In the present section, we would like to compare the relative performances of the three available codes, rmcdhf_mpi, rmcdhf_mem_mpi and rmcdhf_mpi_FD, to perform MCDHF calculations. Here we choose two examples, i.e., Mg VII and Be I, to illustrate and discuss the improvements in efficiency obtained with the two new codes, i.e., rmcdhf_mem_mpi and rmcdhf_mpi_FD. The calculations are all performed using the Linux server with two Intel(R) Xeon(R) Gold 6 278C CPU (2.60 GHz) and 52 cores, except in some cases for which the used CPU is explicitly given. In this comparative work, we carefully checked that the results obtained with the three codes were identical. Throughout the present work, the reported CPU times are all wall-clock times, as they are more meaningful for the end-users.
3.3.1. Mg VII
In our recent work on C-like ions [
16], large-scale MCDHF-RCI calculations were performed for the
states in C-like ions from O III to Mg VII. Electron correlation effects were accounted for by using large configuration state function expansions, built from the orbital sets with principal quantum numbers
. A consistent atomic data set including both energies and transition data with spectroscopic accuracy was produced for the lowest hundreds of states of C-like ions from O III to Mg VII. Here we take Mg VII as an example to investigate the performances of
rmcdhf_mpi,
rmcdhf_mem_mpi and
rmcdhf_mpi_FD programs.
In the MCDHF calculations of [
16] aiming at the orbital optimisation, the CSF expansions were generated by SD-excitations up to
orbitals from all possible
with
configurations. (More details can be found in [
16].) The MCDHF calculations were performed layer by layer using the following sequence of active sets (AS):
Here the test calculations are carried out only for the even states with
J = 0–3. The CSF expansions using the above AS orbitals, as well as the number of targeted levels for each block, are listed in
Table 1. To keep the calculations tractable, only two SCF iterations are performed, taking the converged radial functions from [
16] as the initial estimation. The zero- and first-order partition techniques [
4,
44], often referred to as ‘Zero-First’ methods [
45], are employed. The zero-space contains the CSFs with orbitals up to
, the numbers of which are also reported in
Table 1. The corresponding sizes of
mcp.XXX files are, respectively, about 5.2, 11, 19, 29, and 41 GB in the
through
calculations.
The CPU times for these MCDHF calculations using the
and
orbital sets are reported in
Table 2 and
Table 3, respectively. To show the MPI performance, the calculations are carried out using various
numbers of MPI
processes (
np) ranging from 1 to 48. The
rmcdhf_mpi and
rmcdhf_mem_mpi MPI calculations using the
orbitals set are only performed with
, as the calculations with smaller
-values are too time-consuming. The CPU times are presented in the time sequence of Algorithm 2. For MCDHF calculations limited to two iterations, the eigenpairs are searched three times, i.e., once at step (0.3) and twice at step (3). The three rows with label “SetH&Diag” in
Table 2 and
Table 3 report the corresponding CPU times for setting the Hamiltonian matrix (routine
MATRIXmpi) and for its diagonalization (routine
MANEIGmpi), whereas the row with “Sum(SetH&Diag)” reports their sum. Steps (1.1) and (2) of Algorithm 2 are carried out twice in all calculations, as well as step (1.0) in
rmcdhf_mpi_FD calculations. The rows labeled by “SetCof + LAG” and “IMPROV” report, respectively, the CPU times for routines
SETLAGmpi and
IMPROVmpi, i.e., for steps (1.1) and (2) of Algorithm 2, while the row “Update” gives their sum. The rows labeled “Sum(Update)” display the total CPU times needed to update the orbitals twice. The rows “Walltime” represent the total code execution times. The differences between the summed value “Sum(Update)” + “Sum(SetH&Diag)” and the “Walltime” ones represent the CPU times that are not monitored by the former two. It can be seen that these differences are relatively small in cases of
rmcdhf_mpi and
rmcdhf_mem_mpi, implying that most of the time-consuming parts of the codes have been included in the tables, while the relatively large differences in the case of
rmcdhf_mpi_FD could be reduced if the CPU times needed for constructing the sorted
NXA and
NYA arrays in step (0.4) of Algorithm 2 would be taken into account.
In the
rmcdhf_mpi_FD calculations, five kinds of CPU times are additionally recorded, labeled, respectively, “NXA&NYA”, “SetTVCof”, “WithoutMCP”, “WithMCP”, and “SetLAG”. Row “NXA&NYA” reports the CPU times to construct the sorted
NXA and
NYA arrays. The “SetTVCof” displays the CPU times required to perform all the summations of Equations (
9) and (
12) in the newly added routine
SETTVCOF. The “WithoutMCP” and “WithMCP” rows report the CPU times spent in the added routine
SETALLCOF to accumulate the
and
coefficients using Equations (
8) and (
11), and Equations (
9) and (
12), respectively. These three contributions—“SetTVCof”, “WithoutMCP” and “WithMCP”, correspond to the computation effort associated with step (1.0) of Algorithm 2. The “SetLAG” row represents the CPU times required to calculate the off-diagonal Lagrange multipliers
in routine
SETLAGmpi using the calculated
and
coefficients. The “SetCof + LAG” CPU time values correspond approximately to the sum of the four tasks “SetTVCof” + “WithoutMCP” + “WithMCP” + “SetLAG”, as the calculations involving the one-body integral contributions are generally very fast. The “Update” row reports the summed value of “SetCof + LAG” and “IMPROV”, as done above for
rmcdhf_mpi and
rmcdhf_mem_mpi. (The CPU times with the same labels for the different codes can be compared because they are recorded for the same computation tasks.)
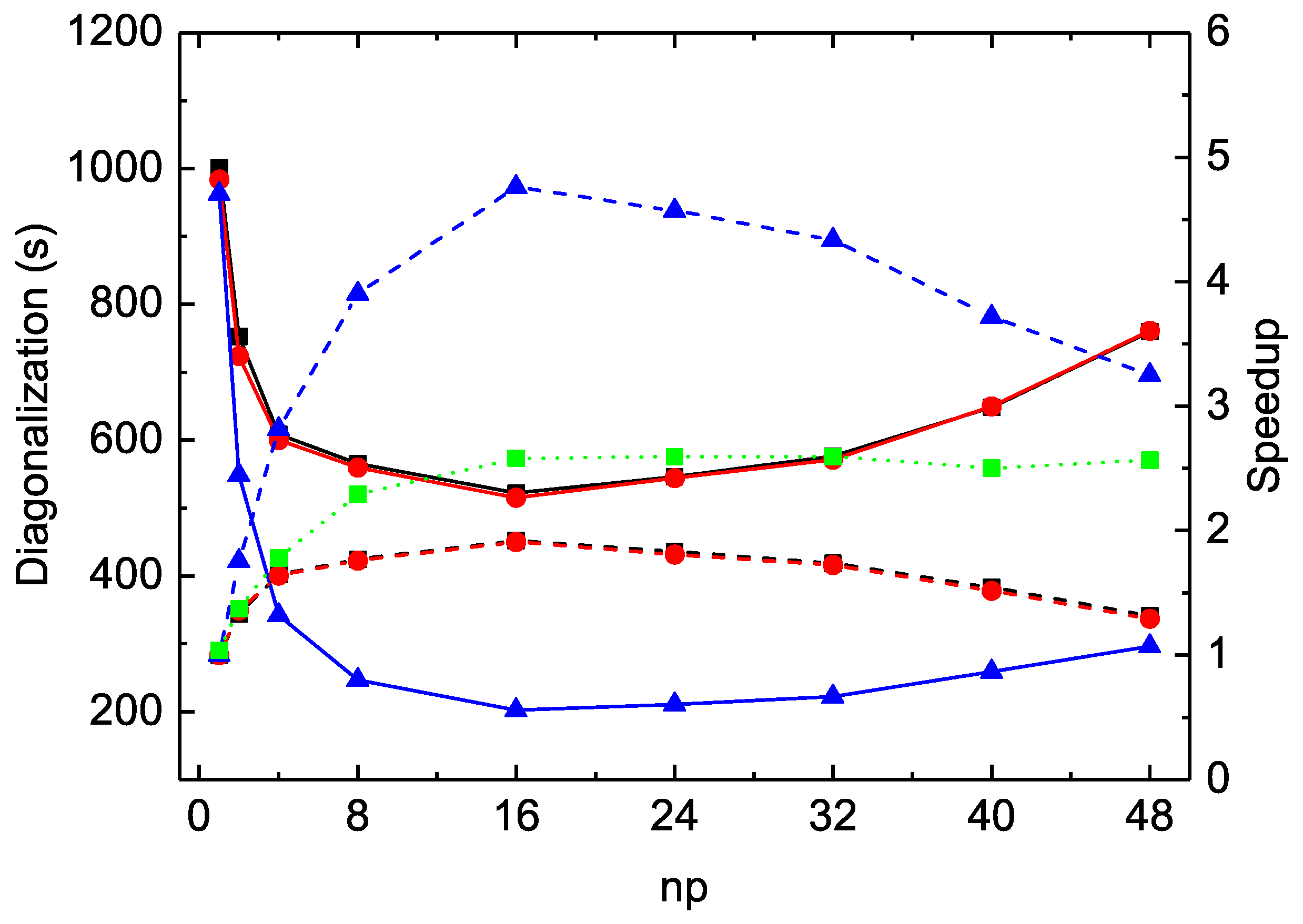
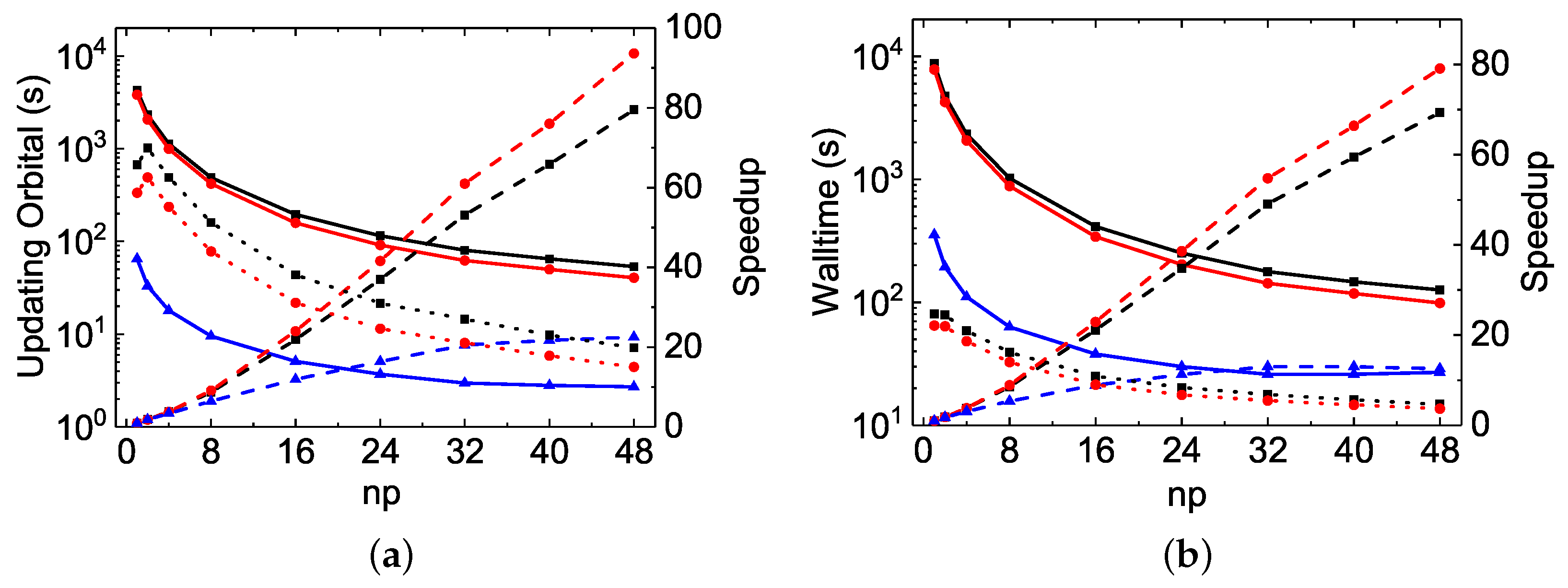
As seen in
Table 2 and
Figure 1 for the
calculations, the MPI performances for diagonalization are unsatisfactory for all three codes. The largest speed-up factors are about
for
rmcdhf_mpi and
rmcdhf_mem_mpi, and
for
rmcdhf_mpi_FD. The optimal numbers of MPI processes used for diagonalization are all around
and the MPI performances deteriorate when
exceeds 24. The CPU times of
rmcdhf_mem_mpi and
rmcdhf_mpi are very similar. Compared to these two codes, the CPU time of
rmcdhf_mpi_FD is reduced by a factor of ≃2.5 with
, thanks to the additional parallelization described in
Section 2. The speed-up efficiency of
rmcdhf_mpi_FD relative to
rmcdhf_mpi increases slightly with the size of the CSF expansion. As seen from the first line of
Table 3, the CPU time gain factor reaches
for the calculations using the
orbital set. It should be noted that the CPU times to set the Hamiltonian matrix are negligible in all three codes, being tens of times shorter than those for the first search of eigenpairs. The eigenpairs are searched three times, and the corresponding CPU times are included in the three rows labeled “SetH&Diag”. As seen in
Table 3, the first “SetH&Diag” CPU time is 945 s in the
rmcdhf_mpi_FD calculation with
, consisting of 14 and 931 s, respectively, for the matrix construction and diagonalization. For the subsequent two “SetH&Diag”, the matrix construction CPU times are still about 14 s, whereas those for diagonalization are, respectively, reduced to 34 and 26 s because the mixing coefficients are already converged. If the present calculations would be initialized by Thomas–Fermi or hydrogen-like approximations, these CPU times should reach about 900 s.
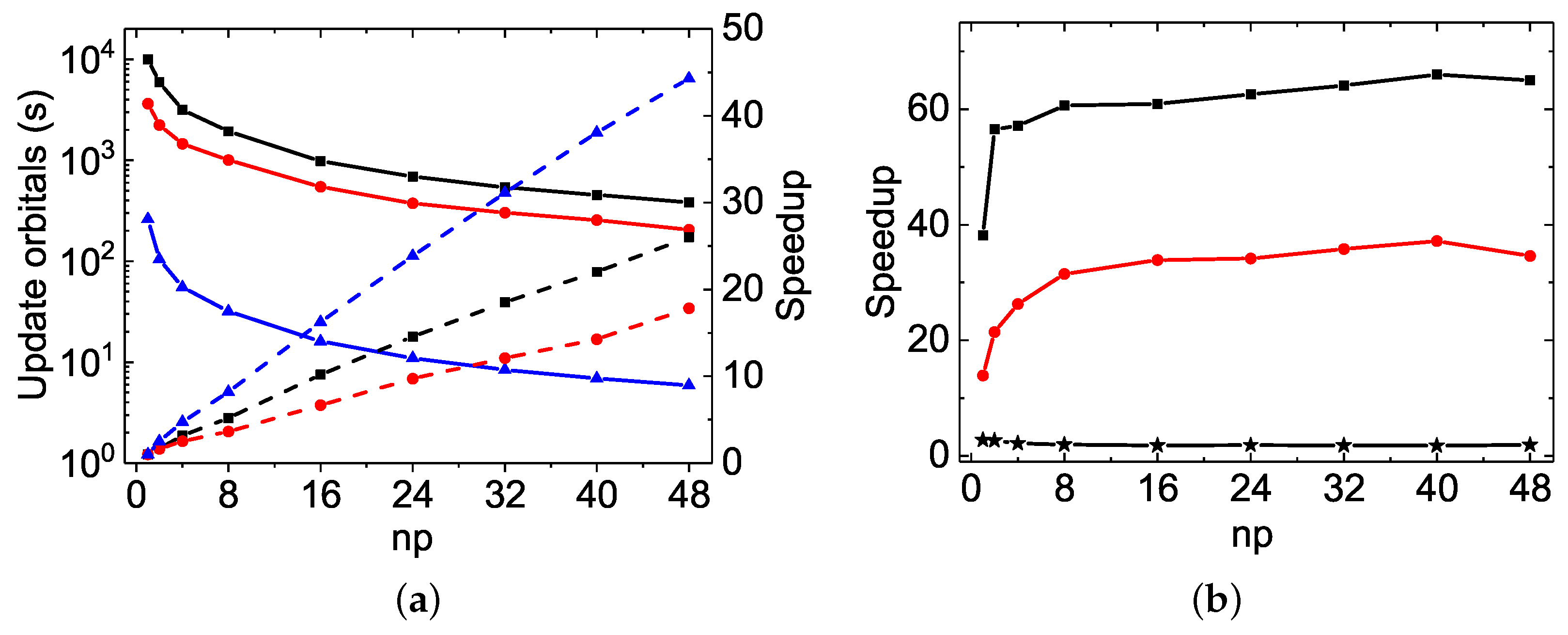
As far as the orbital updating process is concerned, the MPI performances of the three codes scale very well. The linearity is indeed attained even with
or more, as seen in
Figure 2a and
Figure 4b. The speed-up factors with
are, respectively, 26.0, 17.8 and 44.3 for the
rmcdhf_mpi,
rmcdhf_mem_mpi and
rmcdhf_mpi_FD calculations, while it is 43.2 for the
calculation using
rmcdhf_mpi_FD. The slopes obtained by a linear fit of the speed-up factors as a function of
np are, respectively,
,
, and
for the
rmcdhf_mpi,
rmcdhf_mem_mpi, and
rmcdhf_mpi_FD calculations, while it reaches 0.91 for the
rmcdhf_mpi_FD calculation. In the
calculations, compared to
rmcdhf_mpi and
rmcdhf_mem_mpi, the
rmcdhf_mpi_FD CPU times for updating the orbitals with
are, respectively, reduced by factors
and
, as seen in
Figure 2b. The corresponding reduction factors are
and
for the
calculations, as seen in
Figure 4b. These large CPU time-saving factors result from the new strategy developed to calculate the potentials, as implemented in
rmcdhf_mpi_FD, and described in
Section 3.2. Unlike the diagonalization part, the memory version
rmcdhf_mem_mpi brings about some interesting improvements over
rmcdhf_mpi: the orbital updating CPU times are indeed reduced by a factor of 2 and 1.6 for the
and
calculations, respectively.
The MPI performances for the “walltimes” are different from each other among the three codes. As seen in
Table 2 and
Table 3, the orbital updating CPU times are predominant in most of the
rmcdhf_mpi and
rmcdhf_mem_mpi MPI calculations, whereas the diagonalization CPU times dominate in all
rmcdhf_mpi_FD MPI calculations for all
np values. Hence, as seen in
Figure 3a and
Figure 4a, the global MPI performance of
rmcdhf_mpi_FD is similar to the one achieved for diagonalization. The maximum speed-up factors are, respectively, about 6.6 and 7.3 for the
and
calculations, both with
= 16–32, though the “Update” CPU times could be reduced by factors of 44 or 43 with
, as shown above. The speed-ups increase along with
np in the
rmcdhf_mpi and
rmcdhf_mem_mpi calculations, being, respectively, about 13.5 and 7.2 with
. As shown in
Figure 3b, comparing to
rmcdhf_mpi, the walltimes are, respectively, reduced by a factor of 11.2 and 4.3 in
rmcdhf_mpi_FD calculations using
and
, while the reduction factors are, respectively, 15 and 4.8 for the
calculations, as shown in
Figure 4a. The corresponding speed-up factors of
rmcdhf_mpi_FD relative to
rmcdhf_mem_mpi are smaller by a factor of 1.5, as
rmcdhf_mem_mpi is 1.5 times faster than
rmcdhf_mpi, as seen in
Figure 3b.
As mentioned above, the total CPU times for diagonalization reported in
Table 2 and
Table 3 (see rows labeled “Sum(SetH&Diag)”) are dominated by the first diagonalization, as the initial radial functions are taken from the converged calculations. In SCF calculations initialized by Tomas–Fermi or screened Hydrogenic approximations, more computation efforts have to be devoted to subsequent diagonalization during the SCF iterations. It is obvious that the limited MPI performance for diagonalization is the bottleneck in
rmcdhf_mpi_FD calculations. As seen in
Table 2 and
Table 3 and
Figure 3a and
Figure 4a, more CPU time is required if
np exceeds the optimal number of cores for diagonalization, which is generally in the range of 16–32. In
rmcdhf_mpi and
rmcdhf_mem_mpi calculations, the inefficiency of the orbital-updating procedure is another bottleneck, though this limitation may be alleviated by using more cores to perform the SCF calculations. However, this kind of alleviation would be eventually prohibited by the limited MPI performance of diagonalization. As seen in
Table 3 and
Figure 4a for the
rmcdhf_mpi and
rmcdhf_mem_mpi calculations, the walltimes with
are longer than those with
, though the CPU times for updating the orbitals are still reduced significantly in the former calculation.
3.3.2. Be I
To further understand the inefficiency of the orbital updating process in both
rmcdhf_mpi and
rmcdhf_mem_mpi codes, the second test case is carried out for a rather simple system, i.e., Be I. The calculations are performed to target the lowest 99 levels arising from the configurations
with
. The 99 levels are distributed over 15
blocks, i.e.,
, with the largest numbers of 12 for
and
blocks. The MCDHF calculations are performed simultaneously for both even and odd parity states. The largest CSF space contains 55 166 CSFs formed by SD excitation up to
orbitals from all the targeted states distributed over the above 15
blocks, with the largest size of 4 868 for
. The orbitals are also optimized with a layer-by-layer strategy. The CPU times recorded for the calculations using
and
orbital sets are given in
Table 4 and
Table 5. These calculations are hereafter labeled
and
. The corresponding sizes of
mcp.XXX files are, respectively, 760 MB and 19 GB. As the
rmcdhf_mpi and
rmcdhf_mem_mpi calculations are time-consuming, they are only performed with
.
In comparison to the Mg VII test case considered in
Section 3.3.1 (see
Table 1), the CSF expansions for Be I are much smaller, and fewer levels are targeted. Hence, fewer computational efforts are expected for the construction of a Hamiltonian matrix and the subsequent diagonalization. This is true for the diagonalization parts of all the calculations using various
MPI processes. For example, the CPU times for searching for eigenpairs are tens of times smaller than those for building the Hamiltonian matrix, representing 14s out of 150s, as reported by the first “SetH&Diag” value given in
Table 5 for the
rmcdhf_mpi_FD calculation with
. These CPU times are negligible (<1 s) for the following two diagonalizations. Unlike the cases considered for Mg VII, the CPU times for setting the Hamiltonian matrix predominate in the three “SetH&Diag” values, being all around 136s in the
calculations using
, as shown in
Table 5. These large differences in CPU time distributions between our Mg II and Be I test cases arise from the fact that the
expansion in Be I is built on a rather large set of orbitals, consisting of 171 Dirac one-electron orbitals, whereas the
expansion in Mg VII involves only 88 ones. The number of Slater integrals
possibly contributing to matrix elements is, therefore, much larger in Be I (95 451 319) than in Mg VII (6 144 958). Consequently, the three codes report very similar “SetH&Diag” and “Sum(SetH&Diag)” CPU times and all attain the maximum speed-up factors of about 10 around
, as seen in
Table 4.
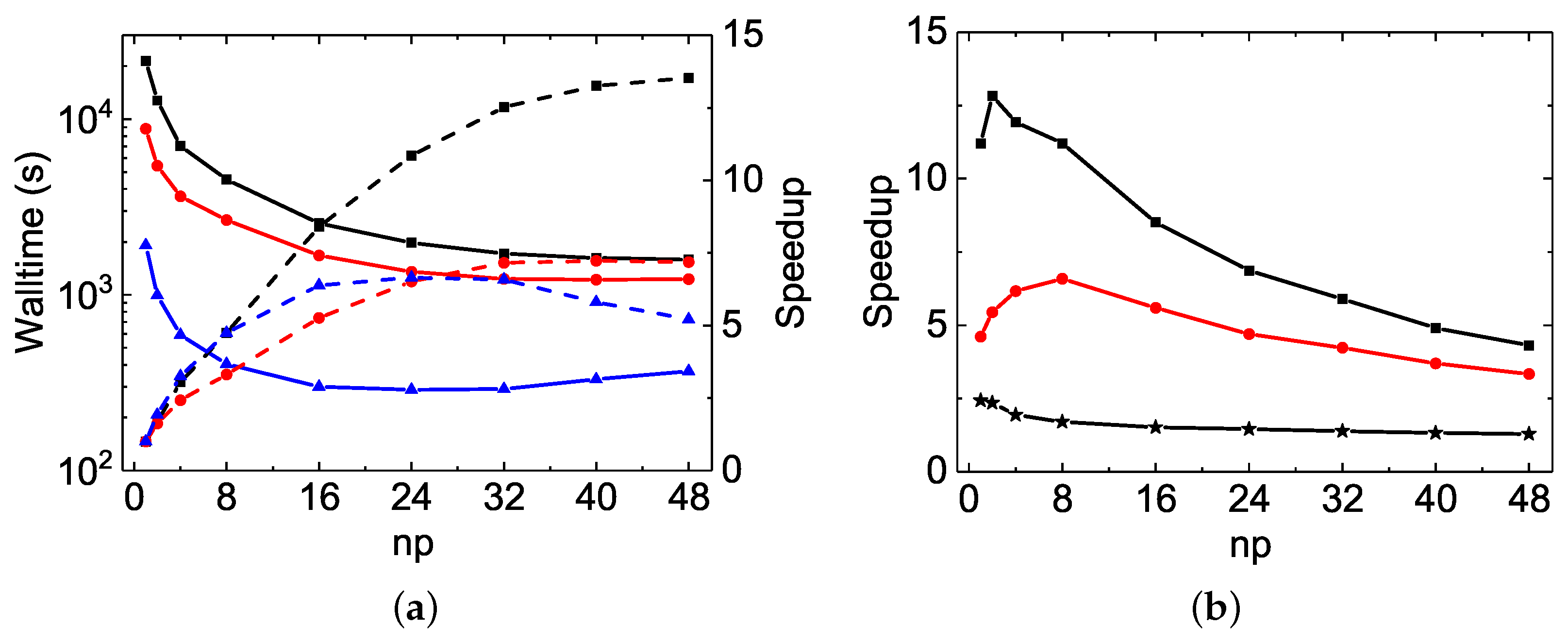
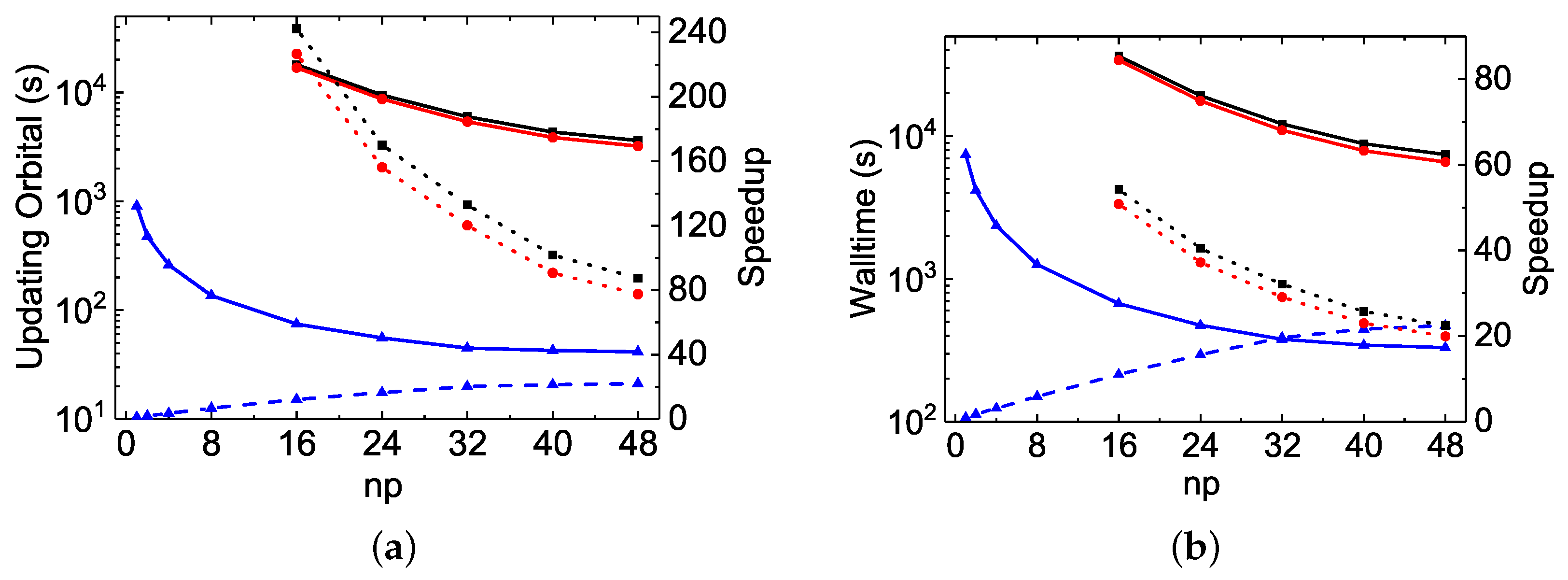
The MPI performances of the Be I
calculations are shown in
Figure 5. In general, a perfect MPI scaling is a speed-up factor equal to
. With respect to this, the speed-up factors observed in
rmcdhf_mpi and
rmcdhf_mem_mpi are unusual, being much larger than the corresponding
values. For example, the speed-up factors of
rmcdhf_mpi and
rmcdhf_mem_mpi for the orbital updating calculations are 79.5 and 93.6 at
, while the corresponding slopes obtained from the linear fit of the speed-up factors as a function of
are about 1.7 and 2.0, respectively. The corresponding reductions at
for the code running times are 69.4 and 79.1, while the slopes are about 1.5 and 1.7, respectively. These reductions should be even larger for the
calculations. A detailed analysis shows that the inefficiency of the sequential search method largely accounts for the above unexpected MPI performances. As mentioned in
Section 3.2, the labels
LABYk and
LABXk are constructed and stored sequentially in
NYA(:,a) and
NXA(:,a) arrays, respectively. In the subsequent accumulations of the
and
coefficients, the sequential search method is employed to match the labels. As mentioned above, a large number of Slater integrals contribute to the Hamiltonian matrix elements in calculations using a large set of orbitals. They are also involved in the calculations of the potential. In general, the number of
terms is much larger than the number of
terms. For example, the largest number of the former is 196 513 for all the
orbitals while there are at most 3 916
terms for all the
orbitals in the
calculations. Similarly, for the
calculations, there are at most 2 191 507
terms and 17 328
terms, both for the
orbitals. These values correspond to the largest sizes of the one-dimension vectors
NXA(:,a) and
NYA(:,a). The sequential search from a large list is obviously more inefficient than from a small list, as the time complexity is
. In the MPI calculations with small
np values, for example
, the sequential search of the
LABXk from the
NXA(:,a) lists of over two million elements is very time-consuming. It is obvious that the sizes of
NXA(:,a) in each MPI process decrease as
increases, alleviating the inefficiency of the sequential search method, as also shown in Mg VII calculations. For example, when
, the size of
in each MPI process is reduced to 528 802 in the
calculations, and consequently, the unusually high speed-up factors are attained with both
rmcdhf_mpi and
rmcdhf_mem_mpi.
In
rmcdhf_mpi_FD, the above inefficiency is removed by using the binary search strategy from the sorted arrays
NXA(:,a) and
NYA(:,a), and this code benefits from some other improvements, as discussed in
Section 3.2. As seen in
Figure 5a and
Figure 6a, the speed-up factors for updating the orbitals increase slightly along with
and attain the value of about 22 for both the
and
rmcdhf_mpi_FD calculations with
, while the corresponding reductions for the code running times are, respectively, 12.5 and 22.5, as seen in
Figure 5b and
Figure 6b. It should be mentioned that in the
rmcdhf_mpi and
rmcdhf_mem_mpi calculations, the “Sum(SetH&Diag)” CPU time values are all less than the “Sum(Update)” ones, as seen in
Table 4 and
Table 5. However, it is the opposite for the
rmcdhf_mpi_FD calculations, with all “Sum(SetH&Diag)” CPU time values still larger than the “Sum(Update)” ones, as in Mg VII calculations. Comparing to
rmcdhf_mpi, the code
rmcdhf_mpi_FD reduces the CPU times required for updating the orbitals by a factor lying in the range of 38–20, with
= 16–48 for the
MCDHF calculations, while for the
calculations, the corresponding reduction factors are in the range of 242–287. The corresponding reduction factor ranges for the code running times are, respectively, 11–4.7 and 54–22.5, as seen in
Figure 5b and
Figure 6b. One can conclude that the larger the scale of the calculations, the larger the CPU time reduction factors. Moreover, the lower the number of cores used, the larger the reduction factors obtained with
rmcdhf_mpi_FD. These features become highly relevant for extremely large-scale MCDHF calculations if they have to be performed using a small number of cores due to the limited performance of diagonalization, as discussed in the above section.
3.3.3. Possible Further Improvements for rmcdhf_mpi_FD
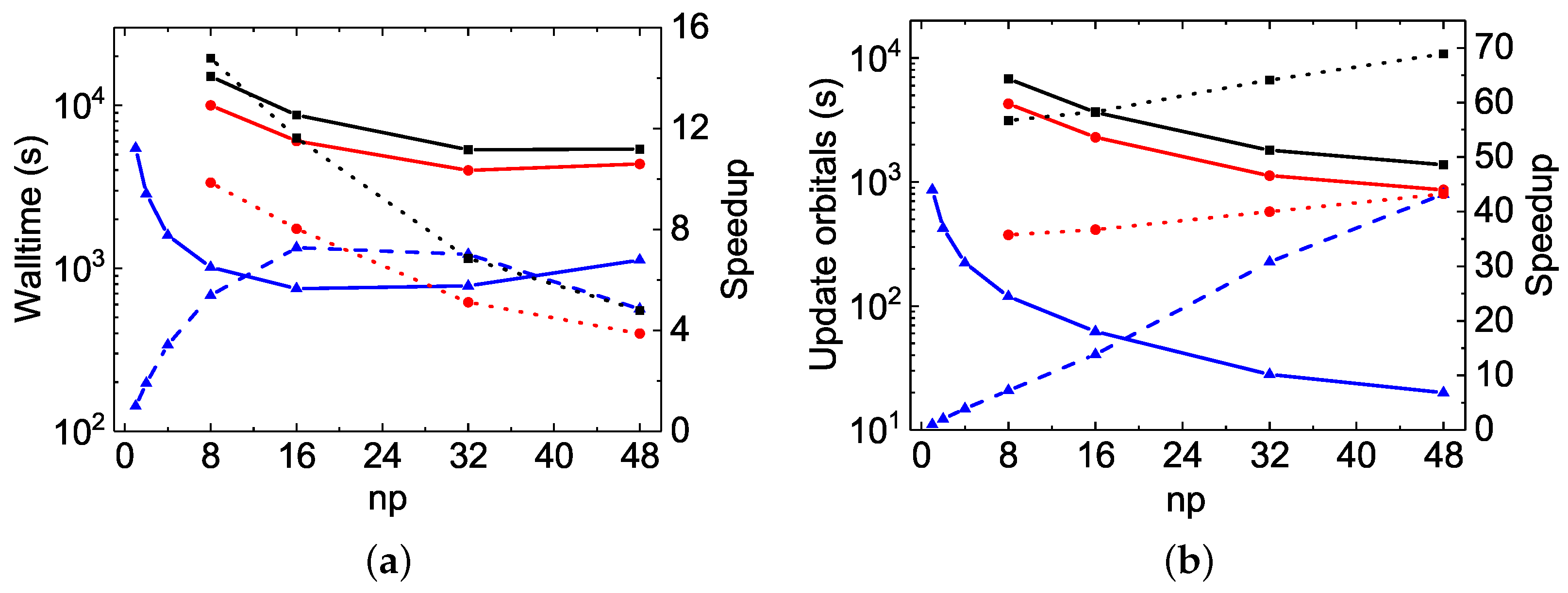
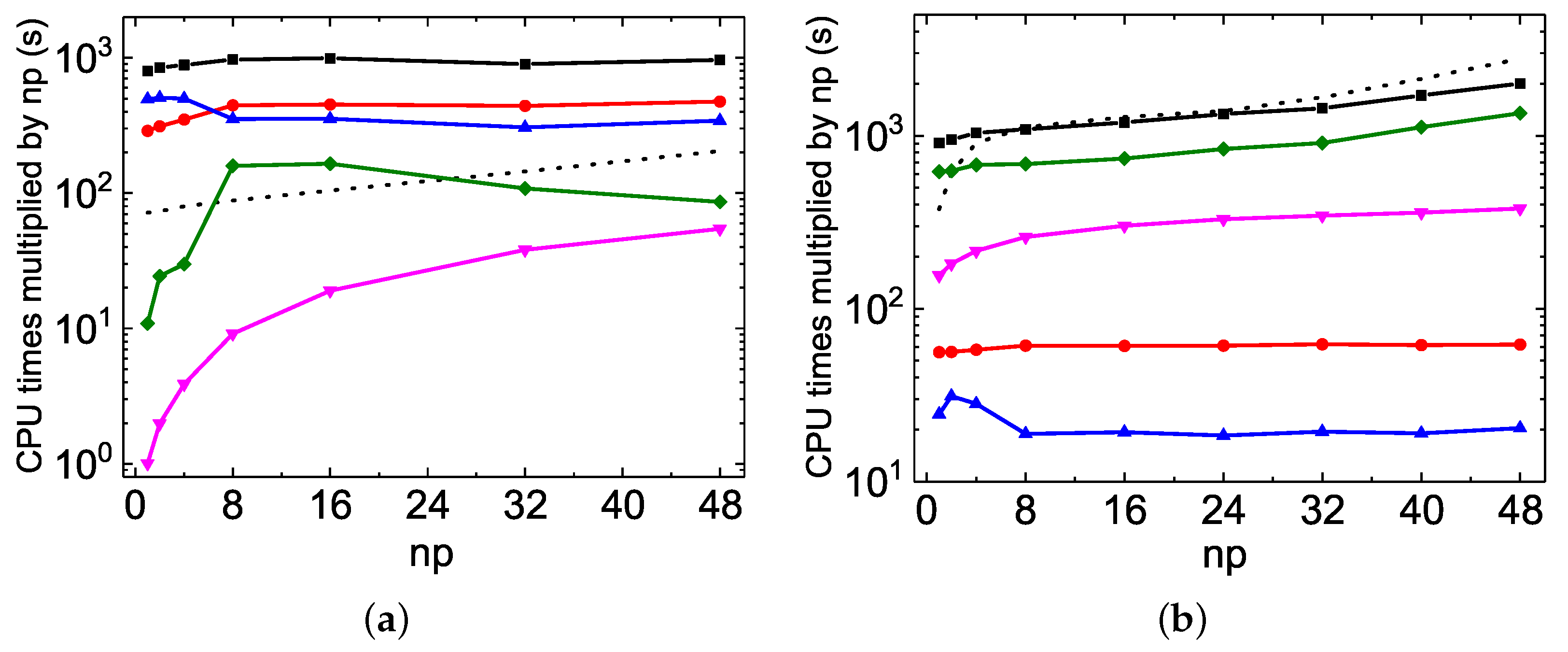
As discussed above, the MPI performances of
rmcdhf_mpi_FD for updating orbitals scale well in Mg VII calculations. The speed-up factors roughly follow a scaling law of ≃
(see
Figure 2b and
Figure 4b). For the Be I calculations, however, as illustrated by
Figure 5a and
Figure 6a, the speed-up factor increases slightly with
to attain a maximum value of 22. The partial CPU times for the orbital updating process, labeled “SetTVCof”, “WithoutMCP”, “WithMCP”, and “SetLAG”, are plotted in
Figure 7, together with the total updating time labeled “Update”, for the Mg VII
and for the Be I
calculations (these labels have been explained in
Section 3.3.1.). The partial CPU times labeled “IMPROV” are not reported here, as they are generally negligible. It can be seen that the “SetTVCof” and “WithoutMCP” partial times dominate the total CPU times required for updating orbitals in Mg VII
calculations, and they all scale well along with
. In the Be I
calculations, the partial “SetLAG” CPU times are predominant, and the remaining partial CPU times scale well. However, the scaling for both the partial “SetLAG” and total CPU times is worse than in Mg VII. Extra speed-up for “SetLAG” in Mg VII
calculations is observed. These different scalings can again be attributed to the fact that there is a large amount of
terms contributing to the exchange potentials in Be I
calculations, as mentioned above. For each
term, calculations of relativistic one-dimensional radial integrals
are performed on the grid with hundreds of
r values. All these calculations are serial and are often repetitious for the same
integral associated with different
terms which differ from each other only by
a and/or
c orbitals. This kind of inefficiency could be improved by calculating all the needed
integrals in advance and storing them in arrays. This will be implemented in future versions of
rmcdhf_mpi_FD codes.
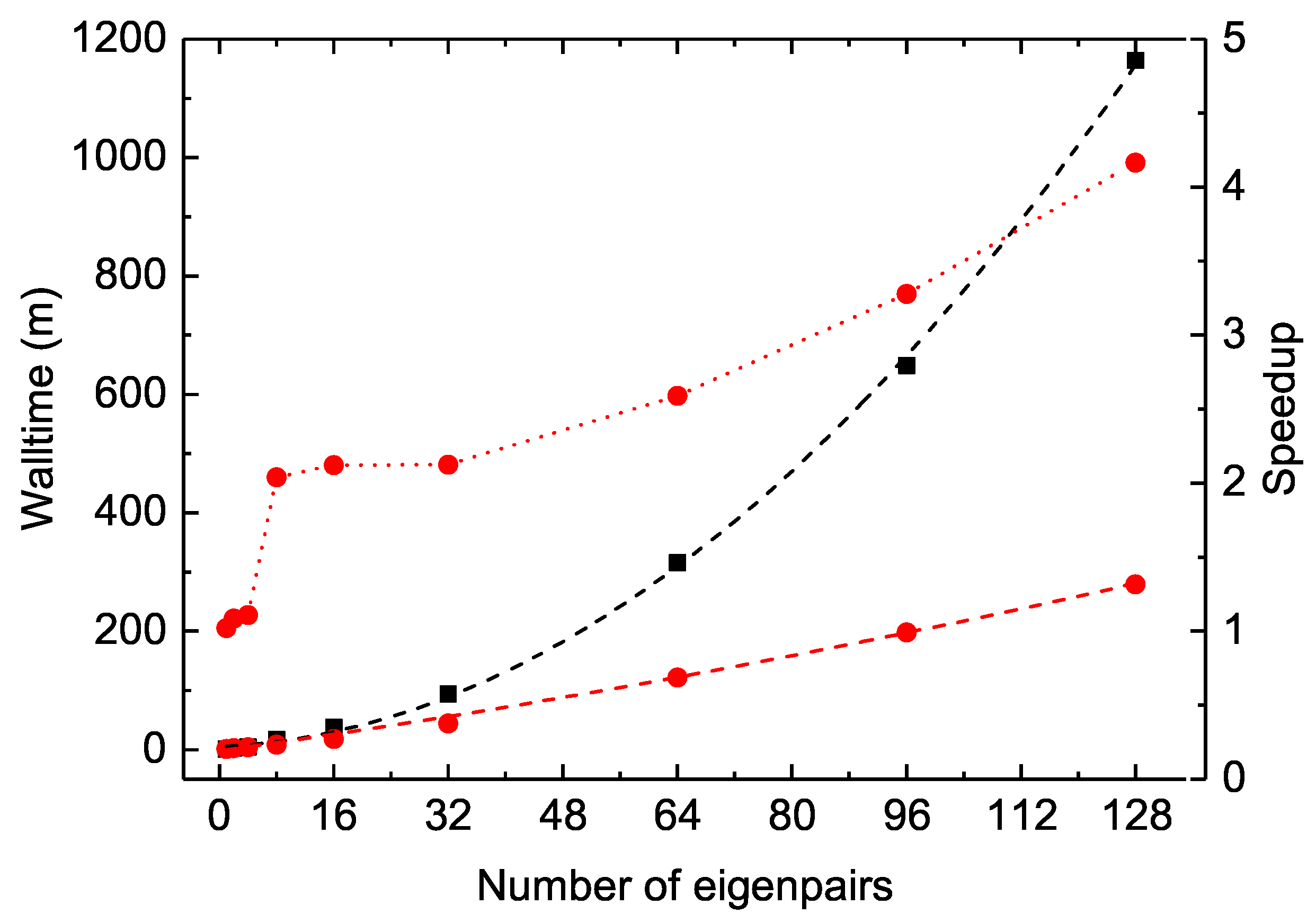
The MPI performances of the construction of
and
arrays are also displayed as dotted lines in
Figure 7. As the distributed
and
values obtained by different MPI processes have to be collected, sorted and then re-distributed, the linear scaling begins to deteriorate at about 32 cores for the Be I
calculations. Fortunately, this construction is realized only once, just before the SCF iterations. This slightly poor MPI performances should not be the bottleneck in large-scale MCDHF calculations.
After a deep investigation of the procedures that could affect the MPI performances of rmcdhf_mpi_FD, one concludes that the poor performances of diagonalization could be the bottleneck for MCDHF calculations based on relatively large expansions consisting of hundreds of thousands of CSFs and targeting dozens of eigenpairs. We will discuss this issue in the next section.


 ,
, 









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}