
Induction of AHR Signaling in Response to the Indolimine Class of Microbial Stress Metabolites
 , , , , , and
, , , , , and
Abstract
:1. Introduction
2. Material and Methods
2.1. Materials
2.2. Cell Culture
2.3. Capillary Immunoblotting Analysis
2.4. RNA Isolation and mRNA Quantitation
2.5. Cell-Based AHR Ligand Competition Binding Assay
2.6. CYP1A1/1B1 Microsomal Assay
2.7. CYP1A1/1B1 Assays Coupled to MS Analysis
2.8. Computational Docking Analysis
2.9. Histone Extraction and Protein Detection
2.10. Viability Assay
2.11. Data Analysis
3. Results
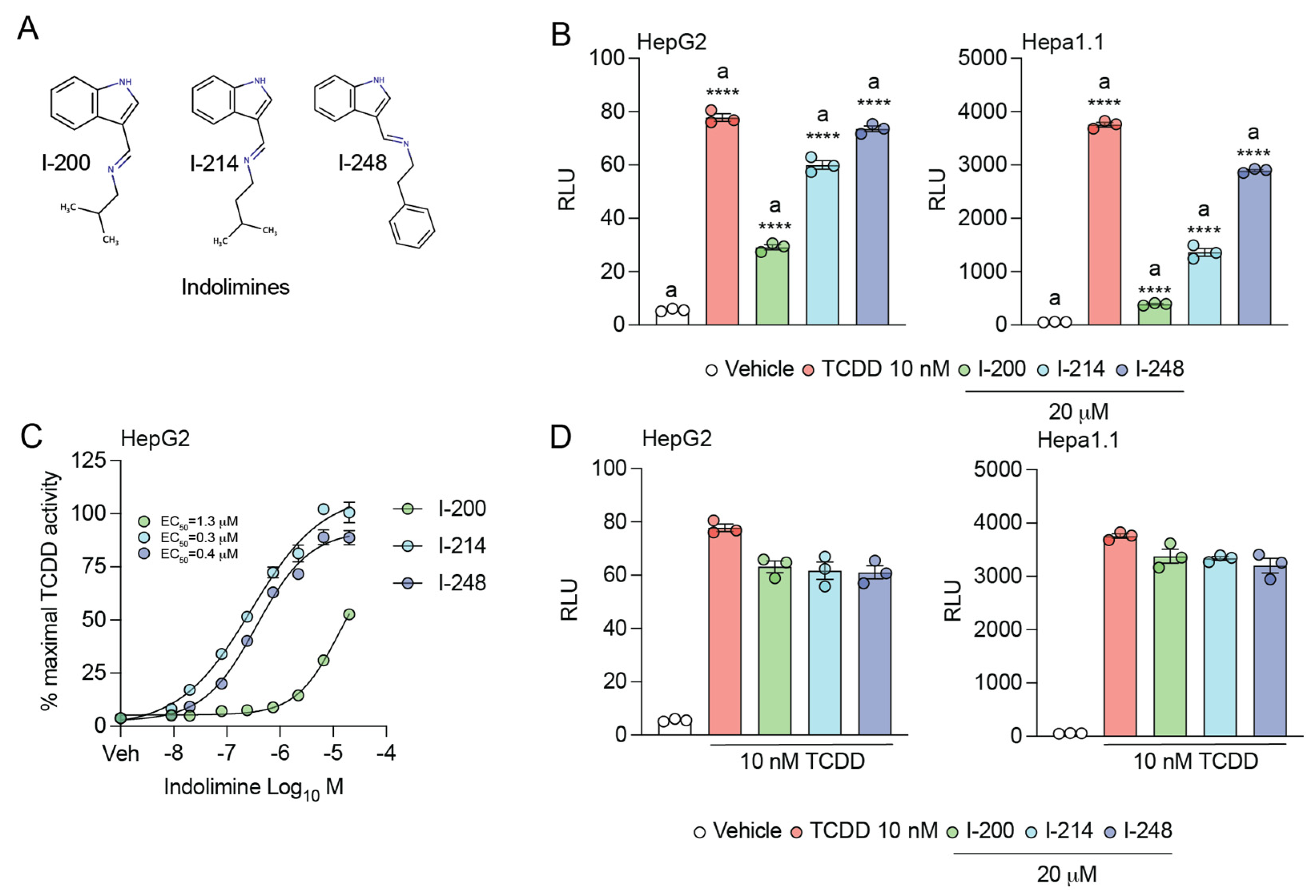
3.1. Indolimines Activate the AHR
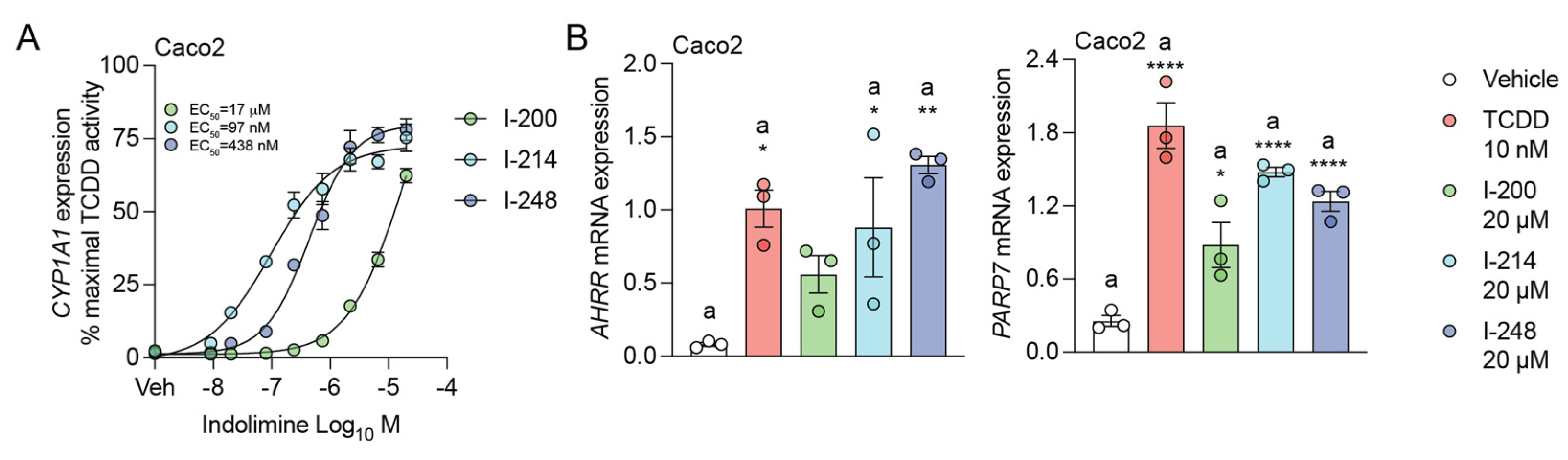
3.2. Indolimines Induce AHR Target Gene Expression
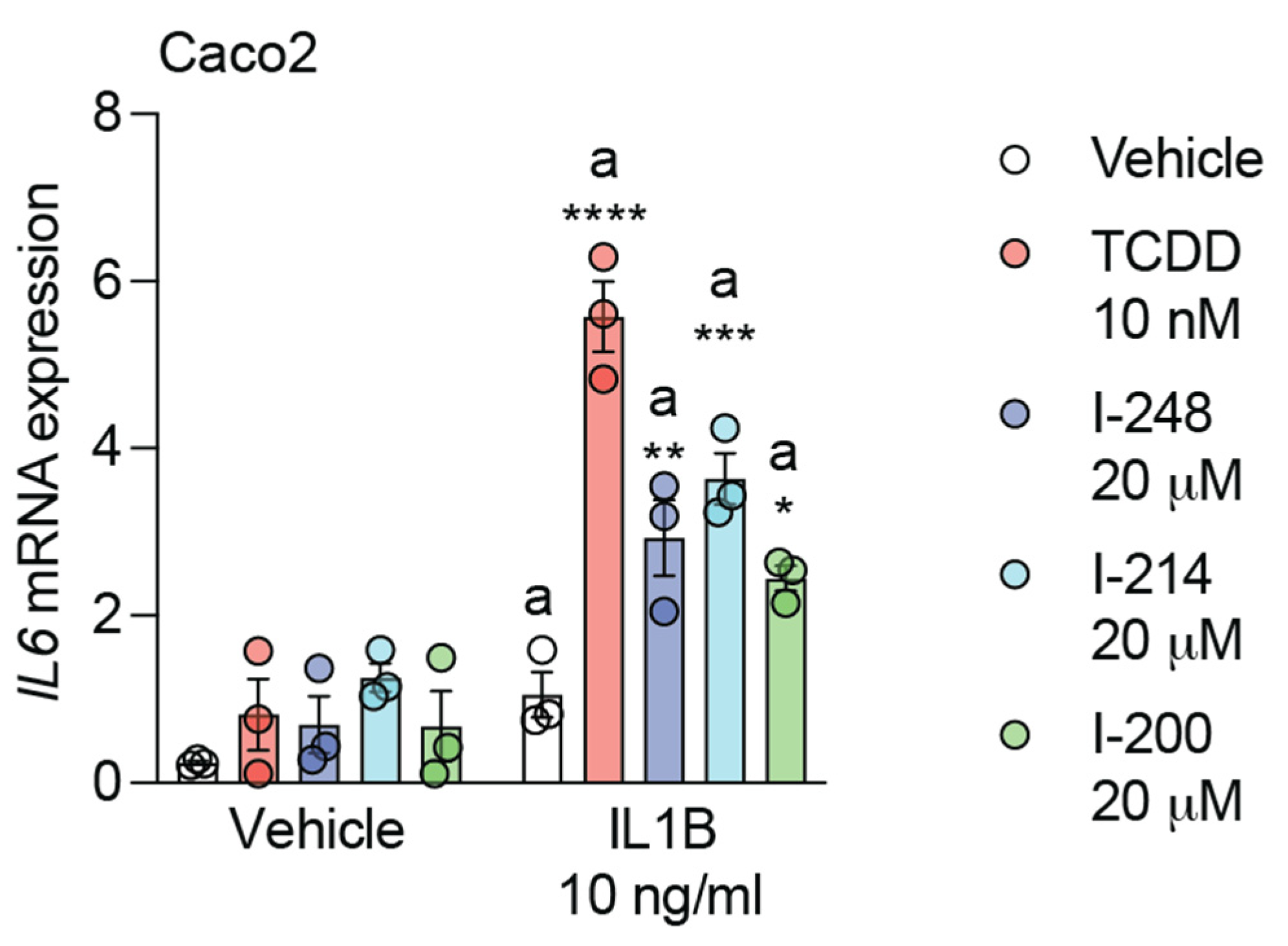
3.3. Indolimines Induce Combinatorial Activation of IL6 Expression
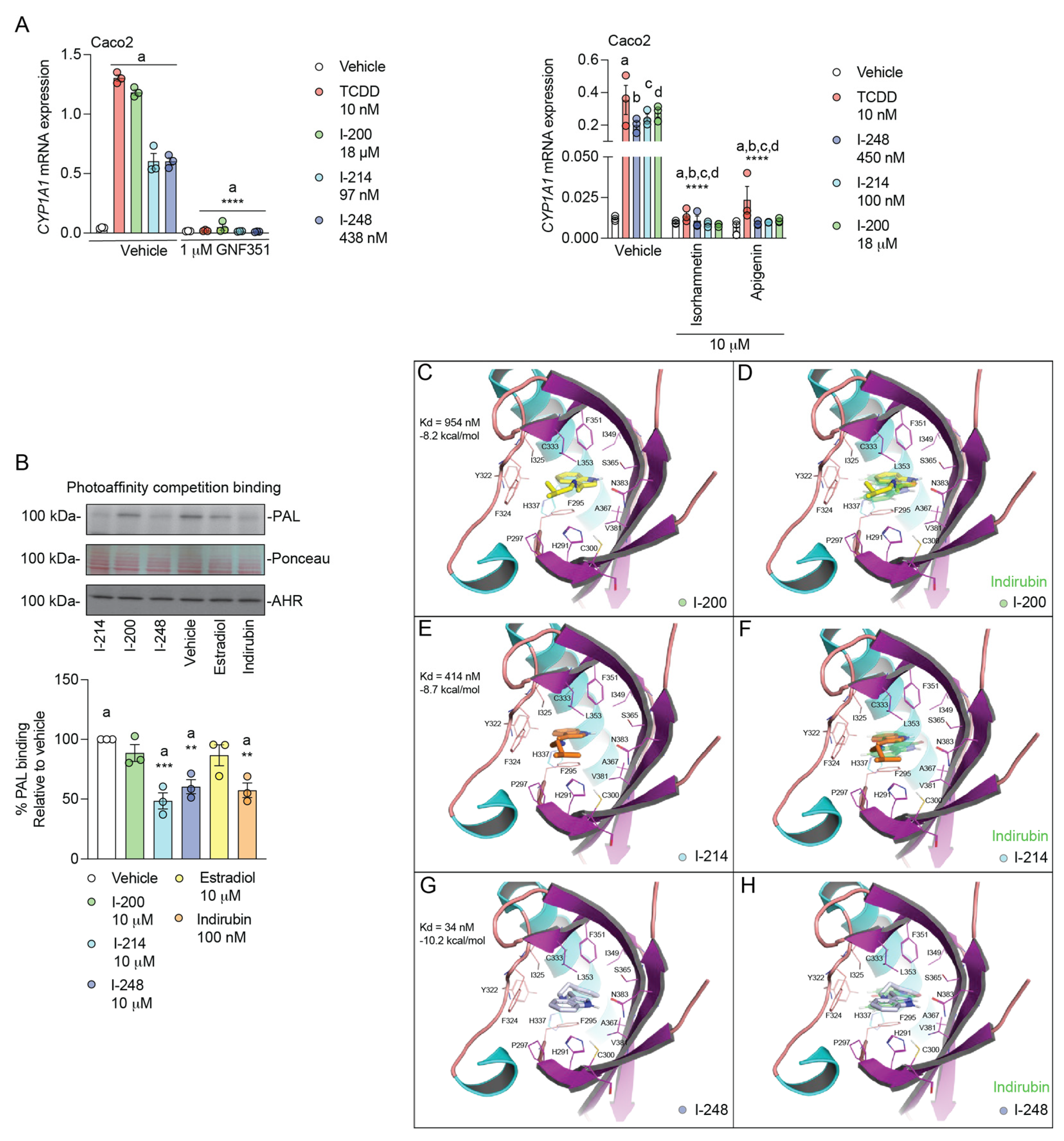
3.4. Indolimines Are Direct AHR Ligands (Modeling Data)
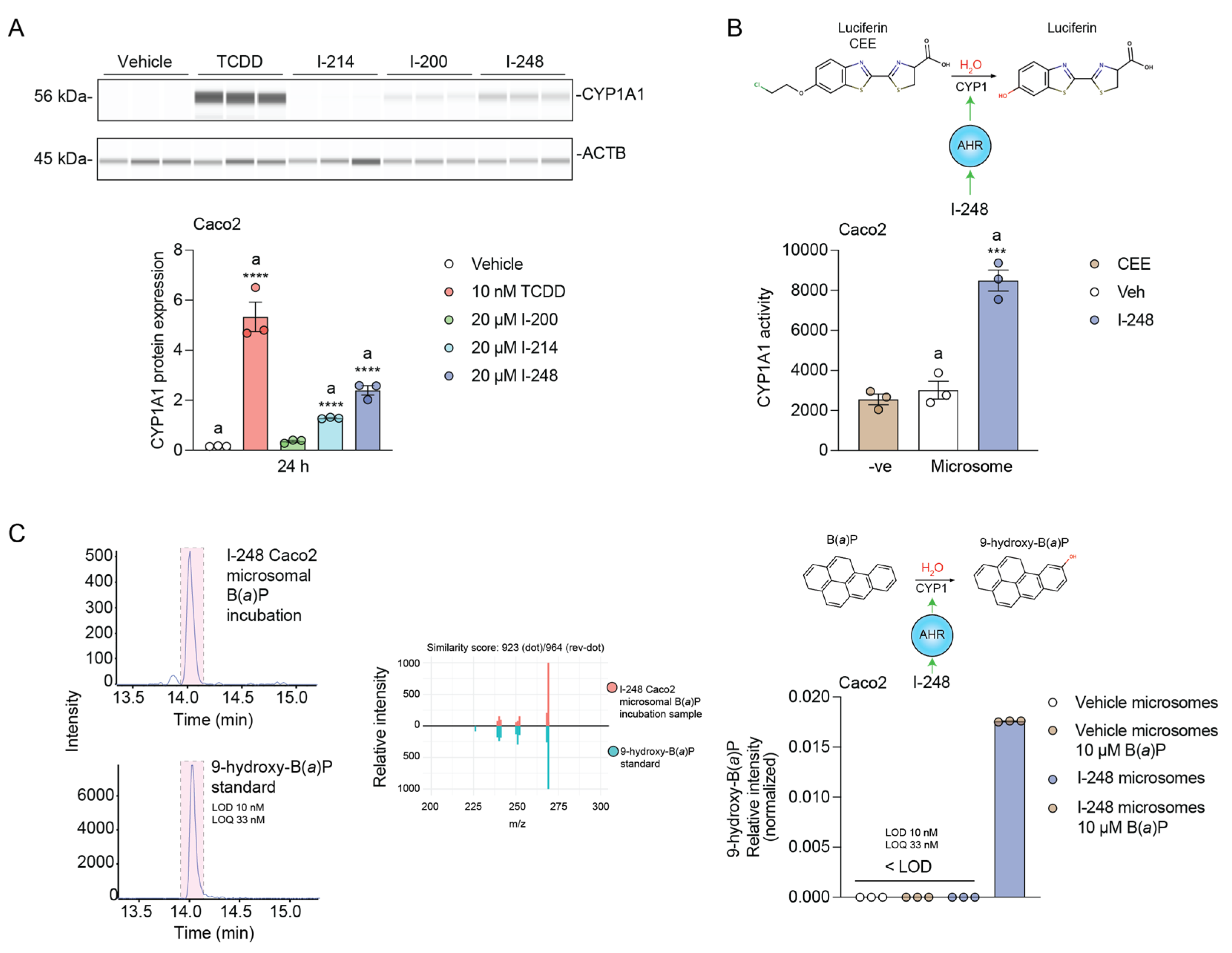
3.5. Indolimines Increase CYP1A1 Protein Expression and Carcinogen Metabolism
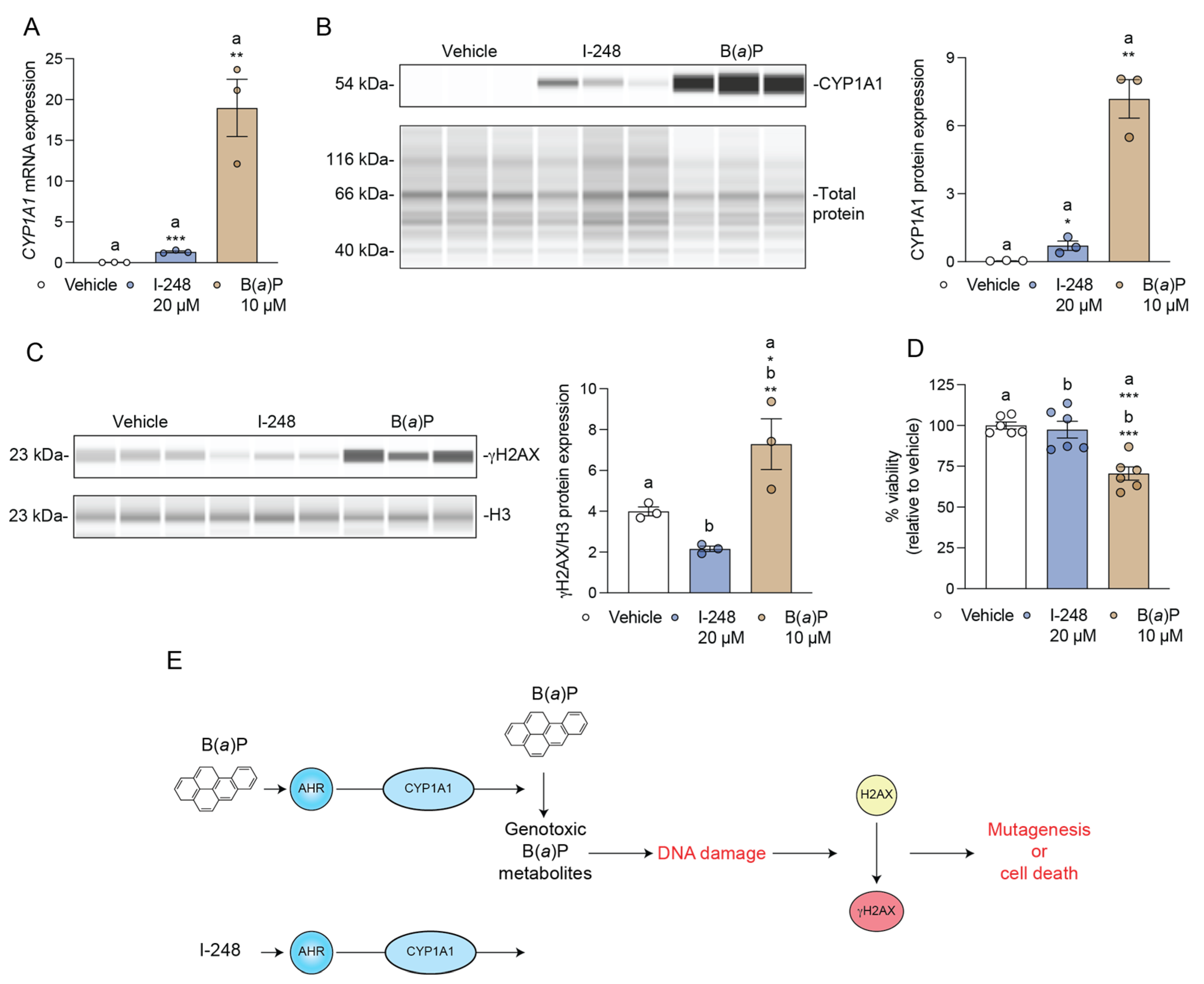
3.6. I-248 Fails to Induce DNA Damage at a Concentration That Induces CYP1A1 Expression
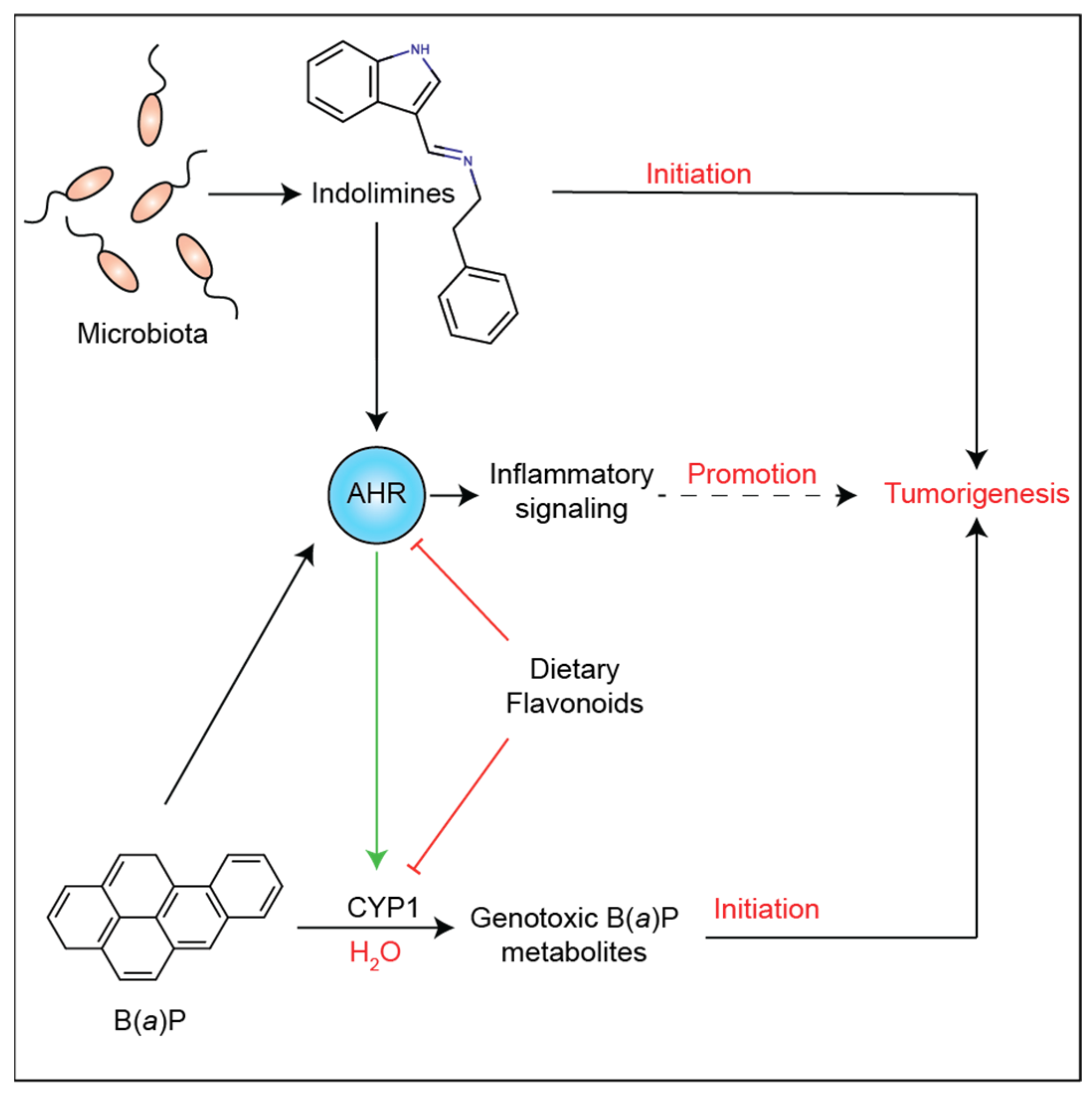
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Wikoff, W.R.; Anfora, A.T.; Liu, J.; Schultz, P.G.; Lesley, S.A.; Peters, E.C.; Siuzdak, G. Metabolomics analysis reveals large effects of gut microflora on mammalian blood metabolites. Proc. Natl. Acad. Sci. USA 2009, 106, 3698–3703. [Google Scholar] [CrossRef]
- Dodd, D.; Spitzer, M.H.; Van Treuren, W.; Merrill, B.D.; Hryckowian, A.J.; Higginbottom, S.K.; Le, A.; Cowan, T.M.; Nolan, G.P.; Fischbach, M.A.; et al. A gut bacterial pathway metabolizes aromatic amino acids into nine circulating metabolites. Nature 2017, 551, 648–652. [Google Scholar] [CrossRef]
- Zhang, B.; Jiang, M.; Zhao, J.; Song, Y.; Du, W.; Shi, J. The Mechanism Underlying the Influence of Indole-3-Propionic Acid: A Relevance to Metabolic Disorders. Front. Endocrinol. 2022, 13, 841703. [Google Scholar] [CrossRef]
- Lymperopoulos, A.; Suster, M.S.; Borges, J.I. Short-Chain Fatty Acid Receptors and Cardiovascular Function. Int. J. Mol. Sci. 2022, 23, 3303. [Google Scholar] [CrossRef]
- Omiecinski, C.J.; Vanden Heuvel, J.P.; Perdew, G.H.; Peters, J.M. Xenobiotic metabolism, disposition, and regulation by receptors: From biochemical phenomenon to predictors of major toxicities. Toxicol. Sci. 2011, 120 (Suppl. S1), S49–S75. [Google Scholar] [CrossRef]
- Beischlag, T.V.; Luis Morales, J.; Hollingshead, B.D.; Perdew, G.H. The aryl hydrocarbon receptor complex and the control of gene expression. Crit. Rev. Eukaryot. Gene Expr. 2008, 18, 207–250. [Google Scholar] [CrossRef]
- van den Bogaard, E.H.; Perdew, G.H. The Enigma of AHR Activation in the Skin: Interplay among Ligands, Metabolism, and Bioavailability. J. Investig. Dermatol. 2021, 141, 1385–1388. [Google Scholar] [CrossRef]
- Stockinger, B.; Shah, K.; Wincent, E. AHR in the intestinal microenvironment: Safeguarding barrier function. Nat. Rev. Gastroenterol. Hepatol. 2021, 18, 559–570. [Google Scholar] [CrossRef]
- Morgan, E.W.; Dong, F.; Annalora, A.J.; Murray, I.A.; Wolfe, T.; Erickson, R.; Gowda, K.; Amin, S.G.; Petersen, K.S.; Kris-Etherton, P.M.; et al. Contribution of Circulating Host and Microbial Tryptophan Metabolites Toward Ah Receptor Activation. Int. J. Tryptophan Res. 2023, 16, 11786469231182510. [Google Scholar] [CrossRef]
- Dong, F.; Hao, F.; Murray, I.A.; Smith, P.B.; Koo, I.; Tindall, A.M.; Kris-Etherton, P.M.; Gowda, K.; Amin, S.G.; Patterson, A.D.; et al. Intestinal microbiota-derived tryptophan metabolites are predictive of Ah receptor activity. Gut Microbes 2020, 12, 1–24. [Google Scholar] [CrossRef]
- Cao, Y.; Oh, J.; Xue, M.; Huh, W.J.; Wang, J.; Gonzalez-Hernandez, J.A.; Rice, T.A.; Martin, A.L.; Song, D.; Crawford, J.M.; et al. Commensal microbiota from patients with inflammatory bowel disease produce genotoxic metabolites. Science 2022, 378, eabm3233. [Google Scholar] [CrossRef] [PubMed]
- Hubbard, T.D.; Murray, I.A.; Perdew, G.H. Indole and Tryptophan Metabolism: Endogenous and Dietary Routes to Ah Receptor Activation. Drug Metab. Dispos. 2015, 43, 1522–1535. [Google Scholar] [CrossRef] [PubMed]
- Poland, A.; Glover, E.; Ebetino, H.; Kende, A. Photoaffinity labelling of the Ah receptor. Food Chem. Toxicol. 1986, 24, 781–787. [Google Scholar] [CrossRef] [PubMed]
- Yagi, H.; Holder, G.M.; Dansette, P.M.; Hernandez, O.; Yeh, H.J.C.; LeMahieu, R.A.; Jerina, D.M. Synthesis and spectral properties of the isomeric hydroxybenzo[a]pyrenes. J. Org. Chem. 1976, 41, 977–985. [Google Scholar] [CrossRef]
- Long, W.P.; Pray-Grant, M.; Tsai, J.C.; Perdew, G.H. Protein kinase C activity is required for aryl hydrocarbon receptor pathway-mediated signal transduction. Mol. Pharmacol. 1998, 53, 691–700. [Google Scholar] [CrossRef]
- John, K.; Lahoti, T.S.; Wagner, K.; Hughes, J.M.; Perdew, G.H. The Ah Receptor Regulates Growth Factor Expression in Head and Neck Squamous Cell Carcinoma Cell Lines. Mol. Carcinog. 2013, 53, 765–776. [Google Scholar] [CrossRef]
- Zhou, X.; Chakraborty, D.; Murray, I.A.; Coslo, D.; Kehs, Z.; Vijay, A.; Ton, C.; Desai, D.; Amin, S.G.; Patterson, A.D.; et al. Aryl Hydrocarbon Receptor Activation Coordinates Mouse Small Intestinal Epithelial Cell Programming. Lab. Investig. 2023, 103, 100012. [Google Scholar] [CrossRef]
- Dong, F.; Annalora, A.J.; Murray, I.A.; Tian, Y.; Marcus, C.B.; Patterson, A.D.; Perdew, G.H. Endogenous Tryptophan-Derived Ah Receptor Ligands are Dissociated from CYP1A1/1B1-Dependent Negative-Feedback. Int. J. Tryptophan Res. 2023, 16, 11786469231182508. [Google Scholar] [CrossRef]
- Dong, F.; Murray, I.A.; Annalora, A.; Coslo, D.M.; Desai, D.; Gowda, K.; Yang, J.; Wang, D.; Koo, I.; Hao, F.; et al. Complex chemical signals dictate Ah receptor activation through the gut-lung axis. FASEB J. 2023, 37, e23010. [Google Scholar] [CrossRef]
- Morris, G.M.; Huey, R.; Lindstrom, W.; Sanner, M.F.; Belew, R.K.; Goodsell, D.S.; Olson, A.J. AutoDock4 and AutoDockTools4: Automated docking with selective receptor flexibility. J. Comput. Chem. 2009, 30, 2785–2791. [Google Scholar] [CrossRef]
- Trott, O.; Olson, A.J. AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem. 2010, 31, 455–461. [Google Scholar] [CrossRef]
- Eberhardt, J.; Santos-Martins, D.; Tillack, A.F.; Forli, S. AutoDock Vina 1.2.0: New Docking Methods, Expanded Force Field, and Python Bindings. J. Chem. Inf. Model. 2021, 61, 3891–3898. [Google Scholar] [CrossRef]
- Gruszczyk, J.; Grandvuillemin, L.; Lai-Kee-Him, J.; Paloni, M.; Savva, C.G.; Germain, P.; Grimaldi, M.; Boulahtouf, A.; Kwong, H.S.; Bous, J.; et al. Cryo-EM structure of the agonist-bound Hsp90-XAP2-AHR cytosolic complex. Nat. Commun. 2022, 13, 7010. [Google Scholar] [CrossRef]
- Schrodinger, L.D.W. PyMOL. 2020. Available online: https://pymol.org (accessed on 17 August 2023).
- Annalora, A.J.; Goodin, D.B.; Hong, W.X.; Zhang, Q.; Johnson, E.F.; Stout, C.D. Crystal structure of CYP24A1, a mitochondrial cytochrome P450 involved in vitamin D metabolism. J. Mol. Biol. 2010, 396, 441–451. [Google Scholar] [CrossRef]
- Velazquez-Libera, J.L.; Duran-Verdugo, F.; Valdes-Jimenez, A.; Nunez-Vivanco, G.; Caballero, J. LigRMSD: A web server for automatic structure matching and RMSD calculations among identical and similar compounds in protein-ligand docking. Bioinformatics 2020, 36, 2912–2914. [Google Scholar] [CrossRef] [PubMed]
- Stourac, J.; Vavra, O.; Kokkonen, P.; Filipovic, J.; Pinto, G.; Brezovsky, J.; Damborsky, J.; Bednar, D. Caver Web 1.0: Identification of tunnels and channels in proteins and analysis of ligand transport. Nucleic Acids Res. 2019, 47, W414–W422. [Google Scholar] [CrossRef]
- Dong, F.; Perdew, G.H. The aryl hyrocarbon receptor as a mediator of host-microbiota interplay. Gut Microbes 2020, 12, 1859812. [Google Scholar] [CrossRef] [PubMed]
- Hollingshead, B.D.; Beischlag, T.V.; Dinatale, B.C.; Ramadoss, P.; Perdew, G.H. Inflammatory signaling and aryl hydrocarbon receptor mediate synergistic induction of interleukin 6 in MCF-7 cells. Cancer Res. 2008, 68, 3609–3617. [Google Scholar] [CrossRef]
- DiNatale, B.C.; Schroeder, J.C.; Francey, L.J.; Kusnadi, A.; Perdew, G.H. Mechanistic insights into the events that lead to synergistic induction of interleukin 6 transcription upon activation of the aryl hydrocarbon receptor and inflammatory signaling. J. Biol. Chem. 2010, 285, 24388–24397. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Wong, P.; Radany, E.; Wong, J.Y. HDAC inhibitor, valproic acid, induces p53-dependent radiosensitization of colon cancer cells. Cancer Biother. Radiopharm. 2009, 24, 689–699. [Google Scholar] [CrossRef]
- Silla Santos, M.H. Biogenic amines: Their importance in foods. Int. J. Food Microbiol. 1996, 29, 213–231. [Google Scholar] [CrossRef]
- Lonvaud-Funel, A. Biogenic amines in wines: Role of lactic acid bacteria. FEMS Microbiol. Lett. 2001, 199, 9–13. [Google Scholar] [CrossRef] [PubMed]
- Huang, Z.; Schoones, T.; Wells, J.M.; Fogliano, V.; Capuano, E. Substrate-Driven Differences in Tryptophan Catabolism by Gut Microbiota and Aryl Hydrocarbon Receptor Activation. Mol. Nutr. Food Res. 2021, 65, e2100092. [Google Scholar] [CrossRef]
- Murray, I.A.; Perdew, G.H. How Ah Receptor Ligand Specificity Became Important in Understanding Its Physiological Function. Int. J. Mol. Sci. 2020, 21, 9614. [Google Scholar] [CrossRef] [PubMed]
- Gronke, K.; Hernandez, P.P.; Zimmermann, J.; Klose, C.S.N.; Kofoed-Branzk, M.; Guendel, F.; Witkowski, M.; Tizian, C.; Amann, L.; Schumacher, F.; et al. Interleukin-22 protects intestinal stem cells against genotoxic stress. Nature 2019, 566, 249–253. [Google Scholar] [CrossRef]
- Furumatsu, K.; Nishiumi, S.; Kawano, Y.; Ooi, M.; Yoshie, T.; Shiomi, Y.; Kutsumi, H.; Ashida, H.; Fujii-Kuriyama, Y.; Azuma, T.; et al. A role of the aryl hydrocarbon receptor in attenuation of colitis. Dig. Dis. Sci. 2011, 56, 2532–2544. [Google Scholar] [CrossRef]
- Xie, G.; Raufman, J.P. Role of the Aryl Hydrocarbon Receptor in Colon Neoplasia. Cancers 2015, 7, 1436–1446. [Google Scholar] [CrossRef]
- Kawajiri, K.; Kobayashi, Y.; Ohtake, F.; Ikuta, T.; Matsushima, Y.; Mimura, J.; Pettersson, S.; Pollenz, R.S.; Sakaki, T.; Hirokawa, T.; et al. Aryl hydrocarbon receptor suppresses intestinal carcinogenesis in ApcMin/+ mice with natural ligands. Proc. Natl. Acad. Sci. USA 2009, 106, 13481–13486. [Google Scholar] [CrossRef] [PubMed]
- Saito, R.; Miki, Y.; Hata, S.; Takagi, K.; Iida, S.; Oba, Y.; Ono, K.; Ishida, T.; Suzuki, T.; Ohuchi, N.; et al. Aryl hydrocarbon receptor in breast cancer-a newly defined prognostic marker. Horm. Cancer 2014, 5, 11–21. [Google Scholar] [CrossRef] [PubMed]
- Richmond, O.; Ghotbaddini, M.; Allen, C.; Walker, A.; Zahir, S.; Powell, J.B. The aryl hydrocarbon receptor is constitutively active in advanced prostate cancer cells. PLoS ONE 2014, 9, e95058. [Google Scholar] [CrossRef]
- Panitz, V.; Koncarevic, S.; Sadik, A.; Friedel, D.; Bausbacher, T.; Trump, S.; Farztdinov, V.; Schulz, S.; Sievers, P.; Schmidt, S.; et al. Tryptophan metabolism is inversely regulated in the tumor and blood of patients with glioblastoma. Theranostics 2021, 11, 9217–9233. [Google Scholar] [CrossRef] [PubMed]
- Lahoti, T.S.; Boyer, J.A.; Kusnadi, A.; Muku, G.E.; Murray, I.A.; Perdew, G.H. Aryl Hydrocarbon Receptor Activation Synergistically Induces Lipopolysaccharide-Mediated Expression of Proinflammatory Chemokine (c-c motif) Ligand 20. Toxicol. Sci. 2015, 148, 229–240. [Google Scholar] [CrossRef]
- Smith, K.J.; Boyer, J.A.; Muku, G.E.; Murray, I.A.; Gowda, K.; Desai, D.; Amin, S.G.; Glick, A.B.; Perdew, G.H. Editor’s Highlight: Ah Receptor Activation Potentiates Neutrophil Chemoattractant (C-X-C Motif) Ligand 5 Expression in Keratinocytes and Skin. Toxicol. Sci. 2017, 160, 83–94. [Google Scholar] [CrossRef] [PubMed]
- Bent, E.H.; Millan-Barea, L.R.; Zhuang, I.; Goulet, D.R.; Frose, J.; Hemann, M.T. Microenvironmental IL-6 inhibits anti-cancer immune responses generated by cytotoxic chemotherapy. Nat. Commun. 2021, 12, 6218. [Google Scholar] [CrossRef]
- Androutsopoulos, V.P.; Tsatsakis, A.M.; Spandidos, D.A. Cytochrome P450 CYP1A1: Wider roles in cancer progression and prevention. BMC Cancer 2009, 9, 187. [Google Scholar] [CrossRef]
- Sekimoto, M.; Sumi, H.; Hosaka, T.; Umemura, T.; Nishikawa, A.; Degawa, M. Aryl hydrocarbon receptor activation and CYP1A induction by cooked food-derived carcinogenic heterocyclic amines in human HepG2 cell lines. Food Chem. Toxicol. 2016, 97, 256–264. [Google Scholar] [CrossRef] [PubMed]
- Burczynski, M.E.; Penning, T.M. Genotoxic polycyclic aromatic hydrocarbon ortho-quinones generated by aldo-keto reductases induce CYP1A1 via nuclear translocation of the aryl hydrocarbon receptor. Cancer Res. 2000, 60, 908–915. [Google Scholar]
- Park, J.H.; Mangal, D.; Frey, A.J.; Harvey, R.G.; Blair, I.A.; Penning, T.M. Aryl hydrocarbon receptor facilitates DNA strand breaks and 8-oxo-2’-deoxyguanosine formation by the aldo-keto reductase product benzo[a]pyrene-7,8-dione. J. Biol. Chem. 2009, 284, 29725–29734. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Substrate | Autodock Vina | ||
|---|---|---|---|
| Structure-based Homology Model | |||
| Human AHR PAS B domain | |||
| Binding Energy a (kcal/mole) | Dissociation Constant b [KD] nM | ||
| Indolimine-200 | Max c | −8.2 | 953.9 |
| Avg d | −7.5 ± 0.65 | 5515 ± 8391 | |
| Indolimine-214 | Max | −8.7 | 413.5 |
| Avg | −8.4 ± 0.34 | 865 ± 572 | |
| Indolimine-248 | Max | −10.2 | 33.7 |
| Avg | −9.9 ± 0.42 | 69.7 ± 57.5 | |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Patel, D.; Murray, I.A.; Dong, F.; Annalora, A.J.; Gowda, K.; Coslo, D.M.; Krzeminski, J.; Koo, I.; Hao, F.; Amin, S.G.; et al. Induction of AHR Signaling in Response to the Indolimine Class of Microbial Stress Metabolites. Metabolites 2023, 13, 985. https://doi.org/10.3390/metabo13090985
Patel D, Murray IA, Dong F, Annalora AJ, Gowda K, Coslo DM, Krzeminski J, Koo I, Hao F, Amin SG, et al. Induction of AHR Signaling in Response to the Indolimine Class of Microbial Stress Metabolites. Metabolites. 2023; 13(9):985. https://doi.org/10.3390/metabo13090985
Chicago/Turabian StylePatel, Dhwani, Iain A. Murray, Fangcong Dong, Andrew J. Annalora, Krishne Gowda, Denise M. Coslo, Jacek Krzeminski, Imhoi Koo, Fuhua Hao, Shantu G. Amin, and et al. 2023. "Induction of AHR Signaling in Response to the Indolimine Class of Microbial Stress Metabolites" Metabolites 13, no. 9: 985. https://doi.org/10.3390/metabo13090985