Carbonic Anhydrases: Role in pH Control and Cancer


Abstract
:1. Introduction
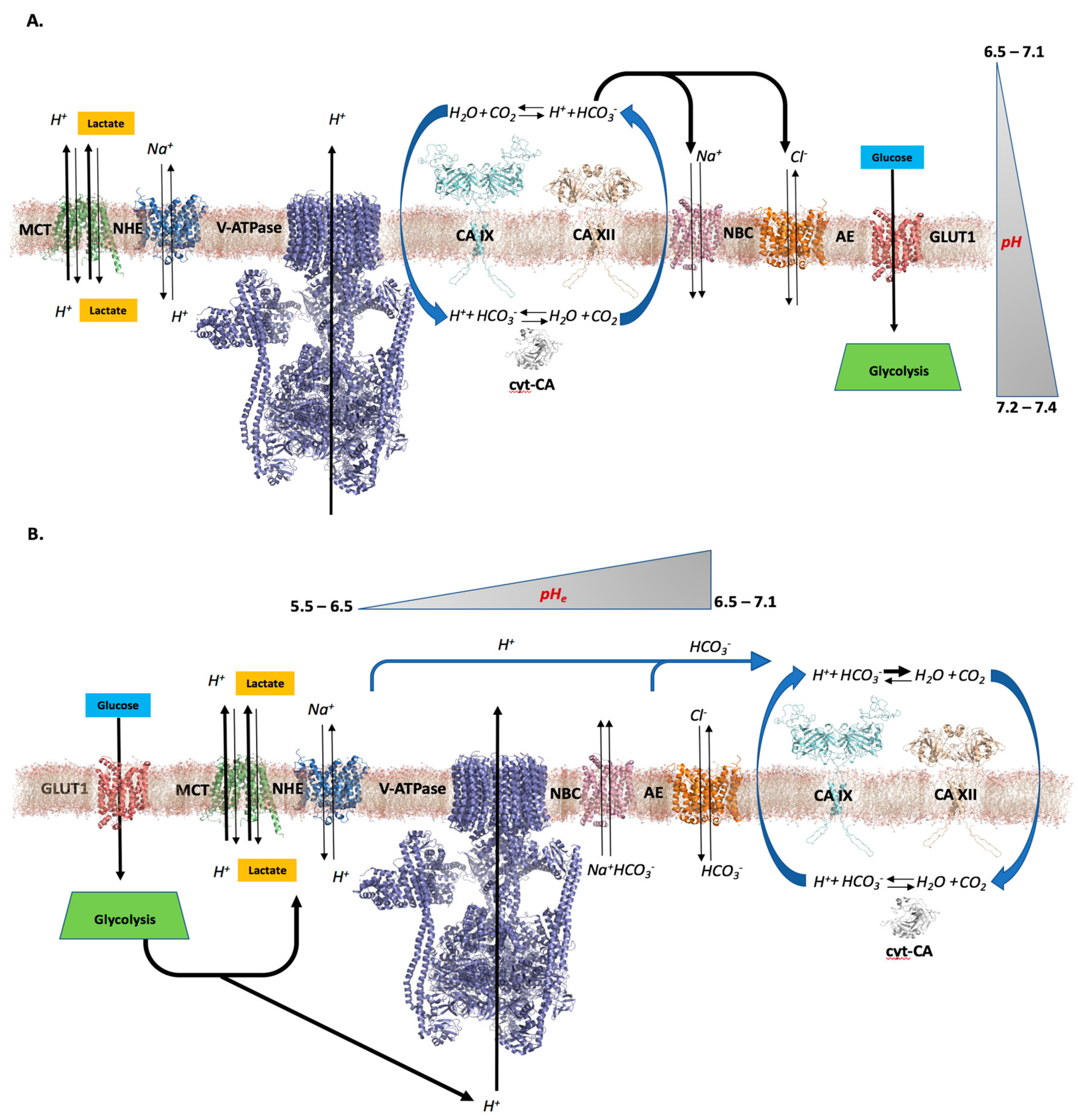
2. pH—The Role of CAs in Tumors
2.1. Differential pH Creates the Ideal Conditions for Tumor Cell Proliferation and Survival
2.2. CAs’ Role in Creating a Tumor Cell pH Differential
2.3. Further Considerations
3. Cytosolic CA Isoforms: Expression, Distribution, and Function
3.1. Expression and Function of CA I and CA II in Normal Cells
3.2. Expression and Function of CA I and CA II in Tumor Cells
3.3. Expression and Function of CA III and CA VII in Normal Cells
3.4. Expression and Function of CA III and CA VII in Tumor Cells
3.5. Expression and Function of CA XIII in Normal and Tumor Cells
4. Mitochondrial CA Isoforms: Expression, Distribution, and Function
4.1. Expression and Function of CA VA and CA VB in Normal Cells
4.2. Expression and Function of CA VA and CA VB in Tumor Cells
5. Secreted CA Isoforms: Expression, Distribution, and Function
Expression and Function of CA VI in Normal and Tumor Cells
6. Membrane Associated CA Isoforms: Expression, Distribution, and Function
6.1. Expression and Function of CA IV and CA XIV in Normal Cells
6.2. Expression and Function of CA IV and CA XIV in Tumor Cells
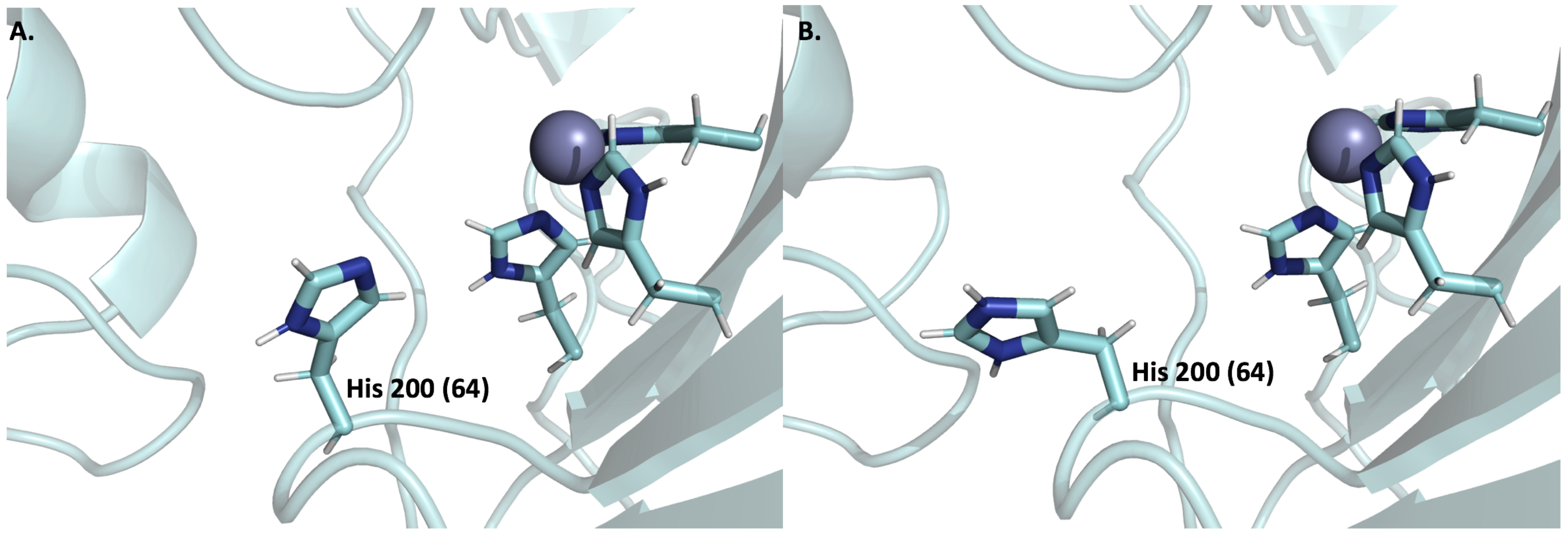
6.3. Expression and Function of CA IX and CA XII in Normal Cells
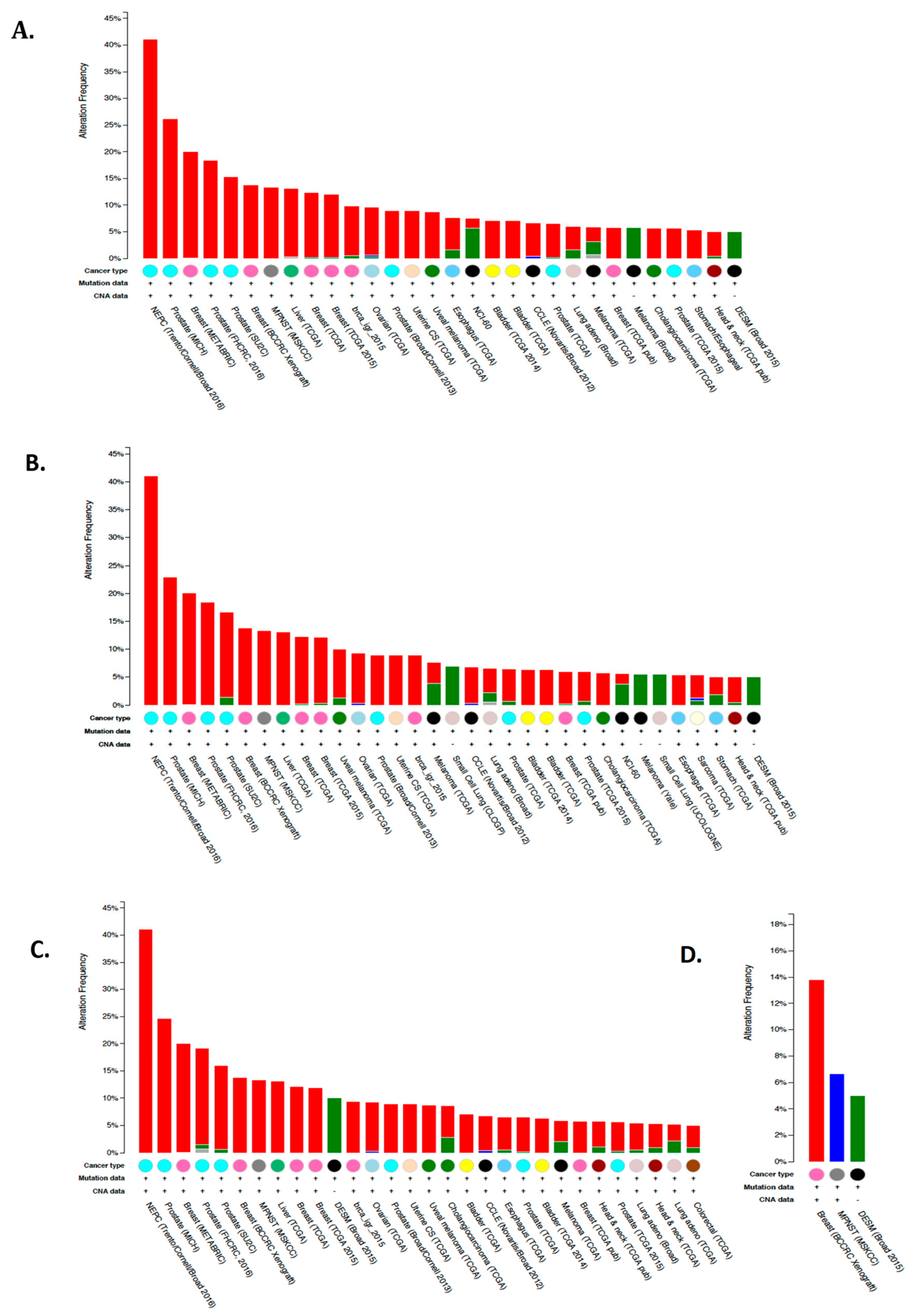
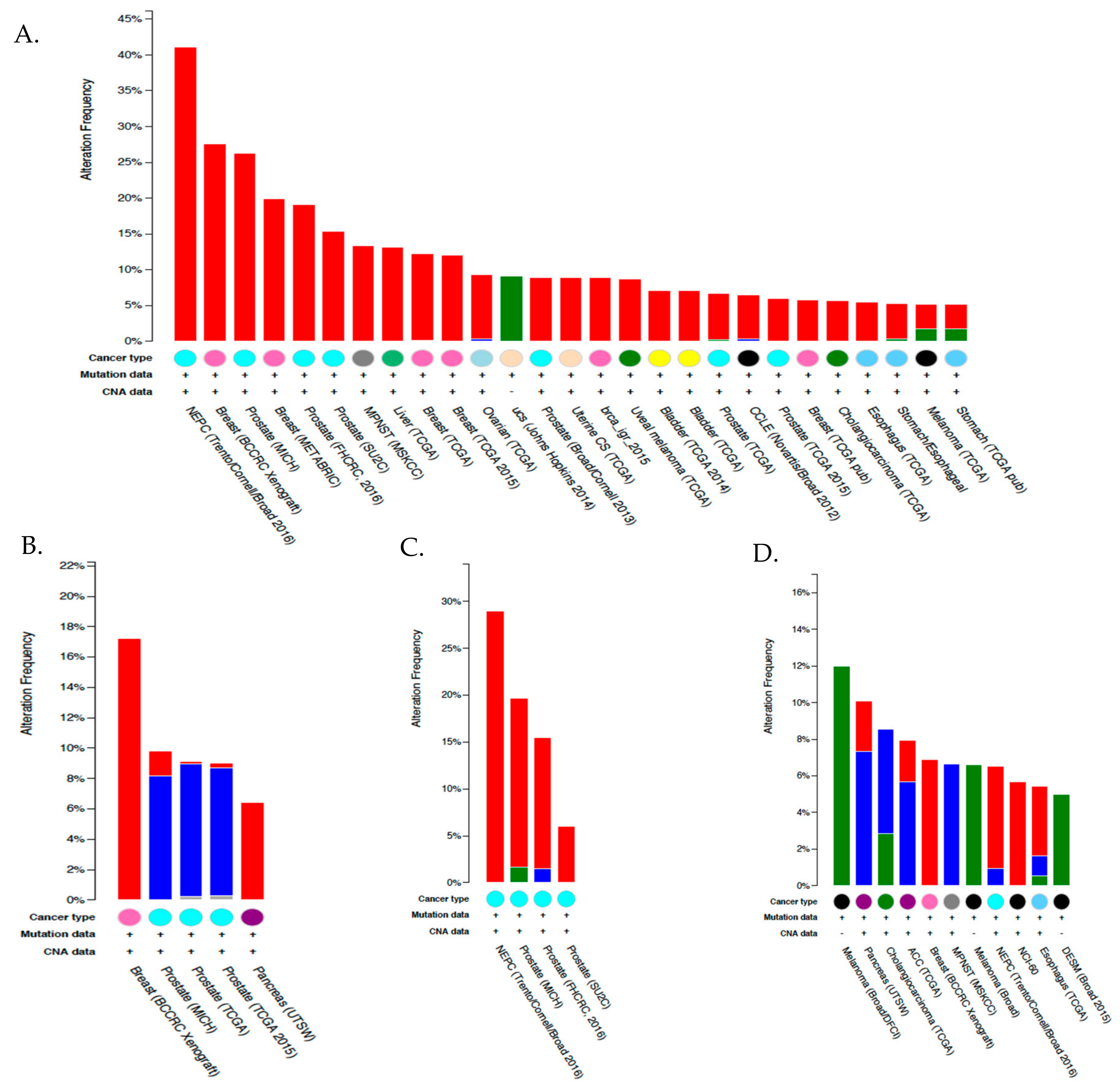
6.4. Expression and Function of CA IX and CA XII in Tumor Cells
7. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Stadie, W.C.; O’Brien, H. The catalytic hydration of carbondioxide and dehydration of carbonic acid by enzyme isolated from red blood cells. J. Biol. Chem. 1933, 103, 521–529. [Google Scholar]
- Supuran, C.T. Structure and function of carbonic anhydrases. Biochem. J. 2016, 473, 2023–2032. [Google Scholar] [CrossRef] [PubMed]
- Frost, S.C.; McKenna, R. (Eds.) Carbonic Anhydrase: Mechanism, Regulation, Links to Disease, and Industrial Applications, Subcellular Biochemistry; Springer: Berlin/Heidelberg, Germany, 2014. [Google Scholar]
- Supuran, C.T. Carbonic anhydrases—An overview. Curr. Pharm. Des. 2008, 14, 603–614. [Google Scholar] [CrossRef] [PubMed]
- Del Prete, S.; Vullo, D.; Fisher, G.M.; Andrews, K.T.; Poulsen, S.-A.; Capasso, C.; Supuran, C.T. Discovery of a new family of carbonic anhydrases in the malaria pathogen Plasmodium falciparum—the η-carbonic anhydrases. Bioorg. Med. Chem. Lett. 2014, 24, 4389–4396. [Google Scholar] [CrossRef] [PubMed]
- Nyman, P.; Lindskog, S. Amino acid composition of various forms of bovine and human erythrocyte carbonic anhydrase. Biochim. Biophys. Acta 1964, 85, 141–151. [Google Scholar] [CrossRef]
- Andersson, B.; Nyman, P.O.; Strid, L. Amino acid sequence of human erythrocyte carbonic anhydrase B. Biochem. Biophys. Res. Commun. 1972, 48, 670–677. [Google Scholar] [CrossRef]
- Lindskog, S. Purification and properties of bovine erythrocyte carbonic anhydrase. Biochim. Biophys. Acta 1960, 39, 218–226. [Google Scholar] [CrossRef]
- Nyman, P.O. Purification and properties of carbonic anhydrase from human erythrocytes. Biochim. Biophys. Acta 1961, 52, 1–12. [Google Scholar] [CrossRef]
- Liljas, A.; Kannan, K.K.; Bergstén, P.C.; Waara, I.; Fridborg, K.; Strandberg, B.; Carlbom, U.; Järup, L.; Lövgren, S.; Petef, M. Crystal structure of human carbonic anhydrase C. Nat. New Biol. 1972, 235, 131–137. [Google Scholar] [CrossRef] [PubMed]
- Lin, K.T.; Deutsch, H.F. Human carbonic anhydrases. XI. The complete primary structure of carbonic anhydrase B. J. Biol. Chem. 1973, 248, 1885–1893. [Google Scholar] [PubMed]
- Balckburn, M.N.; Chirgwin, J.M.; Gordon, J.T.; Thomas, K.D.; Parsons, T.F.; Register, A.M.; Schnackerz, K.D.; Noltmann, E.A. Pseudoisoenzymes of rabbit muscle phosphoglucose isomerase. J. Biol. Chem. 1972, 247, 1170–1179. [Google Scholar]
- Fernley, R.T.; Wright, R.D.; Coghlan, J.P. A novel carbonic anhydrase from the ovine parotid gland. FEBS Lett. 1979, 105, 299–302. [Google Scholar] [CrossRef]
- Murakami, H.; Sly, W.S. Purification and characterization of human salivary carbonic anhydrase. J. Biol. Chem. 1987, 262, 1382–1388. [Google Scholar] [PubMed]
- Whitney, P.L.; Briggle, T.V. Membrane-associated carbonic anhydrase purified from bovine lung. J. Biol. Chem. 1982, 257, 12056–12059. [Google Scholar] [PubMed]
- Wistrand, P.J. Properties of membrane-bound carbonic anhydrase. Ann. N. Y. Acad. Sci. 1984, 429, 195–206. [Google Scholar] [CrossRef] [PubMed]
- Carter, N.D.; Fryer, A.; Grant, A.G.; Hume, R.; Strange, R.G.; Wistrand, P.J. Membrane specific carbonic anhydrase (CAIV) expression in human tissues. Biochim. Biophys. Acta 1990, 1026, 113–116. [Google Scholar] [CrossRef]
- Zhu, X.L.; Sly, W.S. Carbonic anhydrase IV from human lung. Purification, characterization, and comparison with membrane carbonic anhydrase from human kidney. J. Biol. Chem. 1990, 265, 8795–8801. [Google Scholar] [PubMed]
- Montgomery, J.C.; Venta, P.J.; Eddy, R.L.; Fukushima, Y.-S.; Shows, T.B.; Tashian, R.E. Characterization of the human gene for a newly discovered carbonic anhydrase, CA VII, and its localization to chromosome 16. Genomics 1991, 11, 835–848. [Google Scholar] [CrossRef]
- Pastoreková, S.; Závadová, Z.; Kostál, M.; Babusíková, O.; Závada, J. A novel quasi-viral agent, MaTu, is a two-component system. Virology 1992, 187, 620–626. [Google Scholar] [CrossRef]
- Opavský, R.; Pastoreková, S.; Zelník, V.; Gibadulinová, A.; Stanbridge, E.J.; Závada, J.; Kettmann, R.; Pastorek, J. Human MN/CA9 gene, a novel member of the carbonic anhydrase family: Structure and exon to protein domain relationships. Genomics 1996, 33, 480–487. [Google Scholar] [CrossRef] [PubMed]
- Türeci, O.; Sahin, U.; Vollmar, E.; Siemer, S.; Göttert, E.; Seitz, G.; Parkkila, A.K.; Shah, G.N.; Grubb, J.H.; Pfreundschuh, M.; et al. Human carbonic anhydrase XII: CDNA cloning, expression, and chromosomal localization of a carbonic anhydrase gene that is overexpressed in some renal cell cancers. Proc. Natl. Acad. Sci. USA 1998, 95, 7608–7613. [Google Scholar] [CrossRef] [PubMed]
- Fujikawa-Adachi, K.; Nishimori, I.; Taguchi, T.; Onishi, S. Human carbonic anhydrase XIV (CA14): CDNA cloning, mRNA expression, and mapping to chromosome 1. Genomics 1999, 61, 74–81. [Google Scholar] [CrossRef] [PubMed]
- Mori, K.; Ogawa, Y.; Ebihara, K.; Tamura, N.; Tashiro, K.; Kuwahara, T.; Mukoyama, M.; Sugawara, A.; Ozaki, S.; Tanaka, I.; et al. Isolation and characterization of CA XIV, a novel membrane-bound carbonic anhydrase from mouse kidney. J. Biol. Chem. 1999, 274, 15701–15705. [Google Scholar] [CrossRef] [PubMed]
- Nagao, Y.; Platero, J.S.; Waheed, A.; Sly, W.S. Human mitochondrial carbonic anhydrase: CDNA cloning, expression, subcellular localization, and mapping to chromosome 16. Proc. Natl. Acad. Sci. USA 1993, 90, 7623–7627. [Google Scholar] [CrossRef] [PubMed]
- Fujikawa-Adachi, K.; Nishimori, I.; Taguchi, T.; Onishi, S. Human mitochondrial carbonic anhydrase VB: CDNA cloning, mRNA expression, subcellular localization, and mapping to chromosome X. J. Biol. Chem. 1999, 274, 21228–21233. [Google Scholar] [CrossRef] [PubMed]
- Idrees, D.; Kumar, S.; Rehman, S.A.A.; Gourinath, S.; Islam, A.; Ahmad, F.; Imtaiyaz Hassan, M. Cloning, expression, purification and characterization of human mitochondrial carbonic anhydrase VA. 3 Biotech 2016, 6. [Google Scholar] [CrossRef] [PubMed]
- Lehtonen, J.; Shen, B.; Vihinen, M.; Casini, A.; Scozzafava, A.; Supuran, C.T.; Parkkila, A.-K.; Saarnio, J.; Kivelä, A.J.; Waheed, A.; et al. Characterization of CA XIII, a novel member of the carbonic anhydrase isozyme family. J. Biol. Chem. 2004, 279, 2719–2727. [Google Scholar] [CrossRef] [PubMed]
- Ozensoy Guler, O.; Capasso, C.; Supuran, C.T. A magnificent enzyme superfamily: Carbonic anhydrases, their purification and characterization. J. Enzyme Inhib. Med. Chem. 2015, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Supuran, C.T. Carbonic anhydrases: Novel therapeutic applications for inhibitors and activators. Nat. Rev. Drug Discov. 2008, 7, 168–181. [Google Scholar] [CrossRef] [PubMed]
- Thiry, A.; Dogné, J.-M.; Supuran, C.T.; Masereel, B. Carbonic anhydrase inhibitors as anticonvulsant agents. Curr. Top. Med. Chem. 2007, 7, 855–864. [Google Scholar] [CrossRef] [PubMed]
- Supuran, C.T. How many carbonic anhydrase inhibition mechanisms exist? J. Enzyme Inhib. Med. Chem. 2016, 31, 345–360. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Tu, C.; Wang, H.; Silverman, D.N.; Frost, S.C. Catalysis and pH control by membrane-associated carbonic anhydrase IX in MDA-MB-231 breast cancer cells. J. Biol. Chem. 2011, 286, 15789–15796. [Google Scholar] [CrossRef] [PubMed]
- Sly, W.S. The membrane carbonic anhydrases: From CO2 transport to tumor markers. EXS 2000, 95–104. [Google Scholar]
- Nordfors, K.; Haapasalo, J.; Korja, M.; Niemelä, A.; Laine, J.; Parkkila, A.-K.; Pastorekova, S.; Pastorek, J.; Waheed, A.; Sly, W.S.; et al. The tumour-associated carbonic anhydrases CA II, CA IX and CA XII in a group of medulloblastomas and supratentorial primitive neuroectodermal tumours: An association of CA IX with poor prognosis. BMC Cancer 2010, 10, 148. [Google Scholar] [CrossRef] [PubMed]
- Lounnas, N.; Rosilio, C.; Nebout, M.; Mary, D.; Griessinger, E.; Neffati, Z.; Chiche, J.; Spits, H.; Hagenbeek, T.J.; Asnafi, V.; et al. Pharmacological inhibition of carbonic anhydrase XII interferes with cell proliferation and induces cell apoptosis in T-cell lymphomas. Cancer Lett. 2013, 333, 76–88. [Google Scholar] [CrossRef] [PubMed]
- Saarnio, J.; Parkkila, S.; Parkkila, A.K.; Haukipuro, K.; Pastoreková, S.; Pastorek, J.; Kairaluoma, M.I.; Karttunen, T.J. Immunohistochemical study of colorectal tumors for expression of a novel transmembrane carbonic anhydrase, MN/CA IX, with potential value as a marker of cell proliferation. Am. J. Pathol. 1998, 153, 279–285. [Google Scholar] [CrossRef]
- Kivelä, A.J.; Parkkila, S.; Saarnio, J.; Karttunen, T.J.; Kivelä, J.; Parkkila, A.K.; Pastoreková, S.; Pastorek, J.; Waheed, A.; Sly, W.S.; et al. Expression of transmembrane carbonic anhydrase isoenzymes IX and XII in normal human pancreas and pancreatic tumours. Histochem. Cell Biol. 2000, 114, 197–204. [Google Scholar] [PubMed]
- Kummola, L.; Hämäläinen, J.M.; Kivelä, J.; Kivelä, A.J.; Saarnio, J.; Karttunen, T.; Parkkila, S. Expression of a novel carbonic anhydrase, CA XIII, in normal and neoplastic colorectal mucosa. BMC Cancer 2005, 5, 41. [Google Scholar] [CrossRef] [PubMed]
- Barnett, D.H.; Sheng, S.; Charn, T.H.; Waheed, A.; Sly, W.S.; Lin, C.-Y.; Liu, E.T.; Katzenellenbogen, B.S. Estrogen receptor regulation of carbonic anhydrase XII through a distal enhancer in breast cancer. Cancer Res. 2008, 68, 3505–3515. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.Q.; Howison, C.M.; Spier, C.; Stopeck, A.T.; Malm, S.W.; Pagel, M.D.; Baker, A.F. Assessment of carbonic anhydrase IX expression and extracellular pH in B-cell lymphoma cell line models. Leuk. Lymphoma 2015, 56, 1432–1439. [Google Scholar] [CrossRef] [PubMed]
- Savile, C.K.; Lalonde, J.J. Biotechnology for the acceleration of carbon dioxide capture and sequestration. Curr. Opin. Biotechnol. 2011, 22, 818–823. [Google Scholar] [CrossRef] [PubMed]
- Bond, G.M.; Stringer, J.; Brandvold, D.K.; Simsek, F.A.; Medina, M.-G.; Egeland, G. Development of integrated system for biomimetic CO2 sequestration using the enzyme carbonic anhydrase. Energy Fuels 2001, 15, 309–316. [Google Scholar] [CrossRef]
- Lee, S.-W.; Park, S.-B.; Jeong, S.-K.; Lim, K.-S.; Lee, S.-H.; Trachtenberg, M.C. On carbon dioxide storage based on biomineralization strategies. Micron 2010, 41, 273–282. [Google Scholar] [CrossRef] [PubMed]
- Kaar, J.L.; Oh, H.-I.; Russell, A.J.; Federspiel, W.J. Towards improved artificial lungs through bio-catalysis. Biomaterials 2007, 28, 3131–3139. [Google Scholar] [CrossRef] [PubMed]
- Sreenivasan, R.; Bassett, E.K.; Hoganson, D.M.; Vacanti, J.P.; Gleason, K.K. Ultra-thin, gas permeable free-standing and composite membranes for microfluidic lung assist devices. Biomaterials 2011, 32, 3883–3889. [Google Scholar] [CrossRef] [PubMed]
- Stadermann, M.; Baxamusa, S.H.; Aracne-Ruddle, C.; Chea, M.; Li, S.; Youngblood, K.; Suratwala, T. Fabrication of Large-area Free-standing Ultrathin Polymer Films. J. Vis. Exp. 2015. [Google Scholar] [CrossRef] [PubMed]
- Pinard, M.A.; Mahon, B.; McKenna, R. Probing the surface of human carbonic anhydrase for clues towards the design of isoform specific inhibitors. BioMed Res. Int. 2015, 2015, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Mboge, M.Y.; McKenna, R.; Frost, S.C. Advances in anti-cancer drug development targeting carbonic anhydrase IX and XII. In Topics in Anti-Cancer Research; Bentham Science Publishers: Sharjah, United Arab Emirates, 2016; Volume 5, pp. 3–42. [Google Scholar]
- Mahon, B.P.; Pinard, M.A.; McKenna, R. Targeting carbonic anhydrase IX activity and expression. Molecules 2015, 20, 2323–2348. [Google Scholar] [CrossRef] [PubMed]
- Mahon, B.P.; Bhatt, A.; Socorro, L.; Driscoll, J.M.; Okoh, C.; Lomelino, C.L.; Mboge, M.Y.; Kurian, J.J.; Tu, C.; Agbandje-McKenna, M.; et al. The structure of carbonic anhydrase IX is adapted for low-pH catalysis. Biochemistry 2016, 55, 4642–4653. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.-S.; Lin, C.-W.; Chuang, C.-Y.; Su, S.-C.; Lin, S.-H.; Yang, S.-F. Carbonic anhydrase IX overexpression regulates the migration and progression in oral squamous cell carcinoma. Tumour Biol. J. Int. Soc. Oncodev. Biol. Med. 2015, 36, 9517–9524. [Google Scholar] [CrossRef] [PubMed]
- Hsieh, M.-J.; Chen, K.-S.; Chiou, H.-L.; Hsieh, Y.-S. Carbonic anhydrase XII promotes invasion and migration ability of MDA-MB-231 breast cancer cells through the p38 MAPK signaling pathway. Eur. J. Cell Biol. 2010, 89, 598–606. [Google Scholar] [CrossRef] [PubMed]
- Pacchiano, F.; Carta, F.; McDonald, P.C.; Lou, Y.; Vullo, D.; Scozzafava, A.; Dedhar, S.; Supuran, C.T. Ureido-substituted benzenesulfonamides potently inhibit carbonic anhydrase IX and show antimetastatic activity in a model of breast cancer metastasis. J. Med. Chem. 2011, 54, 1896–1902. [Google Scholar] [CrossRef] [PubMed]
- Winum, J.-Y.; Carta, F.; Ward, C.; Mullen, P.; Harrison, D.; Langdon, S.P.; Cecchi, A.; Scozzafava, A.; Kunkler, I.; Supuran, C.T. Ureido-substituted sulfamates show potent carbonic anhydrase IX inhibitory and antiproliferative activities against breast cancer cell lines. Bioorg. Med. Chem. Lett. 2012, 22, 4681–4685. [Google Scholar] [CrossRef] [PubMed]
- Mahon, B.P.; McKenna, R. Regulation and role of carbonic anhydrase IX and use as a biomarker and therapeutic target in cancer. Res. Trends Curr. Top. Biochem. Res. 2013, 15, 1–21. [Google Scholar]
- Supuran, C.T. Carbonic Anhydrase inhibition and the management of hypoxic tumors. Metabolites 2017, 7, 48. [Google Scholar] [CrossRef] [PubMed]
- Mulders, P.; Bleumer, I.; Debruyne, F.; Oosterwijk, E. Specific monoclonal antibody-based immunotherapy by targeting the RCC-associated antigen carbonic anhydrase-IX(G250/MN). Urol. Ausg A 2004, 43, S146–S147. [Google Scholar] [CrossRef] [PubMed]
- Davis, I.D.; Wiseman, G.A.; Lee, F.-T.; Gansen, D.N.; Hopkins, W.; Papenfuss, A.T.; Liu, Z.; Moynihan, T.J.; Croghan, G.A.; Adjei, A.A.; et al. A phase I multiple dose, dose escalation study of cG250 monoclonal antibody in patients with advanced renal cell carcinoma. Cancer Immun. 2007, 7, 13. [Google Scholar] [PubMed]
- Stillebroer, A.B.; Boerman, O.C.; Desar, I.M.E.; Boers-Sonderen, M.J.; van Herpen, C.M.L.; Langenhuijsen, J.F.; Smith-Jones, P.M.; Oosterwijk, E.; Oyen, W.J.G.; Mulders, P.F.A. Phase 1 radioimmunotherapy study with lutetium 177-labeled anti-carbonic anhydrase IX monoclonal antibody girentuximab in patients with advanced renal cell carcinoma. Eur. Urol. 2013, 64, 478–485. [Google Scholar] [CrossRef] [PubMed]
- Muselaers, C.H.J.; Boers-Sonderen, M.J.; van Oostenbrugge, T.J.; Boerman, O.C.; Desar, I.M.E.; Stillebroer, A.B.; Mulder, S.F.; van Herpen, C.M.L.; Langenhuijsen, J.F.; Oosterwijk, E.; et al. Phase 2 study of lutetium 177-labeled anti-carbonic anhydrase IX monoclonal antibody girentuximab in patients with advanced renal cell carcinoma. Eur. Urol. 2016, 69, 767–770. [Google Scholar] [CrossRef] [PubMed]
- Dubois, L.J.; Niemans, R.; van Kuijk, S.J.A.; Panth, K.M.; Parvathaneni, N.-K.; Peeters, S.G.J.A.; Zegers, C.M.L.; Rekers, N.H.; van Gisbergen, M.W.; Biemans, R.; et al. New ways to image and target tumour hypoxia and its molecular responses. Radiother. Oncol. 2015, 116, 352–357. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Zhang, G.; Wang, X.; Li, X.-F. Is carbonic anhydrase IX a validated target for molecular imaging of cancer and hypoxia? Future Oncol. 2015, 11, 1531–1541. [Google Scholar] [CrossRef] [PubMed]
- Ahlskog, J.K.J.; Schliemann, C.; Mårlind, J.; Qureshi, U.; Ammar, A.; Pedley, R.B.; Neri, D. Human monoclonal antibodies targeting carbonic anhydrase IX for the molecular imaging of hypoxic regions in solid tumours. Br. J. Cancer 2009, 101, 645–657. [Google Scholar] [CrossRef] [PubMed]
- Chen, F.; Zhuang, X.; Lin, L.; Yu, P.; Wang, Y.; Shi, Y.; Hu, G.; Sun, Y. New horizons in tumor microenvironment biology: Challenges and opportunities. BMC Med. 2015, 13. [Google Scholar] [CrossRef] [PubMed]
- Whiteside, T.L. The tumor microenvironment and its role in promoting tumor growth. Oncogene 2008, 27, 5904–5912. [Google Scholar] [CrossRef] [PubMed]
- Brown, J.M. Tumor hypoxia in cancer therapy. Methods Enzymol. 2007, 435, 297–321. [Google Scholar] [CrossRef] [PubMed]
- Jamali, S.; Klier, M.; Ames, S.; Felipe Barros, L.; McKenna, R.; Deitmer, J.W.; Becker, H.M. Hypoxia-induced carbonic anhydrase IX facilitates lactate flux in human breast cancer cells by non-catalytic function. Sci. Rep. 2015, 5. [Google Scholar] [CrossRef] [PubMed]
- Warburg, O. The chemical constitution of respiratory ferment. Science 1928, 68, 437–443. [Google Scholar] [CrossRef] [PubMed]
- Warburg, O.; Wind, F.; Negelein, E. The metabolism of tumors in the body. J. Gen. Physiol. 1927, 8, 519–530. [Google Scholar] [CrossRef] [PubMed]
- Gatenby, R.A.; Gillies, R.J. Why do cancers have high aerobic glycolysis? Nat. Rev. Cancer 2004, 4, 891–899. [Google Scholar] [CrossRef] [PubMed]
- Newsholme, P.; Lima, M.M.R.; Procopio, J.; Pithon-Curi, T.C.; Doi, S.Q.; Bazotte, R.B.; Curi, R. Glutamine and glutamate as vital metabolites. Braz. J. Med. Biol. Res. 2003, 36, 153–163. [Google Scholar] [CrossRef] [PubMed]
- Brekke, E.; Morken, T.S.; Walls, A.B.; Waagepetersen, H.; Schousboe, A.; Sonnewald, U. Anaplerosis for glutamate synthesis in the neonate and in adulthood. In The Glutamate/GABA-Glutamine Cycle; Springer International Publishing: Basel, Switzerland, 2016; Volume 13, pp. 43–58. [Google Scholar]
- DeBerardinis, R.J.; Mancuso, A.; Daikhin, E.; Nissim, I.; Yudkoff, M.; Wehrli, S.; Thompson, C.B. Beyond aerobic glycolysis: Transformed cells can engage in glutamine metabolism that exceeds the requirement for protein and nucleotide synthesis. Proc. Natl. Acad. Sci. USA 2007, 104, 19345–19350. [Google Scholar] [CrossRef] [PubMed]
- Currie, E.; Schulze, A.; Zechner, R.; Walther, T.C.; Farese, R.V. Cellular fatty acid metabolism and cancer. Cell Metab. 2013, 18, 153–161. [Google Scholar] [CrossRef] [PubMed]
- Pavlova, N.N.; Thompson, C.B. The emerging hallmarks of cancer metabolism. Cell Metab. 2016, 23, 27–47. [Google Scholar] [CrossRef] [PubMed]
- Gillies, R.J.; Raghunand, N.; Karczmar, G.S.; Bhujwalla, Z.M. MRI of the tumor microenvironment. J. Magn. Reson. Imaging JMRI 2002, 16, 430–450. [Google Scholar] [CrossRef] [PubMed]
- Webb, B.A.; Chimenti, M.; Jacobson, M.P.; Barber, D.L. Dysregulated pH: A perfect storm for cancer progression. Nat. Rev. Cancer 2011, 11, 671–677. [Google Scholar] [CrossRef] [PubMed]
- Stüwe, L.; Müller, M.; Fabian, A.; Waning, J.; Mally, S.; Noël, J.; Schwab, A.; Stock, C. pH dependence of melanoma cell migration: Protons extruded by NHE1 dominate protons of the bulk solution. J. Physiol. 2007, 585, 351–360. [Google Scholar] [CrossRef] [PubMed]
- Balkwill, F.R.; Capasso, M.; Hagemann, T. The tumor microenvironment at a glance. J. Cell Sci. 2012, 125, 5591–5596. [Google Scholar] [CrossRef] [PubMed]
- Wilson, W.R.; Hay, M.P. Targeting hypoxia in cancer therapy. Nat. Rev. Cancer 2011, 11, 393–410. [Google Scholar] [CrossRef] [PubMed]
- Dietl, K.; Renner, K.; Dettmer, K.; Timischl, B.; Eberhart, K.; Dorn, C.; Hellerbrand, C.; Kastenberger, M.; Kunz-Schughart, L.A.; Oefner, P.J.; et al. Lactic acid and acidification inhibit TNF secretion and glycolysis of human monocytes. J. Immunol. 2010, 184, 1200–1209. [Google Scholar] [CrossRef] [PubMed]
- Kuwata, F.; Suzuki, N.; Otsuka, K.; Taguchi, M.; Sasai, Y.; Wakino, H.; Ito, M.; Ebihara, S.; Suzuki, K. Enzymatic regulation of glycolysis and gluconeogenesis in rabbit periodontal ligament under various physiological pH conditions. J. Nihon Univ. Sch. Dent. 1991, 33, 81–90. [Google Scholar] [CrossRef] [PubMed]
- Chan, F.K.-M.; Moriwaki, K.; De Rosa, M.J. Detection of necrosis by release of lactate dehydrogenase activity. In Immune Homeostasis; Humana Press: Totowa, NJ, USA, 2013; Volume 979, pp. 65–70. [Google Scholar]
- Gray, J.A. Kinetics of enamel dissolution during formation of incipient caries-like lesions. Arch. Oral Biol. 1966. [Google Scholar] [CrossRef]
- Putney, L.K.; Barber, D.L. Expression profile of genes regulated by activity of the Na-H exchanger NHE1. BMC Genom. 2004, 5, 46. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, J.L.-Y.; Lucas, J.E.; Schroeder, T.; Mori, S.; Wu, J.; Nevins, J.; Dewhirst, M.; West, M.; Chi, J.-T. The genomic analysis of lactic acidosis and acidosis response in human cancers. PLoS Genet. 2008, 4, e1000293. [Google Scholar] [CrossRef] [PubMed]
- Menard, L.; Maughan, D.; Vigoreaux, J. The structural and functional coordination of glycolytic enzymes in muscle: Evidence of a metabolon? Biology 2014, 3, 623–644. [Google Scholar] [CrossRef] [PubMed]
- Campanella, M.E.; Chu, H.; Wandersee, N.J.; Peters, L.L.; Mohandas, N.; Gilligan, D.M.; Low, P.S. Characterization of glycolytic enzyme interactions with murine erythrocyte membranes in wild-type and membrane protein knockout mice. Blood 2008, 112, 3900–3906. [Google Scholar] [CrossRef] [PubMed]
- Stock, C.; Schwab, A. Protons make tumor cells move like clockwork. Pflugers Arch. 2009, 458, 981–992. [Google Scholar] [CrossRef] [PubMed]
- Wilhelm, S.M.; Shao, Z.H.; Housley, T.J.; Seperack, P.K.; Baumann, A.P.; Gunja-Smith, Z.; Woessner, J.F. Matrix metalloproteinase-3 (stromelysin-1). Identification as the cartilage acid metalloprotease and effect of pH on catalytic properties and calcium affinity. J. Biol. Chem. 1993, 268, 21906–21913. [Google Scholar] [PubMed]
- Bourguignon, L.Y.W.; Singleton, P.A.; Diedrich, F.; Stern, R.; Gilad, E. CD44 interaction with Na+-H+ exchanger (NHE1) creates acidic microenvironments leading to hyaluronidase-2 and cathepsin B activation and breast tumor cell invasion. J. Biol. Chem. 2004, 279, 26991–27007. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.-S.; Bellin, R.M.; Walker, D.L.; Patel, B.; Powers, P.; Liu, H.; Garcia-Alvarez, B.; de Pereda, J.M.; Liddington, R.C.; Volkmann, N.; et al. Characterization of an actin-binding site within the talin FERM domain. J. Mol. Biol. 2004, 343, 771–784. [Google Scholar] [CrossRef] [PubMed]
- Moseley, J.B.; Okada, K.; Balcer, H.I.; Kovar, D.R.; Pollard, T.D.; Goode, B.L. Twinfilin is an actin-filament-severing protein and promotes rapid turnover of actin structures in vivo. J. Cell Sci. 2006, 119, 1547–1557. [Google Scholar] [CrossRef] [PubMed]
- Pope, B.J.; Zierler-Gould, K.M.; Kühne, R.; Weeds, A.G.; Ball, L.J. Solution structure of human cofilin: Actin binding, pH sensitivity, and relationship to actin-depolymerizing factor. J. Biol. Chem. 2004, 279, 4840–4848. [Google Scholar] [CrossRef] [PubMed]
- Srivastava, J.; Barreiro, G.; Groscurth, S.; Gingras, A.R.; Goult, B.T.; Critchley, D.R.; Kelly, M.J.S.; Jacobson, M.P.; Barber, D.L. Structural model and functional significance of pH-dependent talin-actin binding for focal adhesion remodeling. Proc. Natl. Acad. Sci. USA 2008, 105, 14436–14441. [Google Scholar] [CrossRef] [PubMed]
- Grey, M.J.; Tang, Y.; Alexov, E.; McKnight, C.J.; Raleigh, D.P.; Palmer, A.G. Characterizing a partially folded intermediate of the villin headpiece domain under non-denaturing conditions: Contribution of His41 to the pH-dependent stability of the N-terminal subdomain. J. Mol. Biol. 2006, 355, 1078–1094. [Google Scholar] [CrossRef] [PubMed]
- McLachlan, G.D.; Cahill, S.M.; Girvin, M.E.; Almo, S.C. Acid-induced equilibrium folding intermediate of human platelet profilin. Biochemistry 2007, 46, 6931–6943. [Google Scholar] [CrossRef] [PubMed]
- Lagadic-Gossmann, D.; Huc, L.; Lecureur, V. Alterations of intracellular pH homeostasis in apoptosis: Origins and roles. Cell Death Differ. 2004, 11, 953–961. [Google Scholar] [CrossRef] [PubMed]
- Matsuyama, S.; Llopis, J.; Deveraux, Q.L.; Tsien, R.Y.; Reed, J.C. Changes in intramitochondrial and cytosolic pH: Early events that modulate caspase activation during apoptosis. Nat. Cell Biol. 2000, 2, 318–325. [Google Scholar] [CrossRef] [PubMed]
- Pouysségur, J.; Franchi, A.; L’Allemain, G.; Paris, S. Cytoplasmic pH, a key determinant of growth factor-induced DNA synthesis in quiescent fibroblasts. FEBS Lett. 1985, 190, 115–119. [Google Scholar] [CrossRef]
- Bower, J.J.; Vance, L.D.; Psioda, M.; Smith-Roe, S.L.; Simpson, D.A.; Ibrahim, J.G.; Hoadley, K.A.; Perou, C.M.; Kaufmann, W.K. Patterns of cell cycle checkpoint deregulation associated with intrinsic molecular subtypes of human breast cancer cells. Npj Breast Cancer 2017, 3. [Google Scholar] [CrossRef] [PubMed]
- Khaled, A.R.; Kim, K.; Hofmeister, R.; Muegge, K.; Durum, S.K. Withdrawal of IL-7 induces Bax translocation from cytosol to mitochondria through a rise in intracellular pH. Proc. Natl. Acad. Sci. USA 1999, 96, 14476–14481. [Google Scholar] [CrossRef] [PubMed]
- Swietach, P.; Vaughan-Jones, R.D.; Harris, A.L.; Hulikova, A. The chemistry, physiology and pathology of pH in cancer. Philos. Trans. R. Soc. B Biol. Sci. 2014. [Google Scholar] [CrossRef] [PubMed]
- Neri, D.; Supuran, C.T. Interfering with pH regulation in tumours as a therapeutic strategy. Nat. Rev. Drug Discov. 2011, 10, 767–777. [Google Scholar] [CrossRef] [PubMed]
- Uda, N.R.; Seibert, V.; Stenner-Liewen, F.; Müller, P.; Herzig, P.; Gondi, G.; Zeidler, R.; van Dijk, M.; Zippelius, A.; Renner, C. Esterase activity of carbonic anhydrases serves as surrogate for selecting antibodies blocking hydratase activity. J. Enzyme Inhib. Med. Chem. 2015, 30, 955–960. [Google Scholar] [CrossRef] [PubMed]
- Verpoorte, J.A.; Mehta, S.; Edsall, J.T. Esterase activities of human carbonic anhydrases B and C. J. Biol. Chem. 1967, 242, 4221–4229. [Google Scholar] [PubMed]
- Lindskog, S.; Coleman, J.E. The catalytic mechanism of carbonic anhydrase. Proc. Natl. Acad. Sci. USA 1973, 70, 2505–2508. [Google Scholar] [CrossRef] [PubMed]
- Al-Samir, S.; Papadopoulos, S.; Scheibe, R.J.; Meißner, J.D.; Cartron, J.-P.; Sly, W.S.; Alper, S.L.; Gros, G.; Endeward, V. Activity and distribution of intracellular carbonic anhydrase II and their effects on the transport activity of anion exchanger AE1/SLC4A1: Role of CAII in the function of AE1. J. Physiol. 2013, 591, 4963–4982. [Google Scholar] [CrossRef] [PubMed]
- Benej, M.; Pastorekova, S.; Pastorek, J. Carbonic anhydrase IX: Regulation and role in cancer. Subcell. Biochem. 2014, 75, 199–219. [Google Scholar] [CrossRef] [PubMed]
- Alterio, V.; Hilvo, M.; Di Fiore, A.; Supuran, C.T.; Pan, P.; Parkkila, S.; Scaloni, A.; Pastorek, J.; Pastorekova, S.; Pedone, C.; et al. Crystal structure of the catalytic domain of the tumor-associated human carbonic anhydrase IX. Proc. Natl. Acad. Sci. USA 2009, 106, 16233–16238. [Google Scholar] [CrossRef] [PubMed]
- Widmann, M.; Trodler, P.; Pleiss, J. The isoelectric region of proteins: A systematic analysis. PLoS ONE 2010, 5, e10546. [Google Scholar] [CrossRef] [PubMed]
- Klier, M.; Andes, F.T.; Deitmer, J.W.; Becker, H.M. Intracellular and extracellular carbonic anhydrases cooperate non-enzymatically to enhance activity of monocarboxylate transporters. J. Biol. Chem. 2014, 289, 2765–2775. [Google Scholar] [CrossRef] [PubMed]
- Becker, H.M.; Klier, M.; Schüler, C.; McKenna, R.; Deitmer, J.W. Intramolecular proton shuttle supports not only catalytic but also noncatalytic function of carbonic anhydrase II. Proc. Natl. Acad. Sci. USA 2011, 108, 3071–3076. [Google Scholar] [CrossRef] [PubMed]
- Svastova, E.; Witarski, W.; Csaderova, L.; Kosik, I.; Skvarkova, L.; Hulikova, A.; Zatovicova, M.; Barathova, M.; Kopacek, J.; Pastorek, J.; et al. Carbonic anhydrase IX interacts with bicarbonate transporters in lamellipodia and increases cell migration via its catalytic domain. J. Biol. Chem. 2012, 287, 3392–3402. [Google Scholar] [CrossRef] [PubMed]
- Innocenti, A.; Vullo, D.; Scozzafava, A.; Casey, J.R.; Supuran, C.T. Carbonic anhydrase inhibitors. Interaction of isozymes I, II, IV, V, and IX with carboxylates. Bioorg. Med. Chem. Lett. 2005, 15, 573–578. [Google Scholar] [CrossRef] [PubMed]
- Swietach, P.; Patiar, S.; Supuran, C.T.; Harris, A.L.; Vaughan-Jones, R.D. The role of carbonic anhydrase 9 in regulating extracellular and intracellular pH in three-dimensional tumor cell growths. J. Biol. Chem. 2009, 284, 20299–20310. [Google Scholar] [CrossRef] [PubMed]
- Giffard, R.G.; Monyer, H.; Christine, C.W.; Choi, D.W. Acidosis reduces NMDA receptor activation, glutamate neurotoxicity, and oxygen-glucose deprivation neuronal injury in cortical cultures. Brain Res. 1990, 506, 339–342. [Google Scholar] [CrossRef]
- The PyMOL Molecular Graphics System, Version 2.0 Schrödinger, LLC. Available online: https://pymol.org/2/ (accessed on 21 February 2018).
- Whittington, D.A.; Waheed, A.; Ulmasov, B.; Shah, G.N.; Grubb, J.H.; Sly, W.S.; Christianson, D.W. Crystal structure of the dimeric extracellular domain of human carbonic anhydrase XII, a bitopic membrane protein overexpressed in certain cancer tumor cells. Proc. Natl. Acad. Sci. USA 2001, 98, 9545–9550. [Google Scholar] [CrossRef] [PubMed]
- Zhao, J.; Benlekbir, S.; Rubinstein, J.L. Electron cryomicroscopy observation of rotational states in a eukaryotic V-ATPase. Nature 2015, 521, 241–245. [Google Scholar] [CrossRef] [PubMed]
- Arakawa, T.; Kobayashi-Yurugi, T.; Alguel, Y.; Iwanari, H.; Hatae, H.; Iwata, M.; Abe, Y.; Hino, T.; Ikeda-Suno, C.; Kuma, H.; et al. Crystal structure of the anion exchanger domain of human erythrocyte band 3. Science 2015, 350, 680–684. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y. Structure and mechanism of the glycerol-3-phosphate transporter from escherichia coli. Science 2003, 301, 616–620. [Google Scholar] [CrossRef] [PubMed]
- Deng, D.; Xu, C.; Sun, P.; Wu, J.; Yan, C.; Hu, M.; Yan, N. Crystal structure of the human glucose transporter GLUT1. Nature 2014, 510, 121–125. [Google Scholar] [CrossRef] [PubMed]
- Avvaru, B.S.; Kim, C.U.; Sippel, K.H.; Gruner, S.M.; Agbandje-McKenna, M.; Silverman, D.N.; McKenna, R. A short, Strong hydrogen bond in the active site of human carbonic anhydrase II. Biochemistry 2010, 49, 249–251. [Google Scholar] [CrossRef] [PubMed]
- Michalczyk, R.; Unkefer, C.J.; Bacik, J.-P.; Schrader, T.E.; Ostermann, A.; Kovalevsky, A.Y.; McKenna, R.; Fisher, S.Z. Joint neutron crystallographic and NMR solution studies of Tyr residue ionization and hydrogen bonding: Implications for enzyme-mediated proton transfer. Proc. Natl. Acad. Sci. USA 2015, 112, 5673–5678. [Google Scholar] [CrossRef] [PubMed]
- Chiang, W.L.; Chu, S.C.; Lai, J.C.; Yang, S.F.; Chiou, H.L.; Hsieh, Y.S. Alternations in quantities and activities of erythrocyte cytosolic carbonic anhydrase isoenzymes in glucose-6-phosphate dehydrogenase-deficient individuals. Clin. Chim. Acta Int. J. Clin. Chem. 2001, 314, 195–201. [Google Scholar] [CrossRef]
- Maren, T.H.; Swenson, E.R. A comparative study of the kinetics of the Bohr effect in vertebrates. J. Physiol. 1980, 303, 535–547. [Google Scholar] [CrossRef] [PubMed]
- Swenson, E.R. Respiratory and renal roles of carbonic anhydrase in gas exchange and acid-base regulation. In The Carbonic Anhydrases; Birkhäuser Basel: Basel, Switzerland, 2000; pp. 281–341. [Google Scholar]
- Brown, B.F.; Quon, A.; Dyck, J.R.B.; Casey, J.R. Carbonic anhydrase II promotes cardiomyocyte hypertrophy. Can. J. Physiol. Pharmacol. 2012, 90, 1599–1610. [Google Scholar] [CrossRef] [PubMed]
- Kuo, W.-H.; Yang, S.-F.; Hsieh, Y.-S.; Tsai, C.-S.; Hwang, W.-L.; Chu, S.-C. Differential expression of carbonic anhydrase isoenzymes in various types of anemia. Clin. Chim. Acta Int. J. Clin. Chem. 2005, 351, 79–86. [Google Scholar] [CrossRef] [PubMed]
- Gilmour, K.M. Perspectives on carbonic anhydrase. Comp. Biochem. Physiol. A Mol. Integr. Physiol. 2010, 157, 193–197. [Google Scholar] [CrossRef] [PubMed]
- Becker, H.M.; Deitmer, J.W. Carbonic anhydrase II increases the activity of the human electrogenic Na+/HCO3− cotransporter. J. Biol. Chem. 2007, 282, 13508–13521. [Google Scholar] [CrossRef] [PubMed]
- Becker, H.M.; Deitmer, J.W. Nonenzymatic proton handling by carbonic anhydrase II during H+-lactate cotransport via monocarboxylate transporter 1. J. Biol. Chem. 2008, 283, 21655–21667. [Google Scholar] [CrossRef] [PubMed]
- Stridh, M.H.; Alt, M.D.; Wittmann, S.; Heidtmann, H.; Aggarwal, M.; Riederer, B.; Seidler, U.; Wennemuth, G.; McKenna, R.; Deitmer, J.W.; et al. Lactate flux in astrocytes is enhanced by a non-catalytic action of carbonic anhydrase II: CAII enhances lactate transport in astrocytes. J. Physiol. 2012, 590, 2333–2351. [Google Scholar] [CrossRef] [PubMed]
- Uhlén, M.; Björling, E.; Agaton, C.; Szigyarto, C.A.-K.; Amini, B.; Andersen, E.; Andersson, A.-C.; Angelidou, P.; Asplund, A.; Asplund, C.; et al. A human protein atlas for normal and cancer tissues based on antibody proteomics. Mol. Cell. Proteom. 2005, 4, 1920–1932. [Google Scholar] [CrossRef] [PubMed]
- Uhlen, M.; Oksvold, P.; Fagerberg, L.; Lundberg, E.; Jonasson, K.; Forsberg, M.; Zwahlen, M.; Kampf, C.; Wester, K.; Hober, S.; et al. Towards a knowledge-based human protein atlas. Nat. Biotechnol. 2010, 28, 1248–1250. [Google Scholar] [CrossRef] [PubMed]
- Uhlen, M.; Fagerberg, L.; Hallstrom, B.M.; Lindskog, C.; Oksvold, P.; Mardinoglu, A.; Sivertsson, A.; Kampf, C.; Sjostedt, E.; Asplund, A.; et al. Tissue-based map of the human proteome. Science 2015, 347. [Google Scholar] [CrossRef] [PubMed]
- Uhlen, M.; Zhang, C.; Lee, S.; Sjöstedt, E.; Fagerberg, L.; Bidkhori, G.; Benfeitas, R.; Arif, M.; Liu, Z.; Edfors, F.; et al. A pathology atlas of the human cancer transcriptome. Science 2017. [Google Scholar] [CrossRef] [PubMed]
- Thul, P.J.; Åkesson, L.; Wiking, M.; Mahdessian, D.; Geladaki, A.; Ait Blal, H.; Alm, T.; Asplund, A.; Björk, L.; Breckels, L.M.; et al. A subcellular map of the human proteome. Science 2017, 356, eaal3321. [Google Scholar] [CrossRef] [PubMed]
- Cerami, E.; Gao, J.; Dogrusoz, U.; Gross, B.E.; Sumer, S.O.; Aksoy, B.A.; Jacobsen, A.; Byrne, C.J.; Heuer, M.L.; Larsson, E.; et al. The cBio Cancer Genomics Portal: An open platform for exploring multidimensional cancer genomics data: Figure 1. Cancer Discov. 2012, 2, 401–404. [Google Scholar] [CrossRef] [PubMed]
- Gao, J.; Aksoy, B.A.; Dogrusoz, U.; Dresdner, G.; Gross, B.; Sumer, S.O.; Sun, Y.; Jacobsen, A.; Sinha, R.; Larsson, E.; et al. Integrative Analysis of Complex Cancer Genomics and Clinical Profiles Using the cBioPortal. Sci. Signal. 2013. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zheng, Y.; Xu, B.; Zhao, Y.; Gu, H.; Li, C.; Wang, Y.; Chang, X. CA1 contributes to microcalcification and tumourigenesis in breast cancer. BMC Cancer 2015, 15. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.; Lu, X.; Zhang, X.; Li, Z.; Li, C. Carbonic anhydrase 1 is a promising biomarker for early detection of non-small cell lung cancer. Tumor Biol. 2016, 37, 553–559. [Google Scholar] [CrossRef] [PubMed]
- Takakura, M.; Yokomizo, A.; Tanaka, Y.; Kobayashi, M.; Jung, G.; Banno, M.; Sakuma, T.; Imada, K.; Oda, Y.; Kamita, M.; et al. Carbonic anhydrase I as a new plasma biomarker for prostate cancer. ISRN Oncol. 2012, 2012, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Zhou, R.; Huang, W.; Yao, Y.; Wang, Y.; Li, Z.; Shao, B.; Zhong, J.; Tang, M.; Liang, S.; Zhao, X.; et al. CA II, a potential biomarker by proteomic analysis, exerts significant inhibitory effect on the growth of colorectal cancer cells. Int. J. Oncol. 2013, 43, 611–621. [Google Scholar] [CrossRef] [PubMed]
- Parkkila, S.; Lasota, J.; Fletcher, J.A.; Ou, W.; Kivelä, A.J.; Nuorva, K.; Parkkila, A.-K.; Ollikainen, J.; Sly, W.S.; Waheed, A.; et al. Carbonic anhydrase II. A novel biomarker for gastrointestinal stromal tumors. Mod. Pathol. 2010, 23, 743–750. [Google Scholar] [CrossRef] [PubMed]
- Järvinen, P.; Kivelä, A.J.; Nummela, P.; Lepistö, A.; Ristimäki, A.; Parkkila, S. Carbonic anhydrase II: A novel biomarker for pseudomyxoma peritonei. APMIS 2017, 125, 207–212. [Google Scholar] [CrossRef] [PubMed]
- Waterman, E.A.; Cross, N.A.; Lippitt, J.M.; Cross, S.S.; Rehman, I.; Holen, I.; Hamdy, F.C.; Eaton, C.L. The antibody MAB8051 directed against osteoprotegerin detects carbonic anhydrase II: Implications for association studies with human cancers. Int. J. Cancer 2007, 121, 1958–1966. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.-C. Overexpression of carbonic anhydrase II and Ki-67 proteins in prognosis of gastrointestinal stromal tumors. World J. Gastroenterol. 2013, 19, 2473. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.-M.; Lin, Y.-M.; Yeh, K.-T.; Chen, M.-K.; Chang, J.-H.; Chen, C.-J.; Chou, M.-Y.; Yang, S.-F.; Chien, M.-H. Expression of carbonic anhydrases I/II and the correlation to clinical aspects of oral squamous cell carcinoma analyzed using tissue microarray: CA I/II and correlation to OSCC. J. Oral Pathol. Med. 2012. [Google Scholar] [CrossRef] [PubMed]
- Harju, A.-K.; Bootorabi, F.; Kuuslahti, M.; Supuran, C.T.; Parkkila, S. Carbonic anhydrase III: A neglected isozyme is stepping into the limelight. J. Enzyme Inhib. Med. Chem. 2013, 28, 231–239. [Google Scholar] [CrossRef] [PubMed]
- Shi, C.; Uda, Y.; Dedic, C.; Azab, E.; Sun, N.; Hussein, A.I.; Petty, C.A.; Fulzele, K.; Mitterberger-Vogt, M.C.; Zwerschke, W.; et al. Carbonic anhydrase III protects osteocytes from oxidative stress. FASEB J. 2018, 32, 440–452. [Google Scholar] [CrossRef] [PubMed]
- Dai, H.-Y.; Hong, C.-C.; Liang, S.-C.; Yan, M.-D.; Lai, G.-M.; Cheng, A.-L.; Chuang, S.-E. Carbonic anhydrase III promotes transformation and invasion capability in hepatoma cells through FAK signaling pathway. Mol. Carcinog. 2008, 47, 956–963. [Google Scholar] [CrossRef] [PubMed]
- Bootorabi, F.; Haapasalo, J.; Smith, E.; Haapasalo, H.; Parkkila, S. Carbonic anhydrase VII—A potential prognostic marker in gliomas. Health 2011, 3, 6–12. [Google Scholar] [CrossRef]
- Hilvo, M.; Innocenti, A.; Monti, S.M.; De Simone, G.; Supuran, C.T.; Parkkila, S. Recent advances in research on the most novel carbonic anhydrases, CA XIII and XV. Curr. Pharm. Des. 2008, 14, 672–678. [Google Scholar] [PubMed]
- Shah, G.N.; Hewett-Emmett, D.; Grubb, J.H.; Migas, M.C.; Fleming, R.E.; Waheed, A.; Sly, W.S. Mitochondrial carbonic anhydrase CA VB: Differences in tissue distribution and pattern of evolution from those of CA VA suggest distinct physiological roles. Proc. Natl. Acad. Sci. USA 2000, 97, 1677–1682. [Google Scholar] [CrossRef] [PubMed]
- Nishimori, I.; Vullo, D.; Innocenti, A.; Scozzafava, A.; Mastrolorenzo, A.; Supuran, C.T. Carbonic Anhydrase Inhibitors. The Mitochondrial Isozyme VB as a new target for sulfonamide and sulfamate inhibitors. J. Med. Chem. 2005, 48, 7860–7866. [Google Scholar] [CrossRef] [PubMed]
- Arechederra, R.L.; Waheed, A.; Sly, W.S.; Supuran, C.T.; Minteer, S.D. Effect of sulfonamides as carbonic anhydrase VA and VB inhibitors on mitochondrial metabolic energy conversion. Bioorg. Med. Chem. 2013, 21, 1544–1548. [Google Scholar] [CrossRef] [PubMed]
- Shah, G.N.; Rubbelke, T.S.; Hendin, J.; Nguyen, H.; Waheed, A.; Shoemaker, J.D.; Sly, W.S. Targeted mutagenesis of mitochondrial carbonic anhydrases VA and VB implicates both enzymes in ammonia detoxification and glucose metabolism. Proc. Natl. Acad. Sci. USA 2013, 110, 7423–7428. [Google Scholar] [CrossRef] [PubMed]
- Poulsen, S.-A.; Wilkinson, B.L.; Innocenti, A.; Vullo, D.; Supuran, C.T. Inhibition of human mitochondrial carbonic anhydrases VA and VB with para-(4-phenyltriazole-1-yl)-benzenesulfonamide derivatives. Bioorg. Med. Chem. Lett. 2008, 18, 4624–4627. [Google Scholar] [CrossRef] [PubMed]
- Lusty, C.J. Carbamoylphosphate synthetase I of rat-liver mitochondria. Purification, properties, and polypeptide molecular weight. Eur. J. Biochem. 1978, 85, 373–383. [Google Scholar] [CrossRef] [PubMed]
- Dodgson, S.J.; Forster, R.E.; Storey, B.T. The role of carbonic anhydrase in hepatocyte metabolism. Ann. N. Y. Acad. Sci. 1984, 429, 516–524. [Google Scholar] [CrossRef] [PubMed]
- Dodgson, S.J.; Forster, R.E. Inhibition of CA V decreases glucose synthesis from pyruvate. Arch. Biochem. Biophys. 1986, 251, 198–204. [Google Scholar] [CrossRef]
- Dodgson, S.J. Inhibition of mitochondrial carbonic anhydrase and ureagenesis: A discrepancy examined. J. Appl. Physiol. 1987, 63, 2134–2141. [Google Scholar] [CrossRef] [PubMed]
- Hazen, S.A.; Waheed, A.; Sly, W.S.; LaNoue, K.F.; Lynch, C.J. Differentiation-dependent expression of CA V and the role of carbonic anhydrase isozymes in pyruvate carboxylation in adipocytes. FASEB J. 1996, 10, 481–490. [Google Scholar] [CrossRef] [PubMed]
- Henry, R.P. Multiple roles of carbonic anhydrase in cellular transport and metabolism. Annu. Rev. Physiol. 1996, 58, 523–538. [Google Scholar] [CrossRef] [PubMed]
- Van Karnebeek, C.D.; Sly, W.S.; Ross, C.J.; Salvarinova, R.; Yaplito-Lee, J.; Santra, S.; Shyr, C.; Horvath, G.A.; Eydoux, P.; Lehman, A.M.; et al. Mitochondrial Carbonic Anhydrase VA Deficiency Resulting from CA5A Alterations presents with hyperammonemia in early childhood. Am. J. Hum. Genet. 2014, 94, 453–461. [Google Scholar] [CrossRef] [PubMed]
- Kivelä, J.; Parkkila, S.; Parkkila, A.-K.; Leinonen, J.; Rajaniemi, H. Salivary carbonic anhydrase isoenzyme VI. J. Physiol. 1999, 520, 315–320. [Google Scholar] [CrossRef] [PubMed]
- Frasseto, F.; Parisotto, T.M.; Peres, R.C.R.; Marques, M.R.; Line, S.R.P.; Nobre Dos Santos, M. Relationship among salivary carbonic anhydrase VI activity and flow rate, biofilm pH and caries in primary dentition. Caries Res. 2012, 46, 194–200. [Google Scholar] [CrossRef] [PubMed]
- Kimoto, M.; Kishino, M.; Yura, Y.; Ogawa, Y. A role of salivary carbonic anhydrase VI in dental plaque. Arch. Oral Biol. 2006, 51, 117–122. [Google Scholar] [CrossRef] [PubMed]
- Feeney, E.L.; Hayes, J.E. Exploring associations between taste perception, oral anatomy and polymorphisms in the carbonic anhydrase (gustin) gene CA6. Physiol. Behav. 2014, 128, 148–154. [Google Scholar] [CrossRef] [PubMed]
- Thatcher, B.J.; Doherty, A.E.; Orvisky, E.; Martin, B.M.; Henkin, R.I. Gustin from human parotid saliva is carbonic anhydrase VI. Biochem. Biophys. Res. Commun. 1998, 250, 635–641. [Google Scholar] [CrossRef] [PubMed]
- Karhumaa, P.; Leinonen, J.; Parkkila, S.; Kaunisto, K.; Tapanainen, J.; Rajaniemi, H. The identification of secreted carbonic anhydrase VI as a constitutive glycoprotein of human and rat milk. Proc. Natl. Acad. Sci. USA 2001, 98, 11604–11608. [Google Scholar] [CrossRef] [PubMed]
- Ogawa, Y.; Matsumoto, K.; Maeda, T.; Tamai, R.; Suzuki, T.; Sasano, H.; Fernley, R.T. Characterization of lacrimal gland carbonic anhydrase VI. J. Histochem. Cytochem. 2002, 50, 821–827. [Google Scholar] [CrossRef] [PubMed]
- Leinonen, J.S.; Saari, K.A.; Seppänen, J.M.; Myllylä, H.M.; Rajaniemi, H.J. Immunohistochemical demonstration of carbonic anhydrase isoenzyme VI (CA VI) expression in rat lower airways and lung. J. Histochem. Cytochem. 2004, 52, 1107–1112. [Google Scholar] [CrossRef] [PubMed]
- Kivelä, J.; Parkkila, S.; Waheed, A.; Parkkila, A.K.; Sly, W.S.; Rajaniemi, H. Secretory carbonic anhydrase isoenzyme (CA VI) in human serum. Clin. Chem. 1997, 43, 2318–2322. [Google Scholar] [PubMed]
- Li, Z.-Q.; Hu, X.-P.; Zhou, J.-Y.; Xie, X.-D.; Zhang, J.-M. Genetic polymorphisms in the carbonic anhydrase VI gene and dental caries susceptibility. Genet. Mol. Res. 2015, 14, 5986–5993. [Google Scholar] [CrossRef] [PubMed]
- Sengul, F.; Kilic, M.; Gurbuz, T.; Tasdemir, S. Carbonic anhydrase VI gene polymorphism rs2274327 relationship between salivary parameters and dental-oral health status in children. Biochem. Genet. 2016, 54, 467–475. [Google Scholar] [CrossRef] [PubMed]
- Dowd, F.J. Saliva and dental caries. Dent. Clin. N. Am. 1999, 43, 579–597. [Google Scholar] [PubMed]
- Shatzman, A.R.; Henkin, R.I. Gustin concentration changes relative to salivary zinc and taste in humans. Proc. Natl. Acad. Sci. USA 1981, 78, 3867–3871. [Google Scholar] [CrossRef] [PubMed]
- Henkin, R.I.; Lippoldt, R.E.; Bilstad, J.; Edelhoch, H. A zinc protein isolated from human parotid saliva. Proc. Natl. Acad. Sci. USA 1975, 72, 488–492. [Google Scholar] [CrossRef] [PubMed]
- Innocenti, A.; Pastorekova, S.; Pastorek, J.; Scozzafava, A.; Simone, G.D.; Supuran, C.T. The proteoglycan region of the tumor-associated carbonic anhydrase isoform IX acts as anintrinsic buffer optimizing CO2 hydration at acidic pH values characteristic of solid tumors. Bioorg. Med. Chem. Lett. 2009, 19, 5825–5828. [Google Scholar] [CrossRef] [PubMed]
- Barker, H.; Aaltonen, M.; Pan, P.; Vähätupa, M.; Kaipiainen, P.; May, U.; Prince, S.; Uusitalo-Järvinen, H.; Waheed, A.; Pastoreková, S.; et al. Role of carbonic anhydrases in skin wound healing. Exp. Mol. Med. 2017, 49, e334. [Google Scholar] [CrossRef] [PubMed]
- Fujikawa-Adachi, K.; Nishimori, I.; Sakamoto, S.; Morita, M.; Onishi, S.; Yonezawa, S.; Hollingsworth, M.A. Identification of carbonic anhydrase IV and VI mRNA expression in human pancreas and salivary glands. Pancreas 1999, 18, 329–335. [Google Scholar] [CrossRef] [PubMed]
- Wistrand, P.J.; Carter, N.D.; Conroy, C.W.; Mahieu, I. Carbonic anhydrase IV activity is localized on the exterior surface of human erythrocytes. Acta Physiol. Scand. 1999, 165, 211–218. [Google Scholar] [CrossRef] [PubMed]
- Sender, S.; Decker, B.; Fenske, C.D.; Sly, W.S.; Carter, N.D.; Gros, G. Localization of carbonic anhydrase IV in rat and human heart muscle. J. Histochem. Cytochem. 1998, 46, 855–861. [Google Scholar] [CrossRef] [PubMed]
- Parkkila, S.; Parkkila, A.K.; Juvonen, T.; Waheed, A.; Sly, W.S.; Saarnio, J.; Kaunisto, K.; Kellokumpu, S.; Rajaniemi, H. Membrane-bound carbonic anhydrase IV is expressed in the luminal plasma membrane of the human gallbladder epithelium. Hepatology 1996, 24, 1104–1108. [Google Scholar] [CrossRef] [PubMed]
- Sender, S.; Gros, G.; Waheed, A.; Hageman, G.S.; Sly, W.S. Immunohistochemical localization of carbonic anhydrase IV in capillaries of rat and human skeletal muscle. J. Histochem. Cytochem. 1994, 42, 1229–1236. [Google Scholar] [CrossRef] [PubMed]
- Fleming, R.E.; Parkkila, S.; Parkkila, A.K.; Rajaniemi, H.; Waheed, A.; Sly, W.S. Carbonic anhydrase IV expression in rat and human gastrointestinal tract regional, cellular, and subcellular localization. J. Clin. Investig. 1995, 96, 2907–2913. [Google Scholar] [CrossRef] [PubMed]
- Purkerson, J.M.; Schwartz, G.J. The role of carbonic anhydrases in renal physiology. Kidney Int. 2007, 71, 103–115. [Google Scholar] [CrossRef] [PubMed]
- Alvarez, B.V.; Loiselle, F.B.; Supuran, C.T.; Schwartz, G.J.; Casey, J.R. Direct extracellular interaction between carbonic anhydrase IV and the human NBC1 sodium/bicarbonate co-transporter. Biochemistry 2003, 42, 12321–12329. [Google Scholar] [CrossRef] [PubMed]
- Alvarez, B.V.; Vithana, E.N.; Yang, Z.; Koh, A.H.; Yeung, K.; Yong, V.; Shandro, H.J.; Chen, Y.; Kolatkar, P.; Palasingam, P.; et al. Identification and characterization of a novel mutation in the carbonic anhydrase IV gene that causes retinitis pigmentosa. Invest. Ophthalmol. Vis. Sci. 2007, 48, 3459–3468. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.; Alvarez, B.V.; Chakarova, C.; Jiang, L.; Karan, G.; Frederick, J.M.; Zhao, Y.; Sauvé, Y.; Li, X.; Zrenner, E.; et al. Mutant carbonic anhydrase 4 impairs pH regulation and causes retinal photoreceptor degeneration. Hum. Mol. Genet. 2005, 14, 255–265. [Google Scholar] [CrossRef] [PubMed]
- Parkkila, S.; Parkkila, A.K.; Rajaniemi, H.; Shah, G.N.; Grubb, J.H.; Waheed, A.; Sly, W.S. Expression of membrane-associated carbonic anhydrase XIV on neurons and axons in mouse and human brain. Proc. Natl. Acad. Sci. USA 2001, 98, 1918–1923. [Google Scholar] [CrossRef] [PubMed]
- Kaunisto, K.; Parkkila, S.; Rajaniemi, H.; Waheed, A.; Grubb, J.; Sly, W.S. Carbonic anhydrase XIV: Luminal expression suggests key role in renal acidification. Kidney Int. 2002, 61, 2111–2118. [Google Scholar] [CrossRef] [PubMed]
- Juel, C.; Lundby, C.; Sander, M.; Calbet, J.A.L.; van Hall, G. Human skeletal muscle and erythrocyte proteins involved in acid-base homeostasis: Adaptations to chronic hypoxia. J. Physiol. 2003, 548, 639–648. [Google Scholar] [CrossRef] [PubMed]
- Vargas, L.A.; Alvarez, B.V. Carbonic anhydrase XIV in the normal and hypertrophic myocardium. J. Mol. Cell. Cardiol. 2012, 52, 741–752. [Google Scholar] [CrossRef] [PubMed]
- Pastorek, J.; Pastoreková, S.; Callebaut, I.; Mornon, J.P.; Zelník, V.; Opavský, R.; Zat’ovicová, M.; Liao, S.; Portetelle, D.; Stanbridge, E.J. Cloning and characterization of MN, a human tumor-associated protein with a domain homologous to carbonic anhydrase and a putative helix-loop-helix DNA binding segment. Oncogene 1994, 9, 2877–2888. [Google Scholar] [PubMed]
- Liao, S.-Y.; Lerman, M.I.; Stanbridge, E.J. Expression of transmembrane carbonic anhydrases, CAIX and CAXII, in human development. BMC Dev. Biol. 2009, 9, 22. [Google Scholar] [CrossRef] [PubMed]
- Ivanov, S.; Shu-Yuan, L.; Ivanova, A.; Danilkovitch-Miagkova, A.; Tarasova, N.; Weirich, G.; Merrill, M.J.; Proescholdt, M.A.; Oldfield, E.H.; Lee, J.; et al. Expression of hypoxia inducible cell surface transmembrane carbonic anhydrases in human cancer. Am. J. Pathol. 2001, 158, 905–919. [Google Scholar] [CrossRef]
- Radvak, P.; Repic, M.; Svastova, E.; Takacova, M.; Csaderova, L.; Strnad, H.; Pastorek, J.; Pastorekova, S.; Kopacek, J. Suppression of carbonic anhydrase IX leads to aberrant focal adhesion and decreased invasion of tumor cells. Oncol. Rep. 2013, 29, 1147–1153. [Google Scholar] [CrossRef] [PubMed]
- Csaderova, L.; Debreova, M.; Radvak, P.; Stano, M.; Vrestiakova, M.; Kopacek, J.; Pastorekova, S.; Svastova, E. The effect of carbonic anhydrase IX on focal contacts during cell spreading and migration. Front. Physiol. 2013, 4. [Google Scholar] [CrossRef] [PubMed]
- Karhumaa, P.; Kaunisto, K.; Parkkila, S.; Waheed, A.; Pastoreková, S.; Pastorek, J.; Sly, W.S.; Rajaniemi, H. Expression of the transmembrane carbonic anhydrases, CA IX and CA XII, in the human male excurrent ducts. Mol. Hum. Reprod. 2001, 7, 611–616. [Google Scholar] [CrossRef] [PubMed]
- Parkkila, S.; Parkkila, A.K.; Saarnio, J.; Kivelä, J.; Karttunen, T.J.; Kaunisto, K.; Waheed, A.; Sly, W.S.; Türeci, O.; Virtanen, I.; et al. Expression of the membrane-associated carbonic anhydrase isozyme XII in the human kidney and renal tumors. J. Histochem. Cytochem. 2000, 48, 1601–1608. [Google Scholar] [CrossRef] [PubMed]
- Lee, M.; Vecchio-Pagán, B.; Sharma, N.; Waheed, A.; Li, X.; Raraigh, K.S.; Robbins, S.; Han, S.T.; Franca, A.L.; Pellicore, M.J.; et al. Loss of carbonic anhydrase XII function in individuals with elevated sweat chloride concentration and pulmonary airway disease. Hum. Mol. Genet. 2016, 25, 1923–1933. [Google Scholar] [CrossRef] [PubMed]
- Feldshtein, M.; Elkrinawi, S.; Yerushalmi, B.; Marcus, B.; Vullo, D.; Romi, H.; Ofir, R.; Landau, D.; Sivan, S.; Supuran, C.T.; et al. Hyperchlorhidrosis caused by homozygous mutation in CA12, encoding carbonic anhydrase XII. Am. J. Hum. Genet. 2010, 87, 713–720. [Google Scholar] [CrossRef] [PubMed]
- Capkova, L.; Koubkova, L.; Kodet, R. Expression of carbonic anhydrase IX (CAIX) in malignant mesothelioma. An immunohistochemical and immunocytochemical study. Neoplasma 2014, 61, 161–169. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.-S.; Chen, M.-K.; Yang, S.-F.; Chang, Y.-C.; Su, S.-C.; Chiou, H.-L.; Chien, M.-H.; Lin, C.-W. Increased expression of carbonic anhydrase IX in oral submucous fibrosis and oral squamous cell carcinoma. Clin. Chem. Lab. Med. 2014, 52, 1367–1377. [Google Scholar] [CrossRef] [PubMed]
- Jomrich, G.; Jesch, B.; Birner, P.; Schwameis, K.; Paireder, M.; Asari, R.; Schoppmann, S.F. Stromal expression of carbonic anhydrase IX in esophageal cancer. Clin. Transl. Oncol. 2014, 16, 966–972. [Google Scholar] [CrossRef] [PubMed]
- Wykoff, C.C.; Beasley, N.J.; Watson, P.H.; Turner, K.J.; Pastorek, J.; Sibtain, A.; Wilson, G.D.; Turley, H.; Talks, K.L.; Maxwell, P.H.; et al. Hypoxia-inducible expression of tumor-associated carbonic anhydrases. Cancer Res. 2000, 60, 7075–7083. [Google Scholar] [PubMed]
- Kopacek, J.; Barathova, M.; Dequiedt, F.; Sepelakova, J.; Kettmann, R.; Pastorek, J.; Pastorekova, S. MAPK pathway contributes to density- and hypoxia-induced expression of the tumor-associated carbonic anhydrase IX. Biochim. Biophys. Acta BBA—Gene Struct. Expr. 2005, 1729, 41–49. [Google Scholar] [CrossRef] [PubMed]
- Kaluz, S.; Kaluzová, M.; Chrastina, A.; Olive, P.L.; Pastoreková, S.; Pastorek, J.; Lerman, M.I.; Stanbridge, E.J. Lowered oxygen tension induces expression of the hypoxia marker MN/carbonic anhydrase IX in the absence of hypoxia-inducible factor 1 alpha stabilization: A role for phosphatidylinositol 3′-kinase. Cancer Res. 2002, 62, 4469–4477. [Google Scholar] [PubMed]
- Swayampakula, M.; McDonald, P.C.; Vallejo, M.; Coyaud, E.; Chafe, S.C.; Westerback, A.; Venkateswaran, G.; Shankar, J.; Gao, G.; Laurent, E.M.N.; et al. The interactome of metabolic enzyme carbonic anhydrase IX reveals novel roles in tumor cell migration and invadopodia/MMP14-mediated invasion. Oncogene 2017, 36, 6244–6261. [Google Scholar] [CrossRef] [PubMed]
- Ditte, P.; Dequiedt, F.; Svastova, E.; Hulikova, A.; Ohradanova-Repic, A.; Zatovicova, M.; Csaderova, L.; Kopacek, J.; Supuran, C.T.; Pastorekova, S.; et al. Phosphorylation of carbonic anhydrase IX controls its ability to mediate extracellular acidification in hypoxic tumors. Cancer Res. 2011, 71, 7558–7567. [Google Scholar] [CrossRef] [PubMed]
- Dorai, T.; Sawczuk, I.S.; Pastorek, J.; Wiernik, P.H.; Dutcher, J.P. The role of carbonic anhydrase IX overexpression in kidney cancer. Eur. J. Cancer 2005, 41, 2935–2947. [Google Scholar] [CrossRef] [PubMed]
- Nasu, K.; Yamaguchi, K.; Takanashi, T.; Tamai, K.; Sato, I.; Ine, S.; Sasaki, O.; Satoh, K.; Tanaka, N.; Tanaka, Y.; et al. Crucial role of carbonic anhydrase IX in tumorigenicity of xenotransplanted adult T-cell leukemia-derived cells. Cancer Sci. 2017, 108, 435–443. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Dong, M.; Sheng, W.; Huang, L. Roles of carbonic anhydrase IX in development of pancreatic cancer. Pathol. Oncol. Res. 2016, 22, 277–286. [Google Scholar] [CrossRef] [PubMed]
- Li, G.; Feng, G.; Zhao, A.; Péoc’h, M.; Cottier, M.; Mottet, N. CA9 as a biomarker in preoperative biopsy of small solid renal masses for diagnosis of clear cell renal cell carcinoma. Biomarkers 2017, 22, 123–126. [Google Scholar] [CrossRef] [PubMed]
- Smith, A.D.; Truong, M.; Bristow, R.; Yip, P.; Milosevic, M.F.; Joshua, A.M. The utility of serum CA9 for prognostication in prostate cancer. Anticancer Res. 2016, 36, 4489–4492. [Google Scholar] [CrossRef] [PubMed]
- De Martino, M.; Lucca, I.; Mbeutcha, A.; Wiener, H.G.; Haitel, A.; Susani, M.; Shariat, S.F.; Klatte, T. Carbonic anhydrase IX as a diagnostic urinary marker for urothelial bladder cancer. Eur. Urol. 2015, 68, 552–554. [Google Scholar] [CrossRef] [PubMed]
- Huang, W.-J.; Jeng, Y.-M.; Lai, H.-S.; Fong, I.-U.; Sheu, F.-Y. B.; Lai, P.-L.; Yuan, R.-H. Expression of hypoxic marker carbonic anhydrase IX predicts poor prognosis in resectable hepatocellular carcinoma. PLoS ONE 2015, 10, e0119181. [Google Scholar] [CrossRef] [PubMed]
- Ilie, M.I.; Hofman, V.; Ortholan, C.; Ammadi, R.E.; Bonnetaud, C.; Havet, K.; Venissac, N.; Mouroux, J.; Mazure, N.M.; Pouysségur, J.; et al. Overexpression of carbonic anhydrase XII in tissues from resectable non-small cell lung cancers is a biomarker of good prognosis. Int. J. Cancer 2011, 128, 1614–1623. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, M.; Matsumoto, T.; Ryuge, S.; Yanagita, K.; Nagashio, R.; Kawakami, Y.; Goshima, N.; Jiang, S.-X.; Saegusa, M.; Iyoda, A.; et al. CAXII is a sero-diagnostic marker for lung cancer. PLoS ONE 2012, 7, e33952. [Google Scholar] [CrossRef] [PubMed]
- Yoo, C.W.; Nam, B.-H.; Kim, J.-Y.; Shin, H.-J.; Lim, H.; Lee, S.; Lee, S.-K.; Lim, M.-C.; Song, Y.-J. Carbonic anhydrase XII expression is associated with histologic grade of cervical cancer and superior radiotherapy outcome. Radiat. Oncol. Lond. Engl. 2010, 5, 101. [Google Scholar] [CrossRef] [PubMed]
- Chien, M.-H.; Ying, T.-H.; Hsieh, Y.-H.; Lin, C.-H.; Shih, C.-H.; Wei, L.-H.; Yang, S.-F. Tumor-associated carbonic anhydrase XII is linked to the growth of primary oral squamous cell carcinoma and its poor prognosis. Oral Oncol. 2012, 48, 417–423. [Google Scholar] [CrossRef] [PubMed]
- Kopecka, J.; Campia, I.; Jacobs, A.; Frei, A.P.; Ghigo, D.; Wollscheid, B.; Riganti, C. Carbonic anhydrase XII is a new therapeutic target to overcome chemoresistance in cancer cells. Oncotarget 2015, 6, 6776–6793. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| I | II | III | IV | VA | VB | VI | VII | IX | XII | XIII | XIV | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| I | - | 154 | 141 | 78 | 126 | 128 | 82 | 132 | 83 | 91 | 154 | 85 |
| II | 60 | - | 152 | 88 | 133 | 138 | 90 | 147 | 85 | 89 | 157 | 96 |
| III | 54 | 58 | - | 82 | 120 | 117 | 87 | 130 | 80 | 86 | 151 | 90 |
| IV | 30 | 33 | 32 | - | 89 | 93 | 97 | 90 | 84 | 91 | 84 | 62 |
| VA | 48 | 51 | 45 | 24 | - | 184 | 93 | 131 | 83 | 84 | 124 | 88 |
| VB | 47 | 52 | 43 | 23 | 59 | - | 82 | 134 | 89 | 79 | 131 | 88 |
| VI | 32 | 33 | 32 | 27 | 28 | 24 | - | 93 | 107 | 104 | 90 | 106 |
| VII | 51 | 56 | 50 | 32 | 48 | 49 | 35 | - | 95 | 103 | 139 | 97 |
| IX | 33 | 34 | 31 | 27 | 32 | 33 | 39 | 37 | - | 101 | 90 | 113 |
| XII | 36 | 34 | 32 | 28 | 32 | 30 | 38 | 33 | 39 | - | 91 | 123 |
| XIII | 59 | 60 | 58 | 28 | 46 | 48 | 33 | 53 | 35 | 35 | - | 98 |
| XIV | 34 | 36 | 34 | 29 | 32 | 29 | 36 | 36 | 44 | 46 | 37 | - |
| Isoform | Kcat (s–1) | KM (mM) | Kcat/KM (M−1 s−1) |
|---|---|---|---|
| I | 2.0 × 105 | 4.0 | 5.0 × 107 |
| II | 1.4 × 106 | 9.3 | 1.5 × 108 |
| III | 1.3 × 104 | 33.3 | 4.0 × 105 |
| IV | 1.1 × 106 | 21.5 | 5.1 × 107 |
| VA | 2.9 × 105 | 10.0 | 2.9 × 107 |
| VB | 9.5 × 105 | 9.7 | 9.8 × 107 |
| VI | 3.4 × 105 | 6.9 | 4.9 × 107 |
| VII | 9.5 × 106 | 11.4 | 8.3 × 107 |
| IX | 1.1 × 106 | 6.9 | 1.6 × 108 |
| XII | 4.2 × 105 | 12.0 | 3.5 × 107 |
| XIII | 1.5 × 105 | 13.8 | 1.1 × 107 |
| XIV | 3.1 × 105 | 7.9 | 3.9 × 107 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mboge, M.Y.; Mahon, B.P.; McKenna, R.; Frost, S.C. Carbonic Anhydrases: Role in pH Control and Cancer. Metabolites 2018, 8, 19. https://doi.org/10.3390/metabo8010019
Mboge MY, Mahon BP, McKenna R, Frost SC. Carbonic Anhydrases: Role in pH Control and Cancer. Metabolites. 2018; 8(1):19. https://doi.org/10.3390/metabo8010019
Chicago/Turabian StyleMboge, Mam Y., Brian P. Mahon, Robert McKenna, and Susan C. Frost. 2018. "Carbonic Anhydrases: Role in pH Control and Cancer" Metabolites 8, no. 1: 19. https://doi.org/10.3390/metabo8010019