

Elucidation and Regulation of Tyrosine Kinase Inhibitor Resistance in Renal Cell Carcinoma Cells from the Perspective of Glutamine Metabolism
 , , ,
, , ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Ethics Approval and Consent to Participate
2.2. Patients’ Background and Their Urine Sample Analysis for LC-MS/MS Analysis
2.3. Cell Lines, Culture Conditions, and WST Assay
2.4. Regulating Glutamine Metabolism
- siRNA#1: UUGAAGAAGCGGAUAAGCAGCUCCC
- siRNA#2: UUUACGAAGUCCAAGGACUGCUGGC
2.5. Cell Proliferation, Wound-Healing, and Two-Chamber Assay
2.6. In Vivo Study of Nude Mouse Model Grafted Sunitinib-Resistant Cells
2.7. Chemicals and Reagents to Evaluate Intracellular Metabolites for LC-MS/MS Analysis
2.8. RNA Extraction and Quantitative Real-Time Polymerase Chain Reaction
2.9. Western Blot Analysis
3. Results
3.1. Association between Urinary Glutamate Concentration and Prognosis in Patients Who Failed Sunitinib
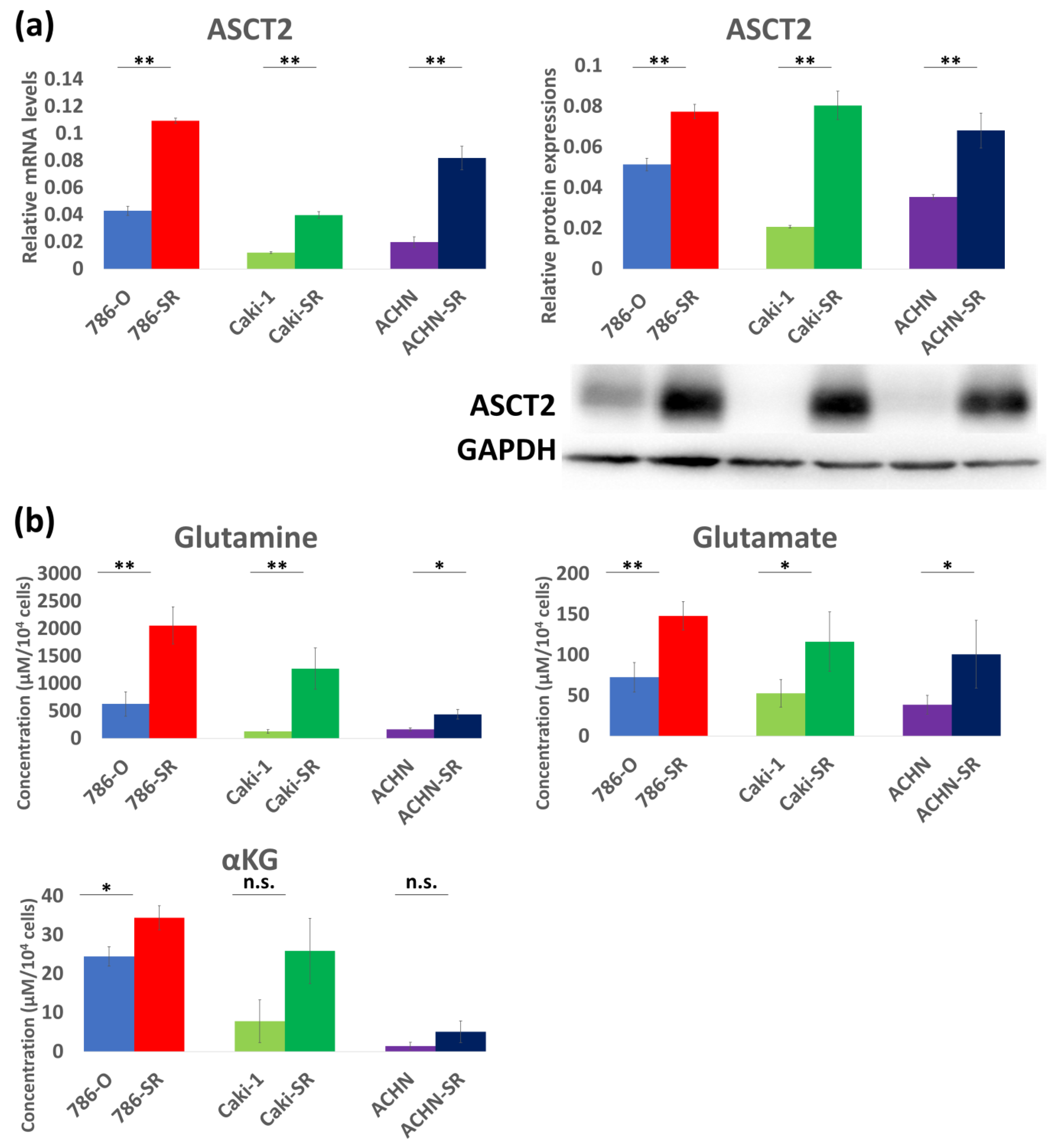
3.2. Expression of Glutamine Transporter in Three Established Sunitinib-Resistant Cell Lines
3.3. Re-Sensitivity to Sunitinib Resulting in Antitumor Effects through Attenuation of Enhanced ASCT2 in Resistant Cells
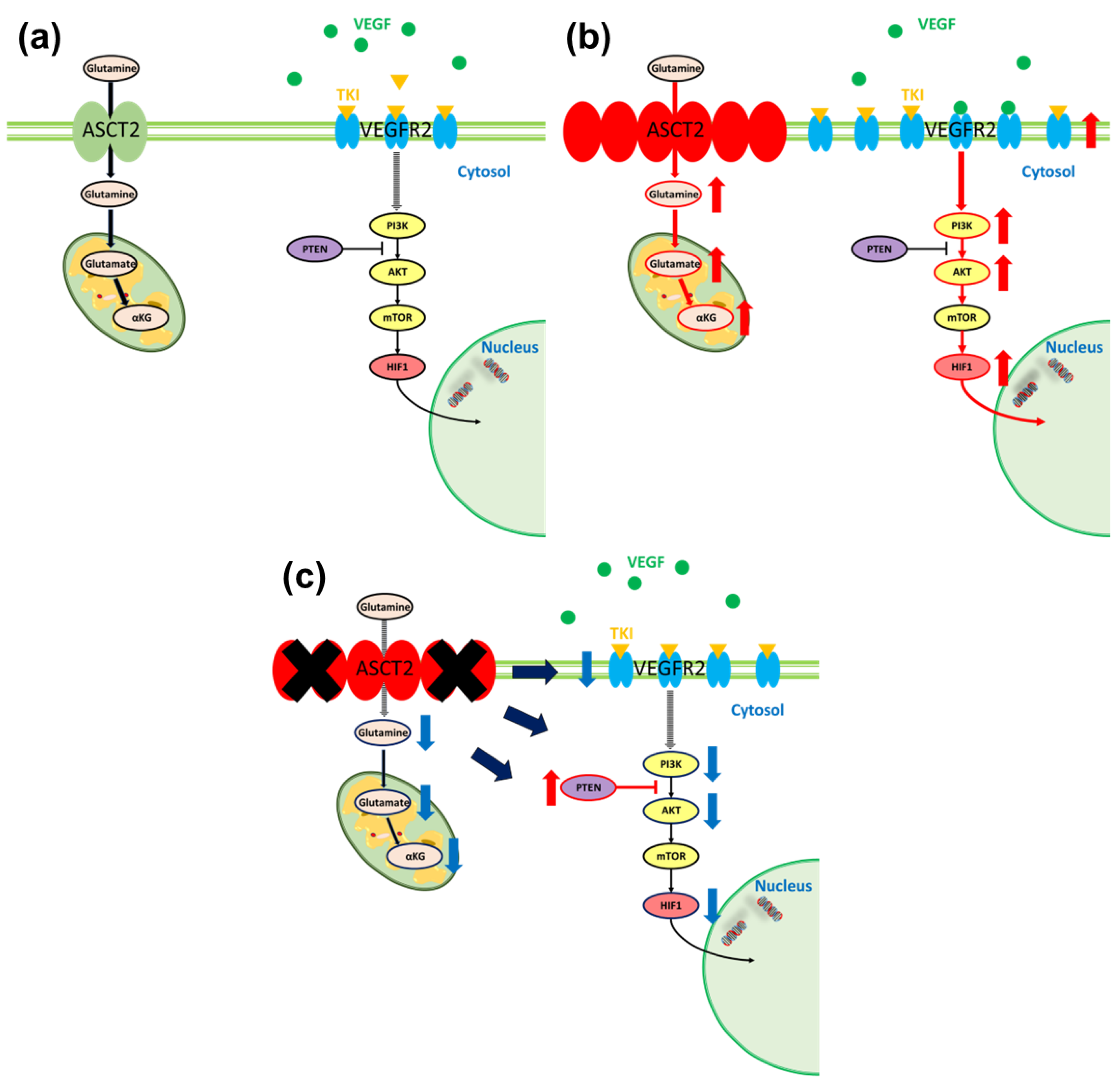
3.4. Relationship between Glutamine Metabolism and VEGF in Sunitinib-Resistant Cells
3.5. Re-Sensitivity to Sunitinib In Vivo by ASCT2 KD
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Abreu, D.; Carvalhal, G.; Gueglio, G.; Tobia, I.; Garcia, P.; Zuñiga, A.; Meza, L.; Bengió, R.; Scorticati, C.; Castillejos, R.; et al. Prognostic Factors in De Novo Metastatic Renal Cell Carcinoma: A Report from the Latin American Renal Cancer Group. JCO Glob. Oncol. 2021, 7, 671–685. [Google Scholar] [CrossRef]
- Ferlay, J.; Soerjomataram, I.; Dikshit, R.; Eser, S.; Mathers, C.; Rebelo, M.; Parkin, D.M.; Forman, D.; Bray, F. Cancer incidence and mortality worldwide: Sources, methods and major patterns in GLOBOCAN 2012. Int. J. Cancer 2015, 136, E359–E386. [Google Scholar] [CrossRef]
- Atkins, M.B.; Tannir, N.M. Current and emerging therapies for first-line treatment of metastatic clear cell renal cell carcinoma. Cancer Treat. Rev. 2018, 70, 127–137. [Google Scholar] [CrossRef] [PubMed]
- Regan, M.M.; Mantia, C.M.; Werner, L.; Tarhini, A.A.; Larkin, J.; Hodi, F.S.; Wolchok, J.; Postow, M.A.; Stwalley, B.; Moshyk, A.; et al. Treatment-free survival over extended follow-up of patients with advanced melanoma treated with immune checkpoint inhibitors in CheckMate 067. J. Immunother. Cancer 2021, 9, e003743. [Google Scholar] [CrossRef] [PubMed]
- Kathuria-Prakash, N.; Drolen, C.; Hannigan, C.A.; Drakaki, A. Immunotherapy and Metastatic Renal Cell Carcinoma: A Review of New Treatment Approaches. Life 2021, 12, 24. [Google Scholar] [CrossRef] [PubMed]
- Motzer, R.J.; Robbins, P.B.; Powles, T.; Albiges, L.; Haanen, J.B.; Larkin, J.; Mu, X.J.; Ching, K.A.; Uemura, M.; Pal, S.K.; et al. Avelumab plus axitinib versus sunitinib in advanced renal cell carcinoma: Biomarker analysis of the phase 3 JAVELIN Renal 101 trial. Nat. Med. 2020, 26, 1733–1741. [Google Scholar] [CrossRef] [PubMed]
- Powles, T.; Plimack, E.R.; Soulières, D.; Waddell, T.; Stus, V.; Gafanov, R.; Nosov, D.; Pouliot, F.; Melichar, B.; Vynnychenko, I.; et al. Pembrolizumab plus axitinib versus sunitinib monotherapy as first-line treatment of advanced renal cell carcinoma (KEYNOTE-426): Extended follow-up from a randomised, open-label, phase 3 trial. Lancet Oncol. 2020, 21, 1563–1573. [Google Scholar] [CrossRef] [PubMed]
- Vermeersch, K.A.; Styczynski, M.P. Applications of metabolomics in cancer research. J. Carcinog. 2013, 12, 9. [Google Scholar] [CrossRef]
- Palmnas, M.S.; Vogel, H.J. The future of NMR metabolomics in cancer therapy: Towards personalizing treatment and developing targeted drugs? Metabolites 2013, 3, 373. [Google Scholar] [CrossRef]
- Sato, T.; Kawasaki, Y.; Maekawa, M.; Takasaki, S.; Morozumi, K.; Sato, M.; Shimada, S.; Kawamorita, N.; Yamashita, S.; Mitsuzuka, K.; et al. Metabolomic Analysis to Elucidate Mechanisms of Sunitinib Resistance in Renal Cell Carcinoma. Metabolites 2020, 11, 1. [Google Scholar] [CrossRef]
- Wang, J.-J.; Siu, M.K.-Y.; Jiang, Y.-X.; Leung, T.H.-Y.; Chan, D.W.; Wang, H.-G.; Ngan, H.Y.-S.; Chan, K.K.-L. A Combination of Glutaminase Inhibitor 968 and PD-L1 Blockade Boosts the Immune Response against Ovarian Cancer. Biomolecules 2021, 11, 1749. [Google Scholar] [CrossRef] [PubMed]
- Gross, M.I.; Demo, S.D.; Dennison, J.B.; Chen, L.; Chernov-Rogan, T.; Goyal, B.; Janes, J.R.; Laidig, G.J.; Lewis, E.R.; Li, J.; et al. Antitumor Antitumor activity of the glutaminase inhibitor CB-839 in triple-negative breast cancer. Mol. Cancer Ther. 2014, 13, 890–901. [Google Scholar] [CrossRef] [PubMed]
- Matés, J.M.; Campos-Sandoval, J.A.; Santos-Jiménez, J.d.L.; Márquez, J. Dysregulation of glutaminase and glutamine synthetase in cancer. Cancer Lett. 2019, 467, 29–39. [Google Scholar] [CrossRef] [PubMed]
- López, R.A.M.; Belenchon, I.R.; Mazuecos-Quirós, J.; Congregado-Ruíz, C.B.; Couñago, F. Update on the treatment of metastatic renal cell carcinoma. World J. Clin. Oncol. 2022, 13, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; Venneti, S.; Nagrath, D. Glutamine metabolism: A Hallmark of Cancer Metabolism. Annu. Rev. Biomed. Eng. 2017, 19, 163–194. [Google Scholar] [CrossRef] [PubMed]
- Sato, T.; Kawasaki, Y.; Maekawa, M.; Takasaki, S.; Shimada, S.; Morozumi, K.; Sato, M.; Kawamorita, N.; Yamashita, S.; Mitsuzuka, K.; et al. Accurate quantification of urinary metabolites for predictive models manifest clinicopathology of renal cell carcinoma. Cancer Sci. 2020, 111, 2570–2578. [Google Scholar] [CrossRef]
- Morozumi, K.; Kawasaki, Y.; Maekawa, M.; Takasaki, S.; Sato, T.; Shimada, S.; Kawamorita, N.; Yamashita, S.; Mitsuzuka, K.; Mano, N.; et al. Predictive model for recurrence of renal cell carcinoma by comparing pre- and postoperative urinary metabolite concentrations. Cancer Sci. 2022, 113, 182–194. [Google Scholar] [CrossRef]
- Huang, D.; Wang, Y.; Thompson, J.W.; Yin, T.; Alexander, P.B.; Qin, D.; Mudgal, P.; Wu, H.; Liang, Y.; Tan, L.; et al. Cancer-cell-derived GABA promotes β-catenin-mediated tumour growth and immunosuppression. Nat. Cell Biol. 2022, 24, 230–241. [Google Scholar] [CrossRef]
- van Geldermalsen, M.; Wang, Q.; Nagarajah, R.; Marshall, A.D.; Thoeng, A.; Gao, D.; Ritchie, W.; Feng, Y.; Bailey, C.G.; Deng, N.; et al. ASCT2/SLC1A5 controls glutamine uptake and tumour growth in triple-negative basal-like breast cancer. Oncogene 2016, 35, 3201–3208. [Google Scholar] [CrossRef]
- Wang, Q.; Beaumont, K.A.; Otte, N.J.; Font, J.; Bailey, C.G.; Van Geldermalsen, M.; Sharp, D.M.; Tiffen, J.C.; Ryan, R.M.; Jormakka, M.; et al. Targeting glutamine transport to suppress melanoma cell growth. Int. J. Cancer 2014, 135, 1060–1071. [Google Scholar] [CrossRef]
- Liu, Y.; Zhao, T.; Li, Z.; Wang, L.; Yuan, S.; Sun, L. The role of ASCT2 in cancer: A review. Eur. J. Pharmacol. 2018, 837, 81–87. [Google Scholar] [CrossRef]
- Chen, C.-Y.; Chen, J.; He, L.; Stiles, B.L. PTEN: Tumor Suppressor and Metabolic Regulator. Front. Endocrinol. 2018, 9, 338. [Google Scholar] [CrossRef]
- Lee, Y.R.; Chen, M.; Lee, J.D.; Zhang, J.; Lin, S.Y.; Fu, T.M.; Chen, H.; Ishikawa, T.; Chiang, S.Y.; Katon, J.; et al. Reactivation of PTEN tumor suppressor for cancer treatment through inhibition of a MYC-WWPI inhibitory pathway. Science 2019, 364, eaau0159. [Google Scholar] [CrossRef]
- Hamadneh, L.; Abuarqoub, R.; Alhusban, A.; Bahader, M. Upregulation of PI3K/AKT/PTEN pathway is correlated with glucose and glutamine metabolic dysfunction during tamoxifen resistance development in MCF-7 cells. Sci. Rep. 2020, 10, 21933. [Google Scholar] [CrossRef]
- Garcia-Cao, I.; Song, M.S.; Hobbs, R.M.; Laurent, G.; Giorgi, C.; de Boer, V.C.; Anastasiou, D.; Ito, K.; Sasaki, A.T.; Rameh, L.; et al. Systemic elevation of PTEN induces a tumor-suppressive metabolic state. Cell 2012, 149, 49–62. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Chen, D.; Piao, H.-L.; Hua, X. PTEN-deficient cells prefer glutamine for metabolic synthesis. Acta Biochim. Biophys. Sin. 2020, 52, 251–258. [Google Scholar] [CrossRef]
- An, H.J.; Song, D.H.; Koh, H.M.; Kim, Y.-M.; Ko, G.H.; Lee, J.-H.; Lee, J.S.; Yang, J.W.; Kim, M.H.; Seo, D.H.; et al. Myoferlin silencing inhibits VEGFR2-mediated proliferation of metastatic clear cell renal cell carcinoma. Sci. Rep. 2019, 9, 12656. [Google Scholar] [CrossRef] [PubMed]
- Roskoski, R. Vascular endothelial growth factor (VEGF) and VEGF receptor inhibitors in the treatment of renal cell carcinomas. Pharmacol. Res. 2017, 120, 116–132. [Google Scholar] [CrossRef]
- Okuro, K.; Fukuhara, A.; Minemura, T.; Hayakawa, T.; Nishitani, S.; Okuno, Y.; Otsuki, M.; Shimomura, I. Glutamine deficiency induces lipolysis in adipocytes. Biochem. Biophys. Res. Commun. 2021, 585, 155–161. [Google Scholar] [CrossRef]
- Hu, X.; Deng, J.; Yu, T.; Chen, S.; Ge, Y.; Zhou, Z.; Guo, Y.; Ying, H.; Zhai, Q.; Chen, Y.; et al. ATF4 Deficiency Promotes Intestinal Inflammation in Mice by Reducing Uptake of Glutamine and Expression of Antimicrobial Peptides. Gastroenterology 2019, 156, 1098–1111. [Google Scholar] [CrossRef] [PubMed]
- Ekmark, L.; Rooyackers, O.; Wernerman, J.; Fläring, U. Plasma glutamine deficiency is associated with multiple organ failure in critically ill children. Amino Acids 2015, 47, 535–542. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| IC50 (μM) | Sunitinib-Sensitive Cells | Sunitinib-Resistance Cells |
|---|---|---|
| 786-O: 4.8 (95%CI 2.1–10.8) | 786-SR:22.1 (95%CI 15.0–29.8) | |
| Caki-1: 2.7 (95%CI 0.6–8.5) | Caki-SR: 14.8 (95%CI 8.7–19.6) | |
| ACHN: 2.1 (95%CI 0.9–6.0) | ACHN-SR: 2.1 (95%CI 9.5–20.1) |
| Glutamine (μM) | Glutamate (μM) | αKG (μM) | |
|---|---|---|---|
| 786-O 786-OK | 629.89 5.18 | 72.49 2.54 | 24.44 0.43 |
| 786-SR 786-SRK | 2058.25 37.95 | 148.25 2.88 | 34.43 0.30 |
| Caki-1 Caki-K | 127.64 3.48 | 52.78 1.35 | 7.81 0.0036 |
| Caki-SR Caki-SRK | 1273.46 24.54 | 116.50 1.61 | 25.87 0.0091 |
| ACHN ACHN-K | 164.83 3.98 | 38.73 1.85 | 1.49 0.0054 |
| ACHN-SR ACHN-SRK | 440.24 14.46 | 100.92 9.52 | 5.09 0.12 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Morozumi, K.; Kawasaki, Y.; Sato, T.; Maekawa, M.; Takasaki, S.; Shimada, S.; Sakai, T.; Yamashita, S.; Mano, N.; Ito, A. Elucidation and Regulation of Tyrosine Kinase Inhibitor Resistance in Renal Cell Carcinoma Cells from the Perspective of Glutamine Metabolism. Metabolites 2024, 14, 170. https://doi.org/10.3390/metabo14030170
Morozumi K, Kawasaki Y, Sato T, Maekawa M, Takasaki S, Shimada S, Sakai T, Yamashita S, Mano N, Ito A. Elucidation and Regulation of Tyrosine Kinase Inhibitor Resistance in Renal Cell Carcinoma Cells from the Perspective of Glutamine Metabolism. Metabolites. 2024; 14(3):170. https://doi.org/10.3390/metabo14030170
Chicago/Turabian StyleMorozumi, Kento, Yoshihide Kawasaki, Tomonori Sato, Masamitsu Maekawa, Shinya Takasaki, Shuichi Shimada, Takanari Sakai, Shinichi Yamashita, Nariyasu Mano, and Akihiro Ito. 2024. "Elucidation and Regulation of Tyrosine Kinase Inhibitor Resistance in Renal Cell Carcinoma Cells from the Perspective of Glutamine Metabolism" Metabolites 14, no. 3: 170. https://doi.org/10.3390/metabo14030170