymes: Lessons from UBE2T and UBE2L3. Biochem. J. 2016, 473, 3401–3419. [Google Scholar] [CrossRef]
- Miller, V.M.; Nelson, R.F.; Gouvion, C.M.; Williams, A.; Rodriguez-Lebron, E.; Harper, S.Q.; Davidson, B.L.; Rebagliati, M.R.; Paulson, H.L. CHIP suppresses polyglutamine aggregation and toxicity in vitro and in vivo. J. Neurosci. 2005, 25, 9152–9161. [Google Scholar] [CrossRef] [Green Version]
- Marín, I. The ubiquilin gene family: Evolutionary patterns and functional insights. BMC Evol. Biol. 2014, 14, 63. [Google Scholar] [CrossRef] [Green Version]
- Renaud, L.; Picher-Martel, V.; Codron, P.; Julien, J.-P. Key role of UBQLN2 in pathogenesis of amyotrophic lateral sclerosis and frontotemporal dementia. Acta Neuropathol. Commun. 2019, 7, 103. [Google Scholar] [CrossRef]
- Ravid, T.; Hochstrasser, M. Diversity of degradation signals in the ubiquitin-proteasome system. Nat. Rev. Mol. Cell Biol. 2008, 9, 679–689. [Google Scholar] [CrossRef] [Green Version]
- Jang, H.H. Regulation of Protein Degradation by Proteasomes in Cancer. J. Cancer Prev. 2018, 23, 153–161. [Google Scholar] [CrossRef]
- Reyes-Turcu, F.E.; Ventii, K.H.; Wilkinson, K.D. Regulation and cellular roles of ubiquitin-specific deubiquitinating enzymes. Annu. Rev. Biochem. 2009, 78, 363–397. [Google Scholar] [CrossRef] [Green Version]
- Amerik, A.Y.; Hochstrasser, M. Mechanism and function of deubiquitinating enzymes. Biochim. Biophys. Acta 2004, 1695, 189–207. [Google Scholar] [CrossRef] [Green Version]
- Bishop, P.; Rocca, D.; Henley, J.M. Ubiquitin C-terminal hydrolase L1 (UCH-L1): Structure, distribution and roles in brain function and dysfunction. Biochem. J. 2016, 473, 2453–2462. [Google Scholar] [CrossRef] [Green Version]
- Setsuie, R.; Wang, Y.L.; Mochizuki, H.; Osaka, H.; Hayakawa, H.; Ichihara, N.; Li, H.; Furuta, A.; Sano, Y.; Sun, Y.J.; et al. Dopaminergic neuronal loss in transgenic mice expressing the Parkinson’s disease-associated UCH-L1 I93M mutant. Neurochem. Int. 2007, 50, 119–129. [Google Scholar] [CrossRef]
- Yao, T.; Song, L.; Xu, W.; DeMartino, G.N.; Florens, L.; Swanson, S.K.; Washburn, M.P.; Conaway, R.C.; Conaway, J.W.; Cohen, R.E. Proteasome recruitment and activation of the Uch37 deubiquitinating enzyme by Adrm1. Nat. Cell Biol. 2006, 8, 994–1002. [Google Scholar] [CrossRef]
- Kim, R.Q.; Sixma, T.K.; van Dijk, W.J.; Sixma, T.K.; Faesen, A.C.; Luna-Vargas, M.P.A.; Geurink, P.P.; Clerici, M.; Merkx, R.; van Dijk, W.J.; et al. Mechanism of USP7/HAUSP activation by its C-Terminal ubiquitin-like domain and allosteric regulation by GMP-synthetase. Mol. Cell 2016, 281, 147–159. [Google Scholar]
- Hu, M.; Li, P.; Song, L.; Jeffrey, P.D.; Chenova, T.A.; Wilkinson, K.D.; Cohen, R.E.; Shi, Y. Structure and mechanisms of the proteasome-associated deubiquitinating enzyme USP14. EMBO J. 2005, 24, 3747–3756. [Google Scholar] [CrossRef]
- Miller, S.; Ortuno, D.; Carlisle, H.J. Does inactivation of USP14 enhance degradation of proteasomal substrates that are associated with neurodegenerative diseases? F1000Research 2016, 5, 137. [Google Scholar]
- Gomes, A.V. Genetics of proteasome diseases. Scientifica 2013, 2013, 637629. [Google Scholar] [CrossRef] [Green Version]
- Luo, G.; Hu, N.; Xia, X.; Zhou, J.; Ye, C. RPN11 deubiquitinase promotes proliferation and migration of breast cancer cells. Mol. Med. Rep. 2017, 16, 331–338. [Google Scholar] [CrossRef]
- Evers, M.M.; Toonen, L.J.A.; van Roon-Mom, W.M.C. Ataxin-3 protein and RNA toxicity in spinocerebellar ataxia type 3: Current insights and emerging therapeutic strategies. Mol. Neurobiol. 2014, 49, 1513–1531. [Google Scholar] [CrossRef] [Green Version]
- Tanaka, K. The proteasome: Overview of structure and functions. Proc. Japan Acad. Ser. B Phys. Biol. Sci. 2009, 85, 12–36. [Google Scholar] [CrossRef] [Green Version]
- Lu, X.; Nowicka, U.; Sridharan, V.; Liu, F.; Randles, L.; Hymel, D.; Dyba, M.; Tarasov, S.G.; Tarasova, N.I.; Zhao, X.Z.; et al. Structure of the Rpn13-Rpn2 complex provides insights for Rpn13 and Uch37 as anticancer targets. Nat. Commun. 2017, 8, 15540. [Google Scholar] [CrossRef]
- Collins, G.A.; Goldberg, A.L. The Logic of the 26S Proteasome. Cell 2017, 169, 792–806. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ciechanover, A.; Schwartz, A.L. The ubiquitin system: Pathogenesis of human diseases and drug targeting. Biochim. Biophys. Acta 2004, 1695, 3–17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kulkarni, P.; Uversky, V.N. Intrinsically Disordered Proteins in Chronic Diseases. Biomolecules 2019, 9, 147. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Giri, R.; Morrone, A.; Toto, A.; Brunori, M.; Gianni, S. Structure of the transition state for the binding of c-Myb and KIX highlights an unexpected order for a disordered system. Proc. Natl. Acad. Sci. USA 2013, 110, 14942–14947. [Google Scholar] [CrossRef] [Green Version]
- Gianni, S.; Morrone, A.; Giri, R.; Brunori, M. A folding-after-binding mechanism describes the recognition between the transactivation domain of c-Myb and the KIX domain of the CREB-binding protein. Biochem. Biophys. Res. Commun. 2012, 428, 205–209. [Google Scholar] [CrossRef]
- Toto, A.; Camilloni, C.; Giri, R.; Brunori, M.; Vendruscolo, M.; Gianni, S. Molecular Recognition by Templated Folding of an Intrinsically Disordered Protein. Sci. Rep. 2016, 6, 21994. [Google Scholar] [CrossRef]
- Sharma, N.; Fonin, A.V.; Shpironok, O.G.; Silonov, S.A.; Turoverov, K.K.; Uversky, V.N.; Kuznetsova, I.M.; Giri, R. Folding perspectives of an intrinsically disordered transactivation domain and its single mutation breaking the folding propensity. Int. J. Biol. Macromol. 2019. [Google Scholar] [CrossRef]
- Gadhave, K.; Giri, R. Amyloid formation by intrinsically disordered trans-activation domain of cMyb. Biochem. Biophys. Res. Commun. 2020, 524, 446–452. [Google Scholar] [CrossRef]
- Habchi, J.; Tompa, P.; Longhi, S.; Uversky, V.N. Introducing protein intrinsic disorder. Chem. Rev. 2014, 114, 6561–6588. [Google Scholar] [CrossRef] [Green Version]
- Perdigão, N.; Heinrich, J.; Stolte, C.; Sabir, K.S.; Buckley, M.J.; Tabor, B.; Signal, B.; Gloss, B.S.; Hammang, C.J.; Rost, B.; et al. Unexpected features of the dark proteome. Proc. Natl. Acad. Sci. USA 2015, 112, 15898–15903. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Basile, W.; Salvatore, M.; Bassot, C.; Elofsson, A. Why do eukaryotic proteins contain more intrinsically disordered regions? PLoS Comput. Biol. 2019, 15, e1007186. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kumar, D.; Sharma, N.; Giri, R. Therapeutic interventions of cancers using intrinsically disordered proteins as drug targets: C-myc as model system. Cancer Inform. 2017, 16, 1176935117699408. [Google Scholar] [CrossRef] [PubMed]
- Aarthy, M.; Kumar, D.; Giri, R.; Singh, S.K. E7 oncoprotein of human papillomavirus: Structural dynamics and inhibitor screening study. Gene 2018, 658, 159–177. [Google Scholar] [CrossRef] [PubMed]
- Kumar, A.; Kumar, P.; Kumari, S.; Uversky, V.N.; Giri, R. Folding and structural polymorphism of p53 C-terminal domain: One peptide with many conformations. Arch. Biochem. Biophys. 2020, 684, 108342. [Google Scholar] [CrossRef] [PubMed]
- Gadhave, K.; Gehi, B.R.; Kumar, P.; Xue, B.; Uversky, V.N.; Giri, R. The dark side of Alzheimer’s disease: Unstructured biology of proteins from the amyloid cascade signaling pathway. Cell. Mol. Life Sci. 2020, 1–46. [Google Scholar] [CrossRef]
- Boomsma, W.; Nielsen, S.V.; Lindorff-Larsen, K.; Hartmann-Petersen, R.; Ellgaard, L. Bioinformatics analysis identifies several intrinsically disordered human E3 ubiquitin-protein ligases. PeerJ 2016, 2016, e1725. [Google Scholar] [CrossRef] [Green Version]
- Bhowmick, P.; Pancsa, R.; Guharoy, M.; Tompa, P. Functional Diversity and Structural Disorder in the Human Ubiquitination Pathway. PLoS ONE 2013, 8, e65443. [Google Scholar] [CrossRef] [Green Version]
- Guharoy, M.; Bhowmick, P.; Tompa, P. Design principles involving protein disorder facilitate specific substrate selection and degradation by the ubiquitin-proteasome system. J. Biol. Chem. 2016, 291, 6723–6731. [Google Scholar] [CrossRef] [Green Version]
- Manasanch, E.E.; Orlowski, R.Z. Proteasome inhibitors in cancer therapy. Nat. Rev. Clin. Oncol. 2017, 14, 417–433. [Google Scholar] [CrossRef]
- Lim, K.H. Diverse Misfolded Conformational Strains and Cross-seeding of Misfolded Proteins Implicated in Neurodegenerative Diseases. Front. Mol. Neurosci. 2019, 12, 158. [Google Scholar] [CrossRef] [PubMed]
- Bateman, A. UniProt: A worldwide hub of protein knowledge. Nucleic Acids Res. 2019, 47, D506–D515. [Google Scholar]
- Lee, I.; Schindelin, H. Structural insights into E1-catalyzed ubiquitin activation and transfer to conjugating enzymes. Cell 2008, 134, 268–278. [Google Scholar] [CrossRef] [Green Version]
- Groen, E.J.N.; Gillingwater, T.H. UBA1: At the Crossroads of Ubiquitin Homeostasis and Neurodegeneration. Trends Mol. Med. 2015, 21, 622–632. [Google Scholar] [CrossRef] [Green Version]
- Valimberti, I.; Tiberti, M.; Lambrughi, M.; Sarcevic, B.; Papaleo, E. E2 superfamily of ubiquitin-conjugating enzymes: Constitutively active or activated through phosphorylation in the catalytic cleft. Sci. Rep. 2015, 5, 14849. [Google Scholar] [CrossRef]
- Kumar, P.; Ambasta, R.K.; Veereshwarayya, V.; Rosen, K.M.; Kosik, K.S.; Band, H.; Mestril, R.; Patterson, C.; Querfurth, H.W. CHIP and HSPs interact with beta-APP in a proteasome-dependent manner and influence Abeta metabolism. Hum. Mol. Genet. 2007, 16, 848–864. [Google Scholar] [CrossRef] [Green Version]
- Stieren, E.S.; El Ayadi, A.; Xiao, Y.; Siller, E.; Landsverk, M.L.; Oberhauser, A.F.; Barral, J.M.; Boehning, D. Ubiquilin-1 is a molecular chaperone for the amyloid precursor protein. J. Biol. Chem. 2011, 286, 35689–35698. [Google Scholar] [CrossRef] [Green Version]
- Williams, K.L.; Warraich, S.T.; Yang, S.; Solski, J.A.; Fernando, R.; Rouleau, G.A.; Nicholson, G.A.; Blair, I.P. UBQLN2/ubiquilin 2 mutation and pathology in familial amyotrophic lateral sclerosis. Neurobiol. Aging 2012, 33, 2527.e3–2527.e10. [Google Scholar] [CrossRef]
- Zhang, K.Y.; Yang, S.; Warraich, S.T.; Blair, I.P. Ubiquilin 2: A component of the ubiquitin-proteasome system with an emerging role in neurodegeneration. Int. J. Biochem. Cell Biol. 2014, 50, 123–126. [Google Scholar] [CrossRef]
- Das, C.; Hoang, Q.Q.; Kreinbring, C.A.; Luchansky, S.J.; Meray, R.K.; Ray, S.S.; Lansbury, P.T.; Ringe, D.; Petsko, G.A. Structural basis for conformational plasticity of the Parkinson’s disease-associated ubiquitin hydrolase UCH-L1. Proc. Natl. Acad. Sci. USA 2006, 103, 4675–4680. [Google Scholar] [CrossRef] [Green Version]
- Fang, Y.; Fu, D.; Tang, W.; Cai, Y.; Ma, D.; Wang, H.; Xue, R.; Liu, T.; Huang, X.; Dong, L.; et al. Ubiquitin C-terminal Hydrolase 37, a novel predictor for hepatocellular carcinoma recurrence, promotes cell migration and invasion via interacting and deubiquitinating PRP19. Biochim. Biophys. Acta 2013, 1833, 559–572. [Google Scholar] [CrossRef] [Green Version]
- Kim, E.; Park, S.; Lee, J.H.; Mun, J.Y.; Choi, W.H.; Yun, Y.; Lee, J.; Kim, J.H.; Kang, M.-J.; Lee, M.J. Dual Function of USP14 Deubiquitinase in Cellular Proteasomal Activity and Autophagic Flux. Cell Rep. 2018, 24, 732–743. [Google Scholar] [CrossRef] [Green Version]
- Blount, J.R.; Tsou, W.-L.; Ristic, G.; Burr, A.A.; Ouyang, M.; Galante, H.; Scaglione, K.M.; Todi, S. V Ubiquitin-binding site 2 of ataxin-3 prevents its proteasomal degradation by interacting with Rad23. Nat. Commun. 2014, 5, 4638. [Google Scholar] [CrossRef] [Green Version]
- Tzvetkov, N.; Breuer, P. Josephin domain-containing proteins from a variety of species are active de-ubiquitination enzymes. Biol. Chem. 2007, 388, 973–978. [Google Scholar] [CrossRef]
- Qiu, X.-B.; Ouyang, S.-Y.; Li, C.-J.; Miao, S.; Wang, L.; Goldberg, A.L. hRpn13/ADRM1/GP110 is a novel proteasome subunit that binds the deubiquitinating enzyme, UCH37. EMBO J. 2006, 25, 5742–5753. [Google Scholar] [CrossRef] [Green Version]
- Byrne, A.; McLaren, R.P.; Mason, P.; Chai, L.; Dufault, M.R.; Huang, Y.; Liang, B.; Gans, J.D.; Zhang, M.; Carter, K.; et al. Knockdown of human deubiquitinase PSMD14 induces cell cycle arrest and senescence. Exp. Cell Res. 2010, 316, 258–271. [Google Scholar] [CrossRef]
- Peng, K.; Radivojac, P.; Vucetic, S.; Dunker, A.K.; Obradovic, Z. Length-dependent prediction of protein intrinsic disorder. BMC Bioinformatics 2006, 7, 208. [Google Scholar] [CrossRef] [Green Version]
- Peng, K.; Vucetic, S.; Radivojac, P.; Brown, C.J.; Dunker, A.K.; Obradovic, Z. Optimizing long intrinsic disorder predictors with protein evolutionary information. J. Bioinform. Comput. Biol. 2005, 3, 35–60. [Google Scholar] [CrossRef]
- Romero, P.; Obradovic, Z.; Li, X.; Garner, E.C.; Brown, C.J.; Dunker, A.K. Sequence complexity of disordered protein. Proteins Struct. Funct. Genet. 2001, 42, 38–48. [Google Scholar] [CrossRef]
- Xue, B.; Dunbrack, R.L.; Williams, R.W.; Dunker, A.K.; Uversky, V.N. PONDR-FIT: A meta-predictor of intrinsically disordered amino acids. Biochim. Biophys. Acta 2010, 1804, 996–1010. [Google Scholar] [CrossRef] [Green Version]
- Dosztányi, Z.; Csizmok, V.; Tompa, P.; Simon, I. IUPred: Web server for the prediction of intrinsically unstructured regions of proteins based on estimated energy content. Bioinformatics 2005, 21, 3433–3434. [Google Scholar] [CrossRef] [Green Version]
- Garg, N.; Kumar, P.; Gadhave, K.; Giri, R. The dark proteome of cancer: Intrinsic disorderedness and functionality of HIF-1α along with its interacting proteins. Prog. Mol. Biol. Transl. Sci. 2019, 166, 371–403. [Google Scholar]
- Kumar, D.; Singh, A.; Kumar, P.; Uversky, V.N.; Rao, C.D.; Giri, R. Understanding the penetrance of intrinsic protein disorder in rotavirus proteome. Int. J. Biol. Macromol. 2019, 144, 892–908. [Google Scholar] [CrossRef]
- Mishra, P.M.; Uversky, V.N.; Giri, R. Molecular Recognition Features in Zika Virus Proteome. J. Mol. Biol. 2018, 430, 2372–2388. [Google Scholar] [CrossRef]
- Giri, R.; Kumar, D.; Sharma, N.; Uversky, V.N. Intrinsically Disordered Side of the Zika Virus Proteome. Front. Cell. Infect. Microbiol. 2016, 6, 144. [Google Scholar] [CrossRef]
- Singh, A.; Kumar, A.; Yadav, R.; Uversky, V.N.; Giri, R. Deciphering the dark proteome of Chikungunya virus. Sci. Rep. 2018, 8, 5822. [Google Scholar] [CrossRef] [Green Version]
- Malhis, N.; Jacobson, M.; Gsponer, J. MoRFchibi SYSTEM: Software tools for the identification of MoRFs in protein sequences. Nucleic Acids Res. 2016, 44, W488–W493. [Google Scholar] [CrossRef] [Green Version]
- Dosztányi, Z.; Mészáros, B.; Simon, I. ANCHOR: Web server for predicting protein binding regions in disordered proteins. Bioinformatics 2009, 25, 2745–2746. [Google Scholar] [CrossRef] [Green Version]
- Disfani, F.M.; Hsu, W.-L.; Mizianty, M.J.; Oldfield, C.J.; Xue, B.; Dunker, A.K.; Uversky, V.N.; Kurgan, L. MoRFpred, a computational tool for sequence-based prediction and characterization of short disorder-to-order transitioning binding regions in proteins. Bioinformatics 2012, 28, i75–i83. [Google Scholar] [CrossRef] [Green Version]
- Jones, D.T.; Cozzetto, D. DISOPRED3: Precise disordered region predictions with annotated protein-binding activity. Bioinformatics 2015, 31, 857–863. [Google Scholar] [CrossRef]
- Giri, R.; Bhardwaj, T.; Shegane, M.; Gehi, B.R.; Kumar, P.; Gadhave, K.; Oldfield, C.J.; Uversky, V.N. When Darkness Becomes a Ray of Light in the Dark Times: Understanding the COVID-19 via the Comparative Analysis of the Dark Proteomes of SARS-CoV-2, Human SARS and Bat SARS-Like Coronaviruses. bioRxiv 2020. [Google Scholar] [CrossRef] [Green Version]
- Singh, A.; Kumar, A.; Uversky, V.N.; Giri, R. Understanding the interactability of chikungunya virus proteins: Via molecular recognition feature analysis. RSC Adv. 2018, 8, 27293–27303. [Google Scholar] [CrossRef] [Green Version]
- Oates, M.E.; Romero, P.; Ishida, T.; Ghalwash, M.; Mizianty, M.J.; Xue, B.; Dosztányi, Z.; Uversky, V.N.; Obradovic, Z.; Kurgan, L.; et al. D2P2: Database of disordered protein predictions. Nucleic Acids Res. 2013, 41, 508–516. [Google Scholar] [CrossRef] [Green Version]
- Szklarczyk, D.; Gable, A.L.; Lyon, D.; Junge, A.; Wyder, S.; Huerta-Cepas, J.; Simonovic, M.; Doncheva, N.T.; Morris, J.H.; Bork, P.; et al. STRING v11: Protein-protein association networks with increased coverage, supporting functional discovery in genome-wide experimental datasets. Nucleic Acids Res. 2019, 47, 607–613. [Google Scholar] [CrossRef] [Green Version]
- Ciechanover, A.; Schwartz, A.L. The ubiquitin-proteasome pathway: The complexity and myriad functions of proteins death. Proc. Natl. Acad. Sci. USA 1998, 95, 2727–2730. [Google Scholar] [CrossRef] [Green Version]
- Wehmer, M.; Sakata, E. Recent advances in the structural biology of the 26S proteasome. Int. J. Biochem. Cell Biol. 2016, 79, 437–442. [Google Scholar] [CrossRef] [Green Version]
- Toh-e, A. Structure and function of the yeast 26S proteasome. Seikagaku 1999, 71, 173–181. [Google Scholar]
- Livneh, I.; Cohen-Kaplan, V.; Cohen-Rosenzweig, C.; Avni, N.; Ciechanover, A. The life cycle of the 26S proteasome: From birth, through regulation and function, and onto its death. Cell Res. 2016, 26, 869–885. [Google Scholar] [CrossRef] [Green Version]
- Marshall, R.S.; Vierstra, R.D. Dynamic Regulation of the 26S Proteasome: From Synthesis to Degradation. Front. Mol. Biosci. 2019, 6, 40. [Google Scholar] [CrossRef]
- Rajagopalan, K.; Mooney, S.M.; Parekh, N.; Getzenberg, R.H.; Kulkarni, P. A majority of the cancer/testis antigens are intrinsically disordered proteins. J. Cell. Biochem. 2011, 112, 3256–3267. [Google Scholar] [CrossRef] [Green Version]
- Ishida, T.; Kinoshita, K. PrDOS: Prediction of disordered protein regions from amino acid sequence. Nucleic Acids Res. 2007, 35, 460–464. [Google Scholar] [CrossRef]
- Walsh, I.; Martin, A.J.M.; Di Domenico, T.; Tosatto, S.C.E. ESpritz: Accurate and fast prediction of protein disorder. Bioinformatics 2012, 28, 503–509. [Google Scholar] [CrossRef] [Green Version]
- Liu, X.; Sun, L.; Gursel, D.B.; Cheng, C.; Huang, S.; Rademaker, A.W.; Khan, S.A.; Yin, J.; Kiyokawa, H. The non-canonical ubiquitin activating enzyme UBA6 suppresses epithelial-mesenchymal transition of mammary epithelial cells. Oncotarget 2017, 8, 87480–87493. [Google Scholar] [CrossRef] [Green Version]
- Liu, H.-Y.; Pfleger, C.M. Mutation in E1, the ubiquitin activating enzyme, reduces Drosophila lifespan and results in motor impairment. PLoS ONE 2013, 8, e32835. [Google Scholar] [CrossRef] [Green Version]
- Ramser, J.; Ahearn, M.E.; Lenski, C.; Yariz, K.O.; Hellebrand, H.; von Rhein, M.; Clark, R.D.; Schmutzler, R.K.; Lichtner, P.; Hoffman, E.P.; et al. Rare missense and synonymous variants in UBE1 are associated with X-linked infantile spinal muscular atrophy. Am. J. Hum. Genet. 2008, 82, 188–193. [Google Scholar] [CrossRef] [Green Version]
- Lv, Z.; Williams, K.M.; Yuan, L.; Atkison, J.H.; Olsen, S.K. Crystal structure of a human ubiquitin E1-ubiquitin complex reveals conserved functional elements essential for activity. J. Biol. Chem. 2018, 293, 18337–18352. [Google Scholar] [CrossRef] [Green Version]
- Xie, S.T. Expression, purification, and crystal structure of N-terminal domains of human ubiquitin-activating enzyme (E1). Biosci. Biotechnol. Biochem. 2014, 78, 1542–1549. [Google Scholar] [CrossRef]
- Stewart, M.D.; Ritterhoff, T.; Klevit, R.E.; Brzovic, P.S. E2 enzymes: More than just middle men. Cell Res. 2016, 26, 423–440. [Google Scholar] [CrossRef] [Green Version]
- Pruneda, J.N.; Littlefield, P.J.; Soss, S.E.; Nordquist, K.A.; Chazin, W.J.; Brzovic, P.S.; Klevit, R.E. Structure of an E3:E2~Ub complex reveals an allosteric mechanism shared among RING/U-box ligases. Mol. Cell 2012, 47, 933–942. [Google Scholar] [CrossRef] [Green Version]
- Wenzel, D.M.; Lissounov, A.; Brzovic, P.S.; Klevit, R.E. UBCH7 reactivity profile reveals parkin and HHARI to be RING/HECT hybrids. Nature 2011, 474, 105–108. [Google Scholar] [CrossRef] [Green Version]
- Cohen, I.; Wiener, R.; Reiss, Y.; Ravid, T. Distinct activation of an E2 ubiquitin-conjugating enzyme by its cognate E3 ligases. Proc. Natl. Acad. Sci. USA 2015, 112, 625–632. [Google Scholar] [CrossRef] [Green Version]
- Schelpe, J.; Monté, D.; Dewitte, F.; Sixma, T.K.; Rucktooa, P. Structure of UBE2Z Enzyme Provides Functional Insight into Specificity in the FAT10 Protein Conjugation Machinery. J. Biol. Chem. 2016, 291, 630–639. [Google Scholar] [CrossRef] [Green Version]
- Williams, K.M.; Qie, S.; Atkison, J.H.; Salazar-Arango, S.; Alan Diehl, J.; Olsen, S.K. Structural insights into E1 recognition and the ubiquitin-conjugating activity of the E2 enzyme Cdc34. Nat. Commun. 2019, 10, 3296. [Google Scholar] [CrossRef]
- Spratt, D.E.; Shaw, G.S. Association of the disordered C-terminus of CDC34 with a catalytically bound ubiquitin. J. Mol. Biol. 2011, 407, 425–438. [Google Scholar] [CrossRef]
- Uchida, C.; Kitagawa, M. RING-, HECT-, and RBR-type E3 Ubiquitin Ligases: Involvement in Human Cancer. Curr. Cancer Drug Targets 2016, 16, 157–174. [Google Scholar] [CrossRef]
- Plechanovová, A.; Jaffray, E.G.; Tatham, M.H.; Naismith, J.H.; Hay, R.T. Structure of a RING E3 ligase and ubiquitin-loaded E2 primed for catalysis. Nature 2012, 489, 115–120. [Google Scholar] [CrossRef] [Green Version]
- Dou, H.; Buetow, L.; Sibbet, G.J.; Cameron, K.; Huang, D.T. BIRC7-E2 ubiquitin conjugate structure reveals the mechanism of ubiquitin transfer by a RING dimer. Nat. Struct. Mol. Biol. 2012, 19, 876–883. [Google Scholar] [CrossRef] [Green Version]
- Lorick, K.L.; Jensen, J.P.; Fang, S.; Ong, A.M.; Hatakeyama, S.; Weissman, A.M. RING fingers mediate ubiquitin-conjugating enzyme (E2)-dependent ubiquitination. Proc. Natl. Acad. Sci. USA 1999, 96, 11364–11369. [Google Scholar] [CrossRef] [Green Version]
- Berndsen, C.E.; Wiener, R.; Yu, I.W.; Ringel, A.E.; Wolberger, C. A conserved asparagine has a structural role in ubiquitin-conjugating enzymes. Nat. Chem. Biol. 2013, 9, 154–156. [Google Scholar] [CrossRef] [Green Version]
- Berndsen, C.E.; Wolberger, C. New insights into ubiquitin E3 ligase mechanism. Nat. Struct. Mol. Biol. 2014, 21, 301–307. [Google Scholar] [CrossRef]
- Kamadurai, H.B.; Qiu, Y.; Deng, A.; Harrison, J.S.; MacDonald, C.; Actis, M.; Rodrigues, P.; Miller, D.J.; Souphron, J.; Lewis, S.M.; et al. Mechanism of ubiquitin ligation and lysine prioritization by a HECT E3. Elife 2013, 2, e00828. [Google Scholar] [CrossRef]
- Pickart, C.M. Echanisms ndPickart, C.M. Echanisms nderlying ubiquitination. Annu. Rev. Biochem. 2001, 70, 503–533. [Google Scholar] [CrossRef]
- Oh, C.; Park, S.; Lee, E.K.; Yoo, Y.J. Downregulation of ubiquitin level via knockdown of polyubiquitin gene Ubb as potential cancer therapeutic intervention. Sci. Rep. 2013, 3, 2623. [Google Scholar] [CrossRef] [Green Version]
- Finch, J.S.; St John, T.; Krieg, P.; Bonham, K.; Smith, H.T.; Fried, V.A.; Bowden, G.T. Overexpression of three ubiquitin genes in mouse epidermal tumors is associated with enhanced cellular proliferation and stress. Cell Growth Differ. 1992, 3, 269–278. [Google Scholar]
- Tank, E.M.H.; True, H.L. Disease-associated mutant ubiquitin causes proteasomal impairment and enhances the toxicity of protein aggregates. PLoS Genet. 2009, 5, e1000382. [Google Scholar] [CrossRef] [Green Version]
- Chen, X.; Petranovic, D. Role of frameshift ubiquitin B protein in Alzheimer’s disease. Wiley Interdiscip. Rev. Syst. Biol. Med. 2016, 8, 300–313. [Google Scholar] [CrossRef]
- Fang, X.; Trexler, C.; Chen, J. Ushering in the cardiac role of Ubiquilin1. J. Clin. Invest. 2018, 128, 5195–5197. [Google Scholar] [CrossRef] [Green Version]
- Ko, H.S.; Uehara, T.; Tsuruma, K.; Nomura, Y. Ubiquilin interacts with ubiquitylated proteins and proteasome through its ubiquitin-associated and ubiquitin-like domains. FEBS Lett. 2004, 566, 110–114. [Google Scholar]
- Whiteley, A.M.; Prado, M.A.; Peng, I.; Abbas, A.R.; Haley, B.; Paulo, J.A.; Reichelt, M.; Katakam, A.; Sagolla, M.; Modrusan, Z.; et al. Ubiquilin1 promotes antigen-receptor mediated proliferation by eliminating mislocalized mitochondrial proteins. Elife 2017, 6, 26435. [Google Scholar] [CrossRef]
- Itakura, E.; Zavodszky, E.; Shao, S.; Wohlever, M.L.; Keenan, R.J.; Hegde, R.S. Ubiquilins Chaperone and Triage Mitochondrial Membrane Proteins for Degradation. Mol. Cell 2016, 63, 21–33. [Google Scholar] [CrossRef] [Green Version]
- Kurlawala, Z.; Dunaway, R.; Shah, P.P.; Gosney, J.A.; Siskind, L.J.; Ceresa, B.P.; Beverly, L.J. Regulation of insulin-like growth factor receptors by Ubiquilin1. Biochem. J. 2017, 474, 4105–4118. [Google Scholar] [CrossRef] [PubMed]
- McKinnon, C.; Tabrizi, S.J. The ubiquitin-proteasome system in neurodegeneration. Antioxid. Redox Signal. 2014, 21, 2302–2321. [Google Scholar] [CrossRef]
- Osaka, M.; Ito, D.; Suzuki, N. Disturbance of proteasomal and autophagic protein degradation pathways by amyotrophic lateral sclerosis-linked mutations in ubiquilin 2. Biochem. Biophys. Res. Commun. 2016, 472, 324–331. [Google Scholar] [CrossRef] [PubMed]
- Hjerpe, R.; Bett, J.S.; Keuss, M.J.; Solovyova, A.; McWilliams, T.G.; Johnson, C.; Sahu, I.; Varghese, J.; Wood, N.; Wightman, M.; et al. UBQLN2 Mediates Autophagy-Independent Protein Aggregate Clearance by the Proteasome. Cell 2016, 166, 935–949. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Picher-Martel, V.; Dutta, K.; Phaneuf, D.; Sobue, G.; Julien, J.-P. Ubiquilin-2 drives NF-κB activity and cytosolic TDP-43 aggregation in neuronal cells. Mol. Brain 2015, 8, 71. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leroy, E.; Boyer, R.; Auburger, G.; Leube, B.; Ulm, G.; Mezey, E.; Harta, G.; Brownstein, M.J.; Jonnalagada, S.; Chernova, T.; et al. The ubiquitin pathway in Parkinson’s disease. Nature 1998, 395, 451–452. [Google Scholar] [CrossRef]
- Gu, Y.; Ding, X.; Huang, J.; Xue, M.; Zhang, J.; Wang, Q.; Yu, H.; Wang, Y.; Zhao, F.; Wang, H.; et al. The deubiquitinating enzyme UCHL1 negatively regulates the immunosuppressive capacity and survival of multipotent mesenchymal stromal cells. Cell Death Dis. 2018, 9, 459. [Google Scholar] [CrossRef]
- Rydning, S.L.; Backe, P.H.; Sousa, M.M.L.; Iqbal, Z.; Øye, A.-M.; Sheng, Y.; Yang, M.; Lin, X.; Slupphaug, G.; Nordenmark, T.H.; et al. Novel UCHL1 mutations reveal new insights into ubiquitin processing. Hum. Mol. Genet. 2017, 26, 1031–1040. [Google Scholar] [CrossRef] [Green Version]
- Choi, J.; Levey, A.I.; Weintraub, S.T.; Rees, H.D.; Gearing, M.; Chin, L.-S.; Li, L. Oxidative modifications and down-regulation of ubiquitin carboxyl-terminal hydrolase L1 associated with idiopathic Parkinson’s and Alzheimer’s diseases. J. Biol. Chem. 2004, 279, 13256–13264. [Google Scholar] [CrossRef] [Green Version]
- Boudreaux, D.A.; Maiti, T.K.; Davies, C.W.; Das, C. Ubiquitin vinyl methyl ester binding orients the misaligned active site of the ubiquitin hydrolase UCHL1 into productive conformation. Proc. Natl. Acad. Sci. USA 2010, 107, 9117–9122. [Google Scholar] [CrossRef] [Green Version]
- Zhou, Z.; Yao, X.; Pang, S.; Chen, P.; Jiang, W.; Shan, Z.; Zhang, Q. The deubiquitinase UCHL5/UCH37 positively regulates Hedgehog signaling by deubiquitinating Smoothened. J. Mol. Cell Biol. 2018, 10, 243–257. [Google Scholar] [CrossRef] [PubMed]
- Ge, J.; Hu, W.; Zhou, H.; Yu, J.; Sun, C.; Chen, W. Ubiquitin carboxyl-terminal hydrolase isozyme L5 inhibits human glioma cell migration and invasion via downregulating SNRPF. Oncotarget 2017, 8, 113635–113649. [Google Scholar] [CrossRef] [PubMed]
- Han, W.; Lee, H.; Han, J.-K. Ubiquitin C-terminal hydrolase37 regulates Tcf7 DNA binding for the activation of Wnt signalling. Sci. Rep. 2017, 7, 42590. [Google Scholar] [CrossRef]
- Chen, X.; Lee, B.-H.; Finley, D.; Walters, K.J. Structure of proteasome ubiquitin receptor hRpn13 and its activation by the scaffolding protein hRpn2. Mol. Cell 2010, 38, 404–415. [Google Scholar] [CrossRef] [Green Version]
- Everett, R.D. A novel ubiquitin-specific protease is dynamically associated with the PML nuclear domain and binds to a herpesvirus regulatory protein. EMBO J. 1997, 16, 1519–1530. [Google Scholar] [CrossRef] [Green Version]
- Holowaty, M.N.; Sheng, Y.; Nguyen, T.; Arrowsmith, C.; Frappier, L. Protein Interaction Domains of the Ubiquitin-specific Protease, USP7/HAUSP. J. Biol. Chem. 2003, 278, 47753–47761. [Google Scholar] [CrossRef] [Green Version]
- Wang, Z.; Kang, W.; You, Y.; Pang, J.; Ren, H.; Suo, Z.; Liu, H.; Zheng, Y. USP7: Novel Drug Target in Cancer Therapy. Front. Pharmacol. 2019, 10, 427. [Google Scholar] [CrossRef] [Green Version]
- Zapata, J.M.; Pawlowski, K.; Haas, E.; Ware, C.F.; Godzik, A.; Reed, J.C. A diverse family of proteins containing tumor necrosis factor receptor-associated factor domains. J. Biol. Chem. 2001, 276, 24242–24252. [Google Scholar] [CrossRef] [Green Version]
- Pfoh, R.; Lacdao, I.K.; Georges, A.A.; Capar, A.; Zheng, H.; Frappier, L.; Saridakis, V. Crystal Structure of USP7 Ubiquitin-like Domains with an ICP0 Peptide Reveals a Novel Mechanism Used by Viral and Cellular Proteins to Target USP7. PLoS Pathog. 2015, 11, e1004950. [Google Scholar] [CrossRef]
- Kim, R.Q.; Sixma, T.K. Regulation of USP7: A High Incidence of E3 Complexes. J. Mol. Biol. 2017, 429, 3395–3408. [Google Scholar] [CrossRef]
- Kim, H.T.; Goldberg, A.L. The deubiquitinating enzyme Usp14 allosterically inhibits multiple proteasomal activities and ubiquitin-independent proteolysis. J. Biol. Chem. 2017, 292, 9830–9839. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hanpude, P.; Bhattacharya, S.; Dey, A.K.; Maiti, T.K. Deubiquitinating enzymes in cellular signaling and disease regulation. IUBMB Life 2015, 67, 544–555. [Google Scholar] [CrossRef]
- Kim, H.T.; Goldberg, A.L. UBL domain of Usp14 and other proteins stimulates proteasome activities and protein degradation in cells. Proc. Natl. Acad. Sci. USA 2018, 115, 11642–11650. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leggett, D.S.; Hanna, J.; Borodovsky, A.; Crosas, B.; Schmidt, M.; Baker, R.T.; Walz, T.; Ploegh, H.; Finley, D. Multiple associated proteins regulate proteasome structure and function. Mol. Cell 2002, 10, 495–507. [Google Scholar] [CrossRef]
- Ye, Y.; Scheel, H.; Hofmann, K.; Komander, D. Dissection of USP catalytic domains reveals five common insertion points. Mol. Biosyst. 2009, 5, 1797–1808. [Google Scholar] [CrossRef] [PubMed]
- Li, F.; Macfarlan, T.; Pittman, R.N.; Chakravarti, D. Ataxin-3 is a histone-binding protein with two independent transcriptional corepressor activities. J. Biol. Chem. 2002, 277, 45004–45012. [Google Scholar] [CrossRef] [Green Version]
- Berke, S.J.S.; Chai, Y.; Marrs, G.L.; Wen, H.; Paulson, H.L. Defining the role of ubiquitin-interacting motifs in the polyglutamine disease protein, ataxin-3. J. Biol. Chem. 2005, 280, 32026–32034. [Google Scholar] [CrossRef] [Green Version]
- Todi, S.V.; Winborn, B.J.; Scaglione, K.M.; Blount, J.R.; Travis, S.M.; Paulson, H.L. Ubiquitination directly enhances activity of the deubiquitinating enzyme ataxin-3. EMBO J. 2009, 28, 372–382. [Google Scholar] [CrossRef]
- Ashkenazi, A.; Bento, C.F.; Ricketts, T.; Vicinanza, M.; Siddiqi, F.; Pavel, M.; Squitieri, F.; Hardenberg, M.C.; Imarisio, S.; Menzies, F.M.; et al. Polyglutamine tracts regulate beclin 1-dependent autophagy. Nature 2017, 545, 108–111. [Google Scholar] [CrossRef] [Green Version]
- Husnjak, K.; Elsasser, S.; Zhang, N.; Chen, X.; Randles, L.; Shi, Y.; Hofmann, K.; Walters, K.J.; Finley, D.; Dikic, I. Proteasome subunit Rpn13 is a novel ubiquitin receptor. Nature 2008, 453, 481–488. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiang, R.T.; Yemelyanova, A.; Xing, D.; Anchoori, R.K.; Hamazaki, J.; Murata, S.; Seidman, J.D.; Wang, T.-L.; Roden, R.B.S. Early and consistent overexpression of ADRM1 in ovarian high-grade serous carcinoma. J. Ovarian Res. 2017, 10, 53. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shi, Y.; Chen, X.; Elsasser, S.; Stocks, B.B.; Tian, G.; Lee, B.-H.; Shi, Y.; Zhang, N.; de Poot, S.A.H.; Tuebing, F.; et al. Rpn1 provides adjacent receptor sites for substrate binding and deubiquitination by the proteasome. Science 2016, 351, aad9421. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Elsasser, S.; Gali, R.R.; Schwickart, M.; Larsen, C.N.; Leggett, D.S.; Müller, B.; Feng, M.T.; Tübing, F.; Dittmar, G.A.G.; Finley, D. Proteasome subunit Rpn1 binds ubiquitin-like protein domains. Nat. Cell Biol. 2002, 4, 725–730. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Huang, J.; Zeng, B.; Yang, D.; Sun, J.; Yin, X.; Lu, M.; Qiu, Z.; Peng, W.; Xiang, T.; et al. PSMD2 regulates breast cancer cell proliferation and cell cycle progression by modulating p21 and p27 proteasomal degradation. Cancer Lett. 2018, 430, 109–122. [Google Scholar] [CrossRef]
- Hamazaki, J.; Sasaki, K.; Kawahara, H.; Hisanaga, S.-I.; Tanaka, K.; Murata, S. Rpn10-Mediated Degradation of Ubiquitinated Proteins Is Essential for Mouse Development. Mol. Cell. Biol. 2007, 27, 6629–6638. [Google Scholar] [CrossRef] [Green Version]
- Jiang, Z.; Zhou, Q.; Ge, C.; Yang, J.; Li, H.; Chen, T.; Xie, H.; Cui, Y.; Shao, M.; Li, J.; et al. Rpn10 promotes tumor progression by regulating hypoxia-inducible factor 1 alpha through the PTEN/Akt signaling pathway in hepatocellular carcinoma. Cancer Lett. 2019, 447, 1–11. [Google Scholar] [CrossRef]
- Chen, X.; Ebelle, D.L.; Wright, B.J.; Sridharan, V.; Hooper, E.; Walters, K.J. Structure of hRpn10 Bound to UBQLN2 UBL Illustrates Basis for Complementarity between Shuttle Factors and Substrates at the Proteasome. J. Mol. Biol. 2019, 431, 939–955. [Google Scholar] [CrossRef]
- Wang, Q.; Young, P.; Walters, K.J. Structure of S5a bound to monoubiquitin provides a model for polyubiquitin recognition. J. Mol. Biol. 2005, 348, 727–739. [Google Scholar] [CrossRef]
- Schweitzer, A.; Aufderheide, A.; Rudack, T.; Beck, F.; Pfeifer, G.; Plitzko, J.M.; Sakata, E.; Schulten, K.; Förster, F.; Baumeister, W. Structure of the human 26S proteasome at a resolution of 3.9 Å. Proc. Natl. Acad. Sci. USA 2016, 113, 7816–7821. [Google Scholar] [CrossRef] [Green Version]
- Mansour, W.; Nakasone, M.A.; von Delbrück, M.; Yu, Z.; Krutauz, D.; Reis, N.; Kleifeld, O.; Sommer, T.; Fushman, D.; Glickman, M.H. Disassembly of Lys11 and mixed linkage polyubiquitin conjugates provides insights into function of proteasomal deubiquitinases Rpn11 and Ubp6. J. Biol. Chem. 2015, 290, 4688–4704. [Google Scholar] [CrossRef] [Green Version]
- Puthiyedth, N.; Riveros, C.; Berretta, R.; Moscato, P. Identification of Differentially Expressed Genes through Integrated Study of Alzheimer’s Disease Affected Brain Regions. PLoS ONE 2016, 11, e0152342. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, L.; Liu, Y.; Wang, B.; Xu, G.; Yang, Z.; Tang, M.; Ma, A.; Jing, T.; Xu, X.; Zhang, X.; et al. POH1 deubiquitinates pro-interleukin-1β and restricts inflammasome activity. Nat. Commun. 2018, 9, 4225. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, C.-H.; Lu, S.-X.; Liu, L.-L.; Li, Y.; Yang, X.; He, Y.-F.; Chen, S.-L.; Cai, S.-H.; Wang, H.; Yun, J.-P. POH1 Knockdown Induces Cancer Cell Apoptosis via p53 and Bim. Neoplasia 2018, 20, 411–424. [Google Scholar] [CrossRef]
- Zhu, R.; Liu, Y.; Zhou, H.; Li, L.; Li, Y.; Ding, F.; Cao, X.; Liu, Z. Deubiquitinating enzyme PSMD14 promotes tumor metastasis through stabilizing SNAIL in human esophageal squamous cell carcinoma. Cancer Lett. 2018, 418, 125–134. [Google Scholar] [CrossRef]
- Song, Y.; Li, S.; Ray, A.; Das, D.S.; Qi, J.; Samur, M.K.; Tai, Y.-T.; Munshi, N.; Carrasco, R.D.; Chauhan, D.; et al. Blockade of deubiquitylating enzyme Rpn11 triggers apoptosis in multiple myeloma cells and overcomes bortezomib resistance. Oncogene 2017, 36, 5631–5638. [Google Scholar] [CrossRef] [Green Version]
- Maytal-Kivity, V.; Reis, N.; Hofmann, K.; Glickman, M.H. MPN+, a putative catalytic motif found in a subset of MPN domain proteins from eukaryotes and prokaryotes, is critical for Rpn11 function. BMC Biochem. 2002, 3, 28. [Google Scholar] [CrossRef] [Green Version]












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sr. No. | Protein/Gene Name | Length (Amino Acids) | Function in UPS | Altered Function of Protein in UPS | Involvement in Diseases | UniProt ID | References |
|---|---|---|---|---|---|---|---|
| 1 | Ubiquitin-like modifier-activating enzyme 1 (UBA1) | 1058 | Catalyzes first step in ubiquitination. Binds with Ub, activates it and transfers it to E2 enzyme | Reduced level of UBA1 affects UPS-mediated protein degradation, missense mutations in UBA1 gene lead to SMAX2 | Neurological disorders such as AD, SMAX2, and HD | P22314 | [21,63,64] |
| 2 | Ubiquitin-conjugating enzyme E2 R2 (UBE2R2) | 238 | Catalyzes second step in ubiquitination. Accepts Ub from E1 enzyme and binds with E3 enzyme | Mutation and dysregulation in UBE2R2 affect UPS function | Dysregulation leads to cancer or neurodegenerative diseases | Q712K3 | [23,65] |
| 3 | E3 ubiquitin-protein ligase enzyme CHIP (STUB1) | 303 | Catalyzes final step of ubiquitination. Binds with target protein and transfers Ub from E2 enzyme to target protein. | Deregulation of E3 enzyme affects UPS-mediated degradation process. | Deregulation leads to cancer and neurodegenerative diseases such as AD, PD, and HD | Q9UNE7 | [23,24,66] |
| 4 | Polyubiquitin-B (UBB) | 229 | Tags target proteins for proteasomal degradation | Frameshift mutation in ubiquitin-B forms UBB+1, which disturbs UPS-mediated protein degradation | UBB+1 accumulation with Aβ in AD and Down’s syndrome | P0CG47 | [18,19] |
| 5 | Ubiquilin-1 (UBQLN1) | 589 | Regulates protein degradation through UPS, autophagy, and ERAD | Defects in UBQLN1 lead to perturbed protein degradation via UPS, UBQLN1 downregulation affects APP processing in AD | Cancer, reduced UBQLN1 level found in AD and other neurodegenerative diseases, PolyQ diseases (HD) | Q9UMX0 | [6,67] |
| 6 | Ubiquilin-2 (UBQLN2) | 624 | Regulates protein degradation via UPS, autophagy, and ERAD | Defects in UBQLN2 lead to perturbed protein degradation, which leads to neurodegenerative diseases | Mutation in UBQLN2 leads to familial amyotrophic lateral sclerosis (ALS) | Q9UHD9 | [26,68,69] |
| 7 | Ubiquitin carboxyl-terminal hydrolase isozyme L1 (UCHL1) | 223 | Processing of ubiquitin precursors and ubiquitinated protein. Maintains pool of mono-Ub | Mutation, dysfunction, and downregulation of UCHL1 affects normal UPS function | Cancer and neurodegenerative diseases such as AD and PD | P09936 | [11,31,70] |
| 8 | Ubiquitin carboxyl-terminal hydrolase isozyme L5 (UCHL5) | 329 | Proteasome-associated DUB that cleaves ‘Lys-48’-linked polyubiquitin chains | Upregulation or downregulation of UCHL5 | Oncogenesis | Q9Y5K5 | [71] |
| 9 | Ubiquitin carboxyl-terminal hydrolase 7 (USP7) | 1102 | Cleaves Ub from polyubiquitin chains of target protein substrate | Poly-Q repeats, mutation, variation in expression level, and dysfunction in USP7 | Dysfunction leads to cancer, metabolic and neurological pathologies | Q93009 | [10] |
| 10 | Ubiquitin carboxyl-terminal hydrolase 14 (USP14) | 494 | Proteasome-associated DUB that cleaves Ub from Poly-Ub protein before degradation by the proteasome. Negatively regulates proteasome activity. | USP14 activation inhibits degradation of pathogenic, neurotoxic proteins | Neurodegenerative diseases such as AD, ALS, PD, and HD. | P54578 | [36,72] |
| 11 | Ataxin-3 (ATXN3) | 361 | DUB that is involved in polyubiquitin chain trimming | PolyQ expansion in ataxin-3 at its C-terminus | Spinocerebellar Ataxia Type 3 (SCA3) | P54252 | [73,74] |
| 12 | Proteasomal ubiquitin receptor (ADRM1) | 407 | Receptor for Ub in RP of 26S proteasome that captures target protein by binding to Ub. It also binds and activates DUB enzyme UCHL5 | - | - | Q16186 | [75] |
| 13 | 26S proteasome non-ATPase regulatory subunit 2 (PSMD2) | 908 | Receptor for Ub in RP of 26S proteasome. | - | - | Q13200 | [41] |
| 14 | 26S proteasome non-ATPase regulatory subunit 4 (PSMD4) | 377 | Receptor for Ub in RP of 26S proteasome, captures target protein by binding to Ub. | - | - | P55036 | [41] |
| 15 | 26S proteasome non-ATPase regulatory subunit 14 (PSMD14) | 310 | PSMD14 is a 19S-proteasome-associated DUB enzyme that deubiquitinates substrate protein during proteasomal degradation. | Upregulation of PSMD14 leads to dysfunction of UPS | Increase in level of PSMD14 leads to carcinogenesis | O00487 | [38,76] |
| Protein | PPID_VSL2 | PPID_VL3 | PPID_VLXT | PPID_FIT | PPID_IUPRED | PPID_MEAN |
|---|---|---|---|---|---|---|
| UBA1 (E1) | 17.86 | 9.45 | 22.59 | 6.14 | 8.13 | 9.74 |
| UBE2R2 (E2) | 48.74 | 42.44 | 42.44 | 28.15 | 24.79 | 37.39 |
| STUB1 (E3) | 41.91 | 37.62 | 46.86 | 34.32 | 19.80 | 37.95 |
| UBB | 30.57 | 17.03 | 36.24 | 7.86 | 11.35 | 10.04 |
| UBQLN1 | 88.96 | 82.17 | 62.99 | 85.06 | 77.76 | 87.10 |
| UBQLN2 | 86.86 | 73.40 | 70.99 | 81.41 | 70.83 | 80.93 |
| UCHL1 | 30.94 | 11.66 | 34.98 | 10.31 | 3.14 | 7.62 |
| UCHL5 | 28.27 | 22.49 | 25.53 | 17.02 | 10.33 | 16.11 |
| USP7 | 23.96 | 10.89 | 19.06 | 11.62 | 11.25 | 11.62 |
| USP14 | 37.25 | 31.17 | 26.32 | 20.85 | 8.91 | 23.48 |
| ATXN3 | 58.17 | 63.16 | 55.12 | 47.65 | 42.38 | 53.74 |
| ADRM1 | 70.52 | 60.93 | 62.16 | 52.83 | 51.35 | 61.92 |
| PSMD2 | 20.37 | 15.53 | 27.97 | 15.86 | 12.22 | 13.77 |
| PSMD4 | 63.93 | 59.15 | 64.46 | 47.75 | 47.21 | 55.17 |
| PSMD14 | 29.68 | 20.97 | 22.26 | 16.45 | 13.23 | 18.71 |
| Protein | MoRFCHiBi_Web | MoRFpred | DISOPRED3 | ANCHOR |
|---|---|---|---|---|
| UBA1 (E1) | 1–13, 1048–1057 | 5–12, 54–60, 423–427, 1051–1058 | 802–817 | 1–16, 23–39 |
| UBE2R2 (E2) | 166–170, 203–217, 219–226 | 11–19, 205–213 | 1–6, 212–238 | 199–238 |
| STUB1 (E3) | - | - | - | 163–169, 198–204, 206–214, 230–239 |
| UBB | 40–50, 116–122, 192–202 | 221–228 | - | - |
| UBQLN1 | 18–39 | 34–45, 318–327 | 1–5, 14–21, 456–474 | 1–44, 49–54, 72–88, 91–113, 142–168, 192–307, 311–350, 355–496, 507–543 |
| UBQLN2 | 10–38 | 30–41, 560–565, 588–592 | 1–19 | 1–38, 87–107, 136–161, 193–208, 218–329, 353–377, 398–456, 498–598 |
| UCHL1 | - | 215–220 | - | - |
| UCHL5 | 324–328 | 168–173 | 1–6, 252–256, 320–329 | - |
| USP7 | 2–26, 1077–1082, 1090–1102 | 262–267, 511–516, 1094–1099 | 1084–1093, 1056–1061, 495–505 | 1–64 |
| USP14 | - | 477–482 | 66–75, 226–232, 489–494 | - |
| ATXN3 | 56–65, 246–255, 285–290, 312–357 | 250–254, 282–292, 342–350 | 1–21, 353–361 | 215–291, 307–355 |
| ADRM1 | 21–30 | 24–28, 399–407 | 1–19, 385–407 | 140–202, 208–318, 347–383, 399–407 |
| PSMD2 | 1–13, 96–102 | 51–62, 614–618 | - | 1–30, 35–79 |
| PSMD4 | 320–345, 365–377 | 201–205, 329–340, 372–377 | 196–203, 359–377 | 201–226, 237–365 |
| PSMD14 | 1–7 | 1–9, 249–255 | 1–12, 16–24 | - |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gadhave, K.; Kumar, P.; Kapuganti, S.K.; Uversky, V.N.; Giri, R. Unstructured Biology of Proteins from Ubiquitin-Proteasome System: Roles in Cancer and Neurodegenerative Diseases. Biomolecules 2020, 10, 796. https://doi.org/10.3390/biom10050796
Gadhave K, Kumar P, Kapuganti SK, Uversky VN, Giri R. Unstructured Biology of Proteins from Ubiquitin-Proteasome System: Roles in Cancer and Neurodegenerative Diseases. Biomolecules. 2020; 10(5):796. https://doi.org/10.3390/biom10050796
Chicago/Turabian StyleGadhave, Kundlik, Prateek Kumar, Shivani K. Kapuganti, Vladimir N. Uversky, and Rajanish Giri. 2020. "Unstructured Biology of Proteins from Ubiquitin-Proteasome System: Roles in Cancer and Neurodegenerative Diseases" Biomolecules 10, no. 5: 796. https://doi.org/10.3390/biom10050796