
Molecular Mechanisms of Western Diet-Induced Obesity and Obesity-Related Carcinogenesis—A Narrative Review


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
- The current global phenomenon of excess body weight.
- Excess energy intake and inadequate energy expenditure in obesity.
- The ‘thrifty gene’ versus the ‘thrifty epigenome’ hypothesis.
- Cell-to-cell communication between adipose tissue and other organs in the development of obesity and metabolic syndrome.
- The role of Western diets, fat and carbohydrate metabolism in obesity and obesity-related carcinogenesis.
- The molecular mechanisms of obesity and relationship with the 14 Hallmarks of Cancer and enabling characteristics.
- Weight loss interventions in the resolution of metabolic syndrome, adipose tissue dysfunction and prevention of future cancers in patients with obesity.
2. Methods
3. Discussion
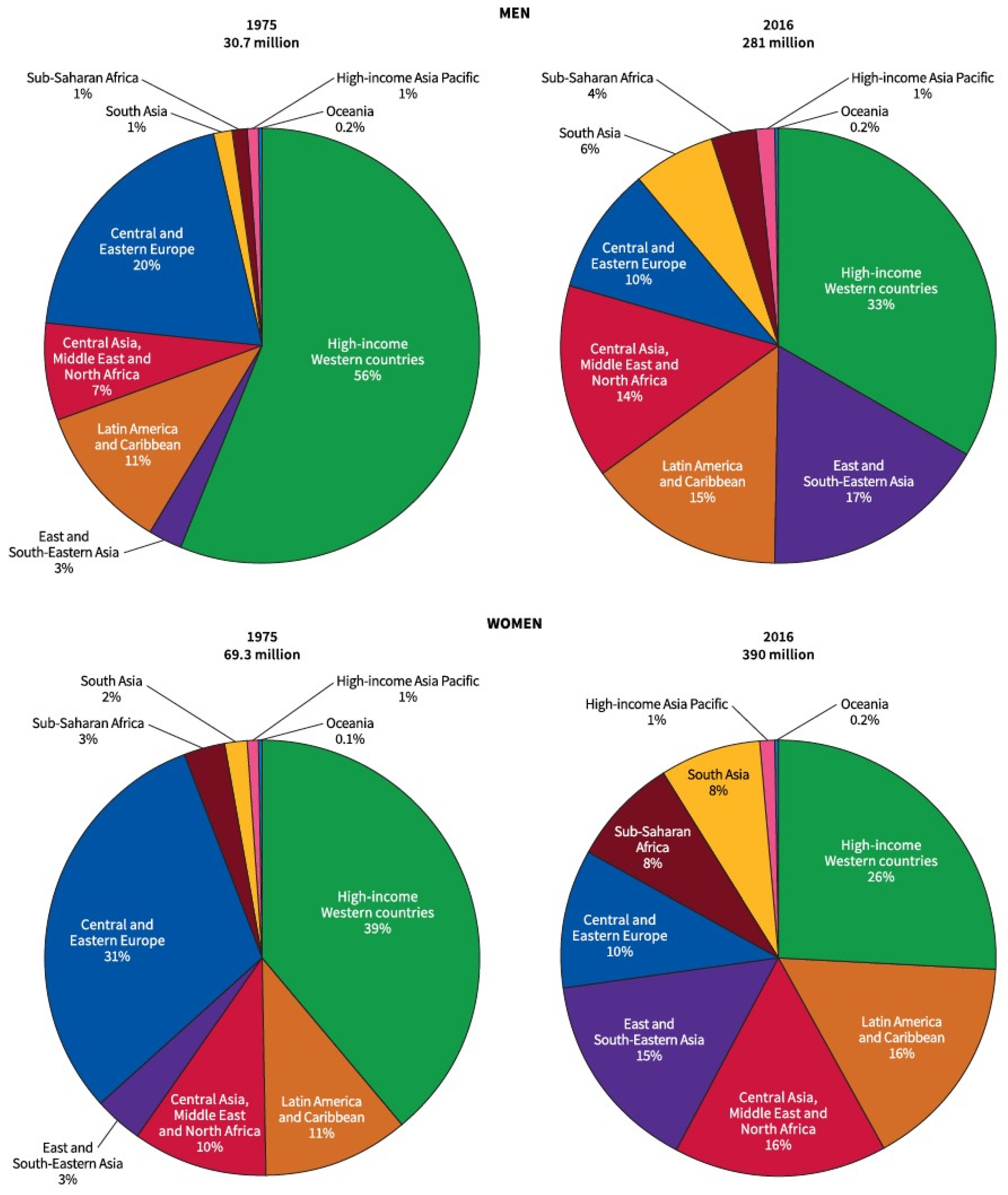
3.1. Epidemiology of Obesity
- Overweight (BMI = 25–29.9 kg/m2).
- Obese (BMI ≥ 30 kg/m2).
- Class I obesity (BMI = 30–34.9 kg/m2),
- Class II obesity (BMI= 35–39.9 kg/m2),
- Class III (BMI ≥ 40 kg/m2).
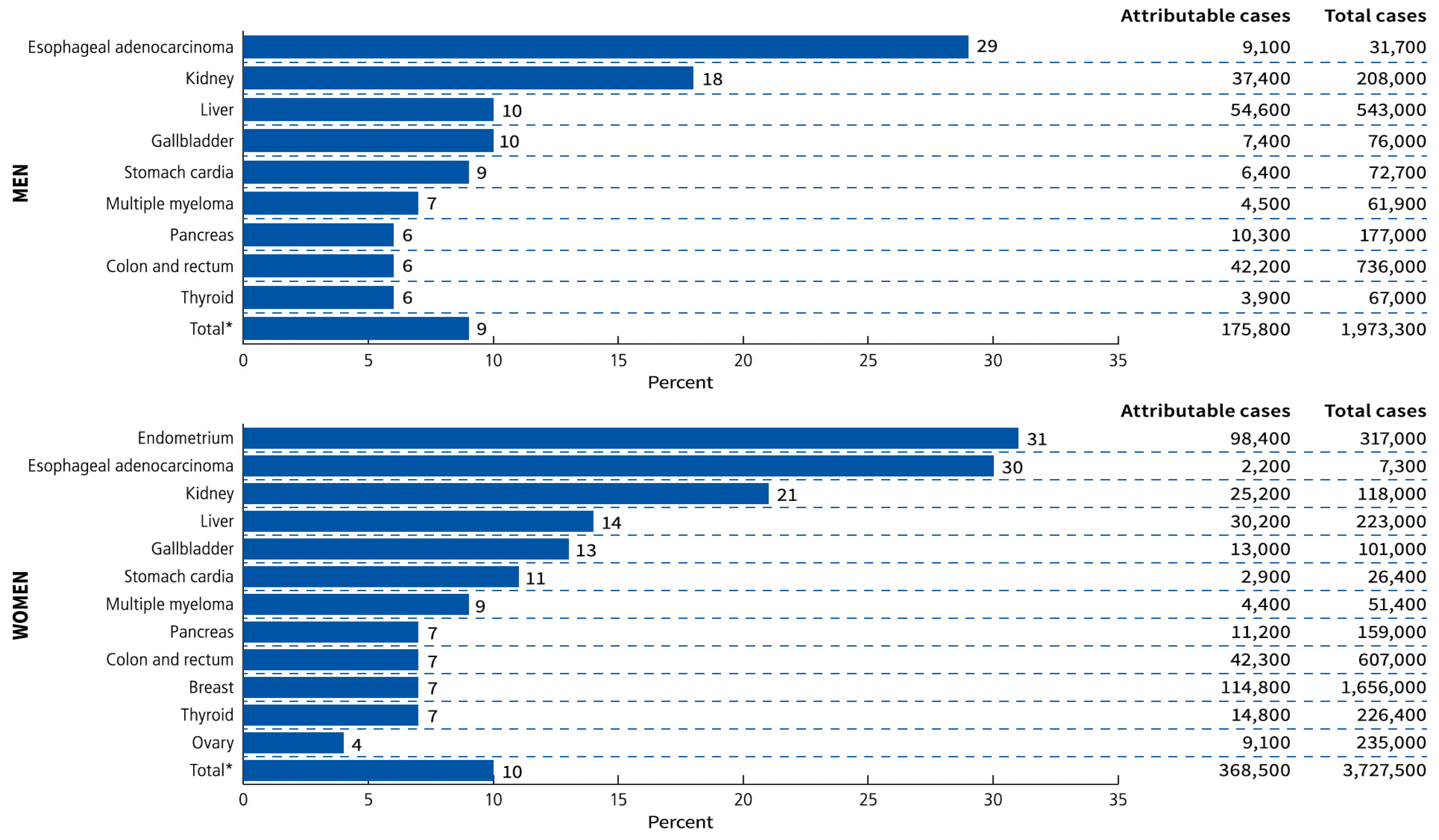
3.2. Obesity-Associated Cancer
3.3. Pathogenesis of Obesity
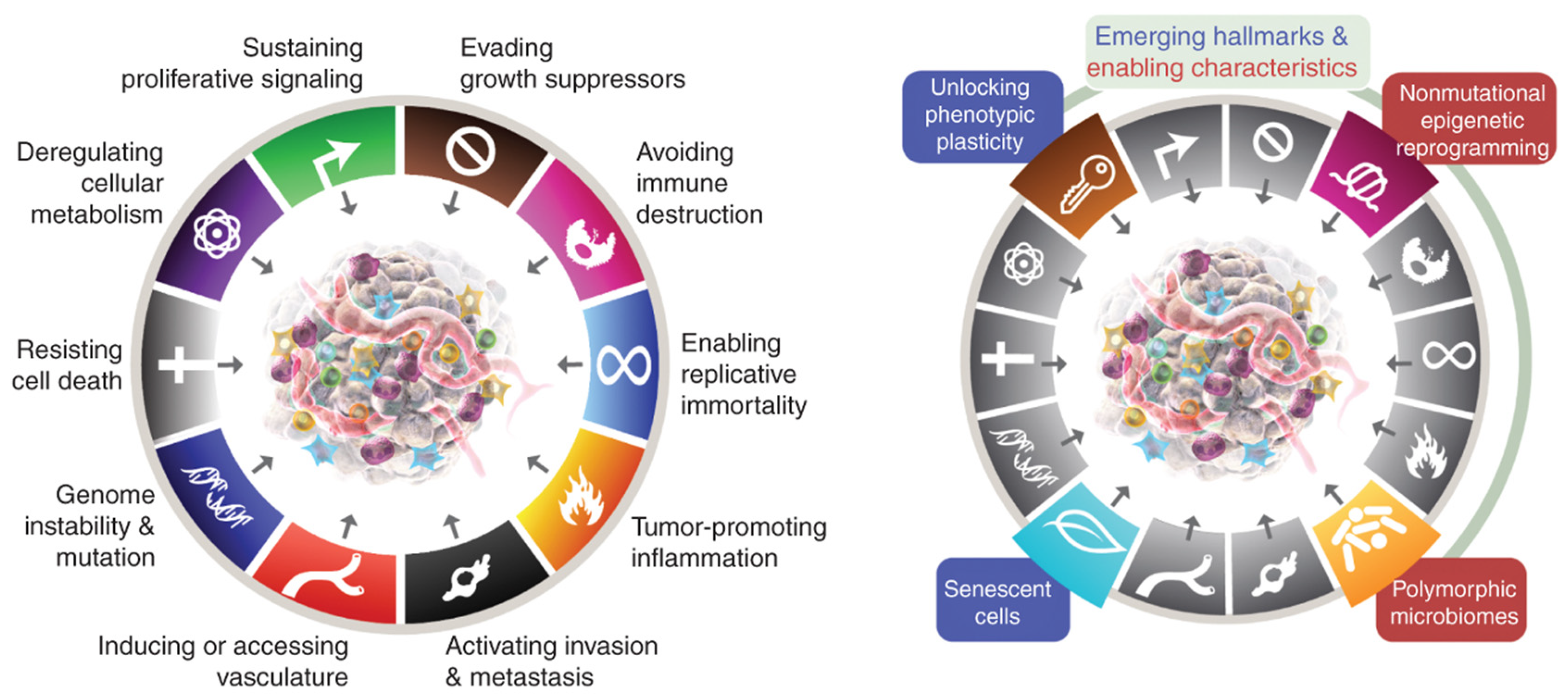
3.4. Hallmarks of Cancer
3.5. Metabolic Syndrome and Cancer
- 1.13 to 6.73 for breast;
- 1.14 to 2.61 for colorectal;
- 1.18 to 2.50 for gastric cardia;
- 1.37 to 2.20 for endometrial;
- 1.59 to 2.13 for pancreas;
- 2.13 to 5.06 for hepatocellular carcinoma [27].
3.6. Metabolic Syndrome and VAT
3.7. Metabolic Syndrome and Colorectal Cancer
- Type 1 (‘sporadic’-MSI-H, CIMP-high, BRAF-mutated, KRAS-wild type),
- Type 2 (MSS/MSI-L, CIMP-high, BRAF-mutated, KRAS-wild type);
- Type 3 (MSS/MSI-L, CIMP-low or negative, BRAF-wild type, KRAS-mutated),
- Type 4 (MSS/MSI-L, CIMP-low or negative, BRAF-wild type, KRAS-wild type);
- Type 5 (‘Lynch syndrome’-MSI-H, CIMP-low or negative, BRAF-wild type, KRAS-wild type).
3.8. Dennis Burkitt, Western Diets, Obesity and CRC
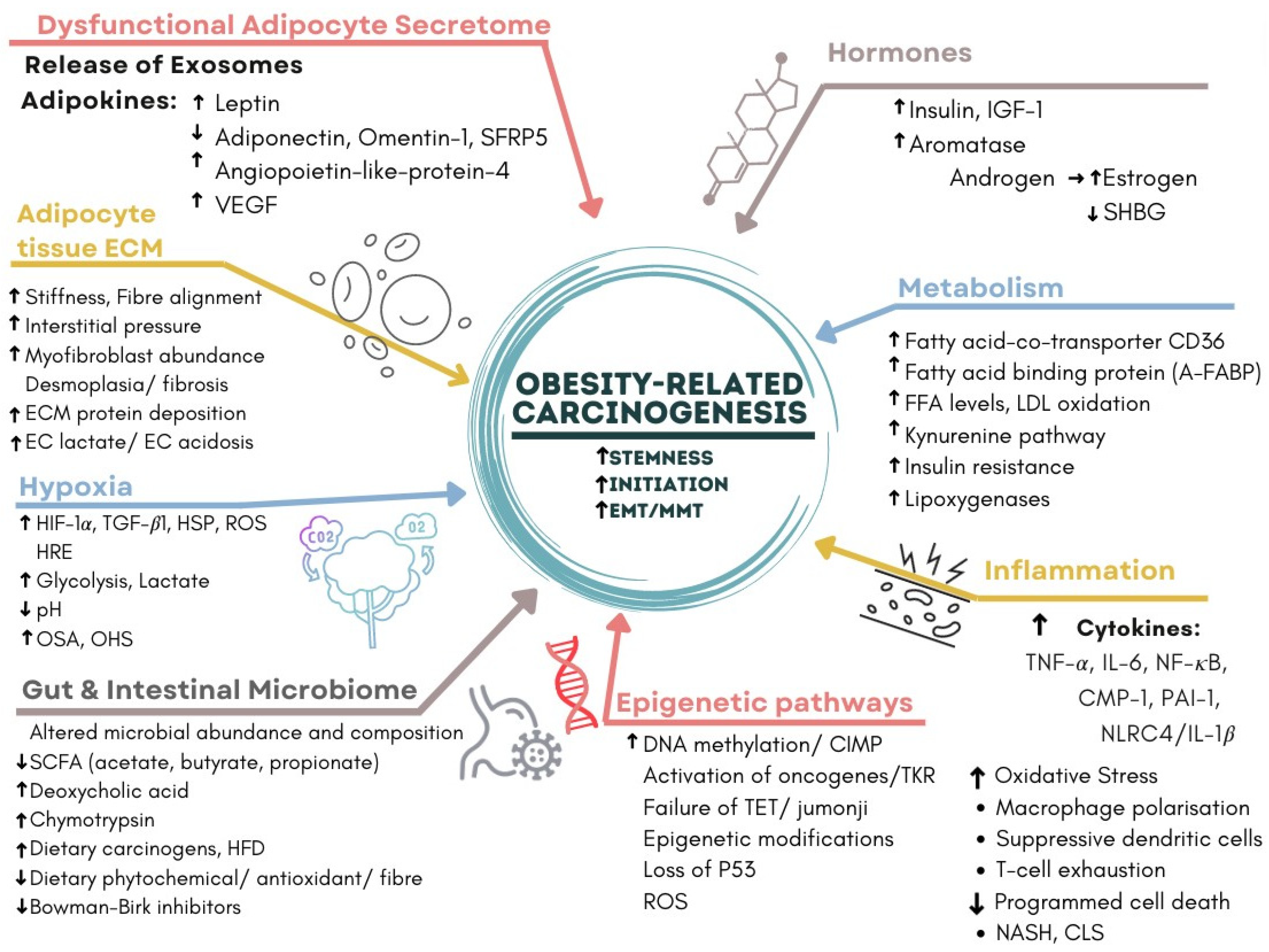
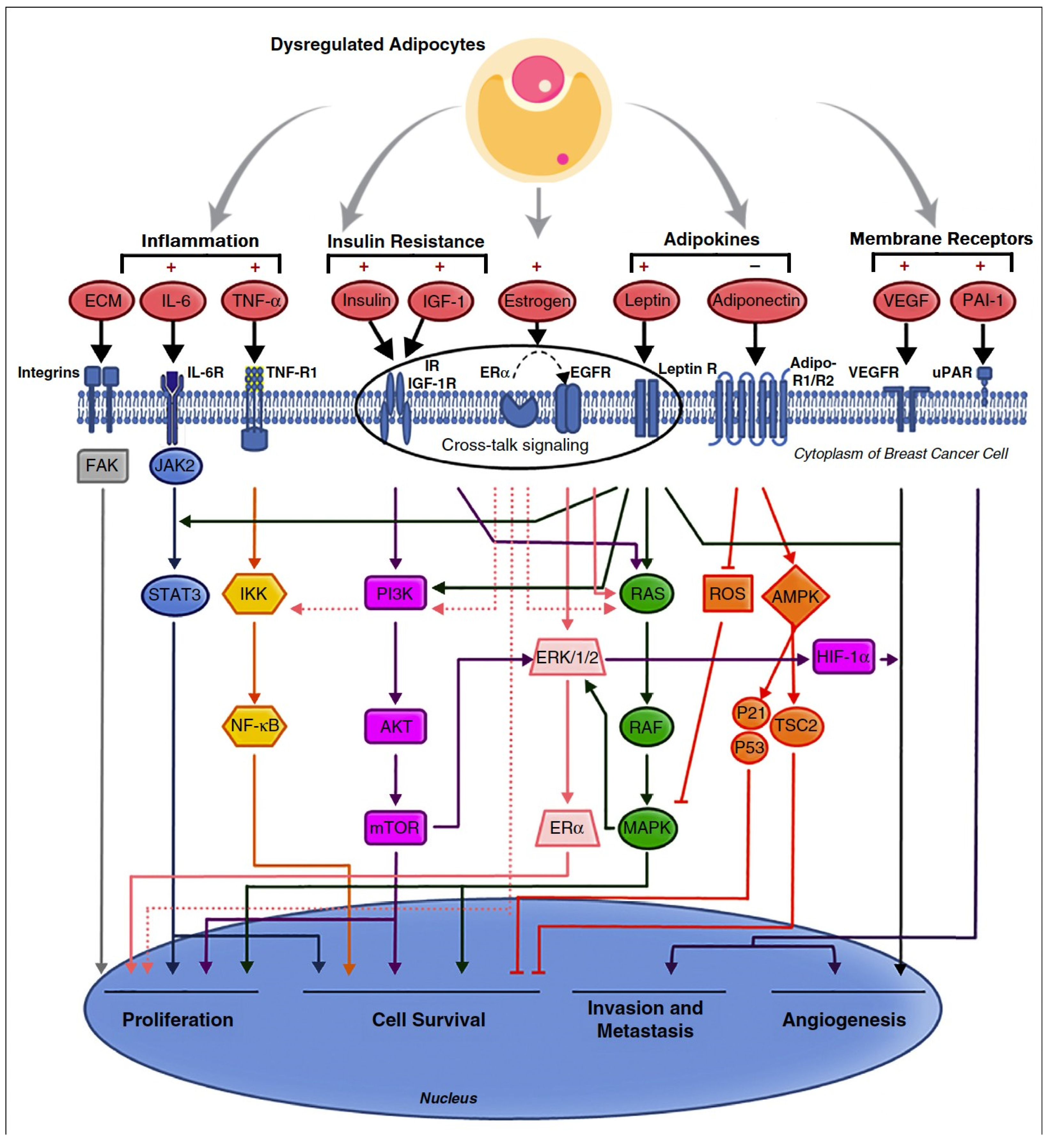
3.9. Molecular Mechanisms of Obesity-Related Carcinogenesis
- (1)
- Dysfunctional Adipocyte secretome: senescence, CLS, adipokines and exosomes.
- (2)
- Hormones: insulin, oestrogen, CLS and aromatase activity.
- (3)
- Inflammation and metabolism: CLS, cytokines and glycolysis.
- (4)
- Hypoxia: HIF-1α, CLS, EMT, angiogenesis, VAT and OSA [50].
- (5)
- WAT and Hepatocyte Extracellular matrix: CLS, NASH, fibrosis and cirrhosis [51].
- (6)
- Epigenetic pathways: DNA methylation, CIMP and loss of tumour suppressors.
- (7)
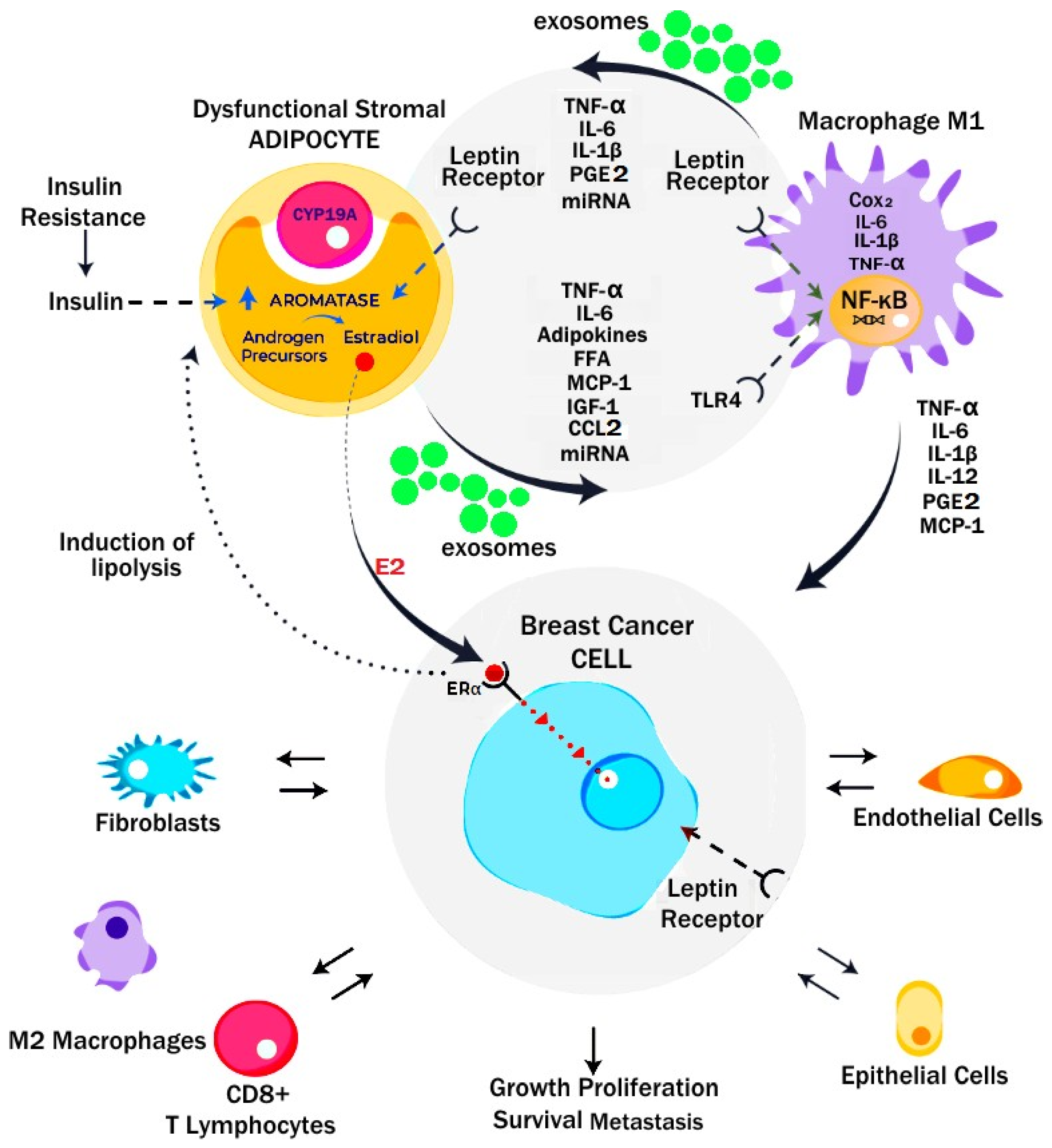
4. Dysfunctional Adipocyte Secretome
4.1. Adipocytes and Fat Storage
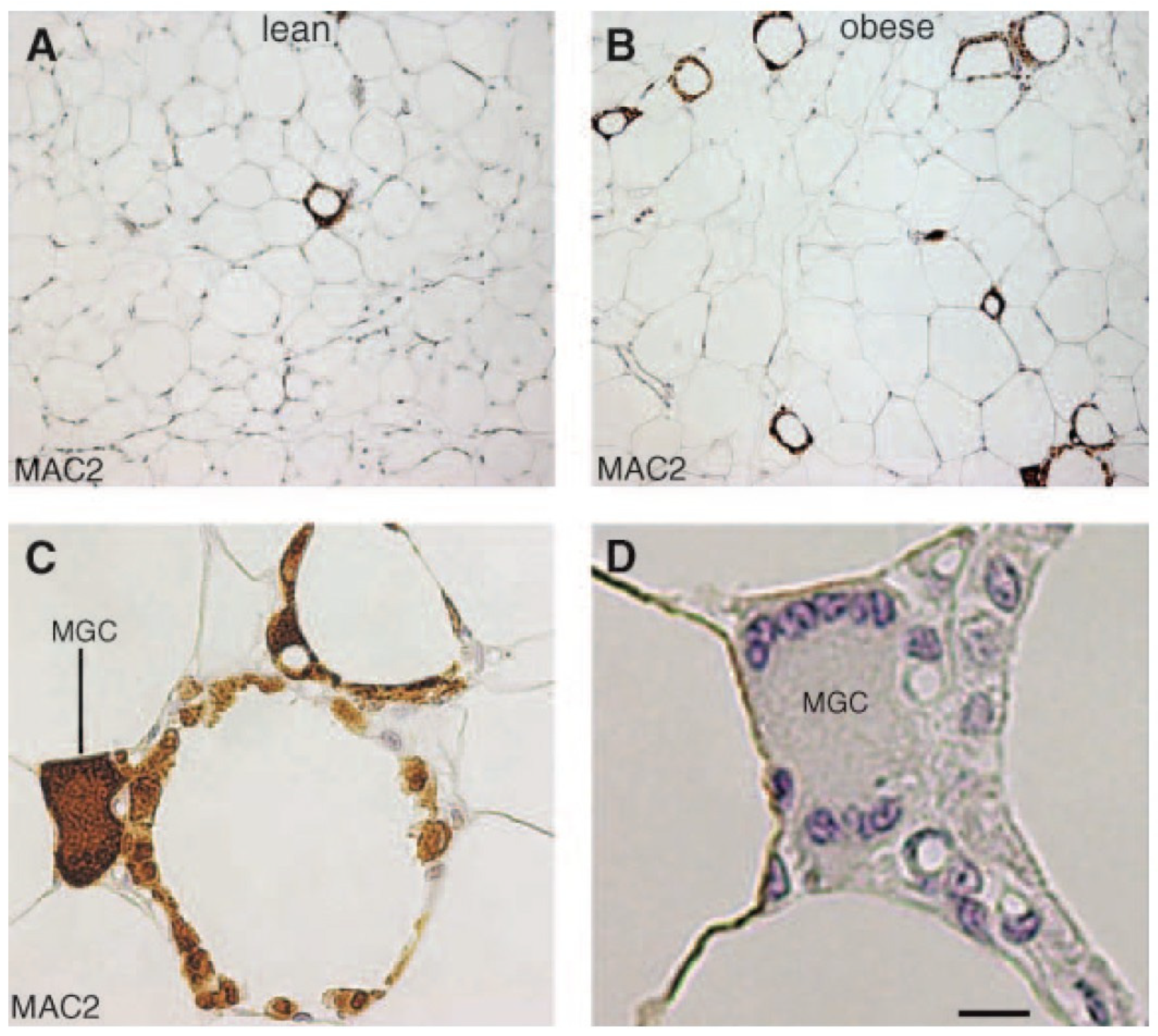
4.2. WAT Senescence
4.3. Prevention and Treatment of WAT Senescence
4.4. Adipokines



4.5. Leptin
4.6. Leptin and Cancer
4.7. Anti-Inflammatory Adipokines
4.8. Adipokines and Cancer
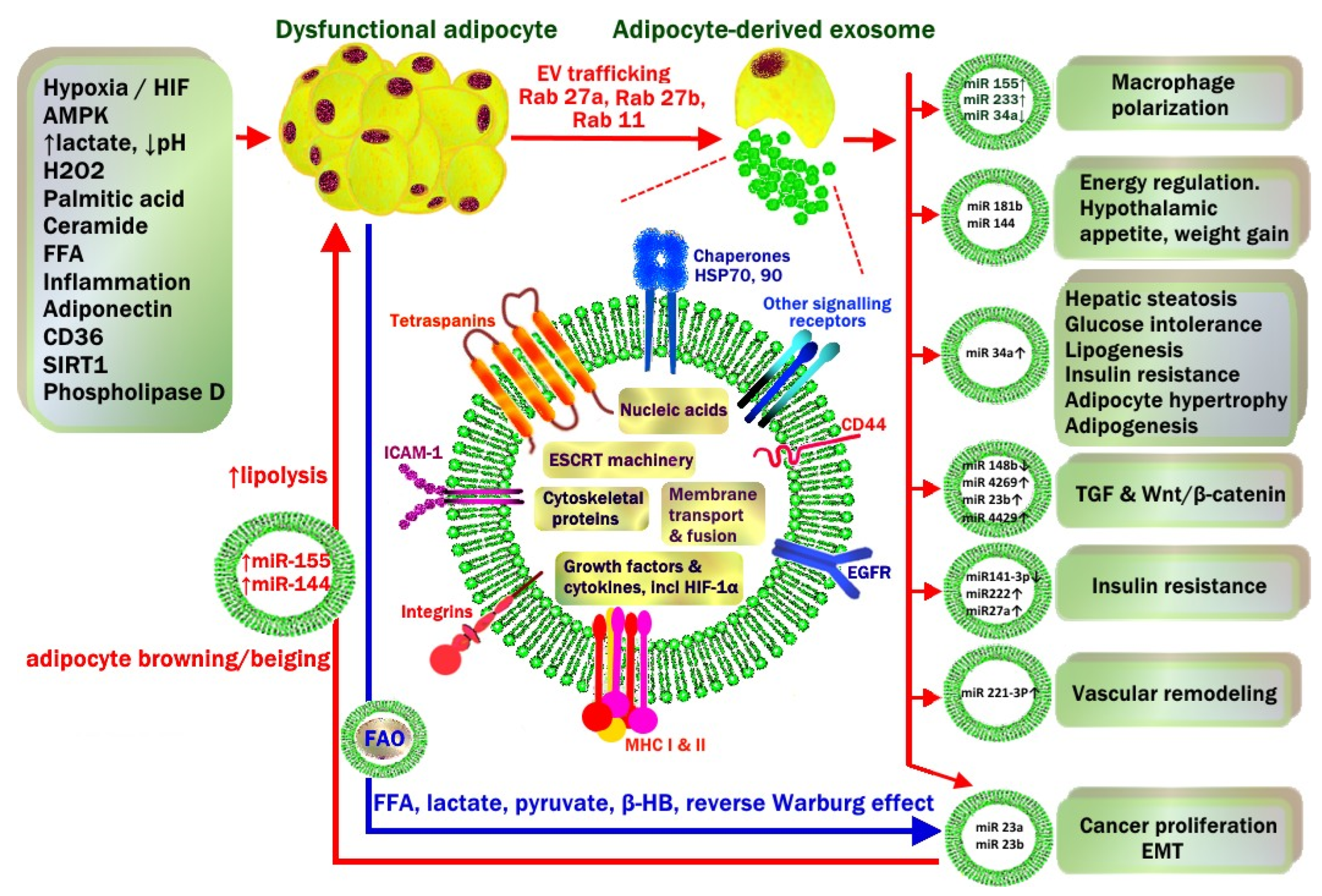
4.9. Adipocyte-Derived Exosomes
4.10. WAT Exosomes in Lean vs. Obese Patients
4.11. Exosomal miRNA
- Activation of TGF-β and Wnt/β-catenin pathways (miRNA-23b, miRNA-4429).
- Macrophage M1 polarisation (miRNA-155) and the suppression of M2 macrophage polarization (miRNA-34a).
- Hepatic steatosis, glucose intolerance, insulin resistance (miRNA-34a).
- Vascular remodelling and vascular smooth muscle proliferation (miRNA-221-3p).
- Hypothalamic POMC neuronal regulation of appetite, energy intake and weight gain (miRNA-181b, miRNA-144).
5. Hormones
5.1. Insulin and Oestrogen
5.2. Obesity and Aromatase
5.3. Crown-like Structures and Breast Cancer
6. Inflammation and Metabolism

6.1. Adipose Tissue Macrophage Polarization
6.2. Metabolically Activated Macrophages and Crown-like Structures
7. Hypoxia
7.1. Hypoxia and Obstructive Sleep Apnoea
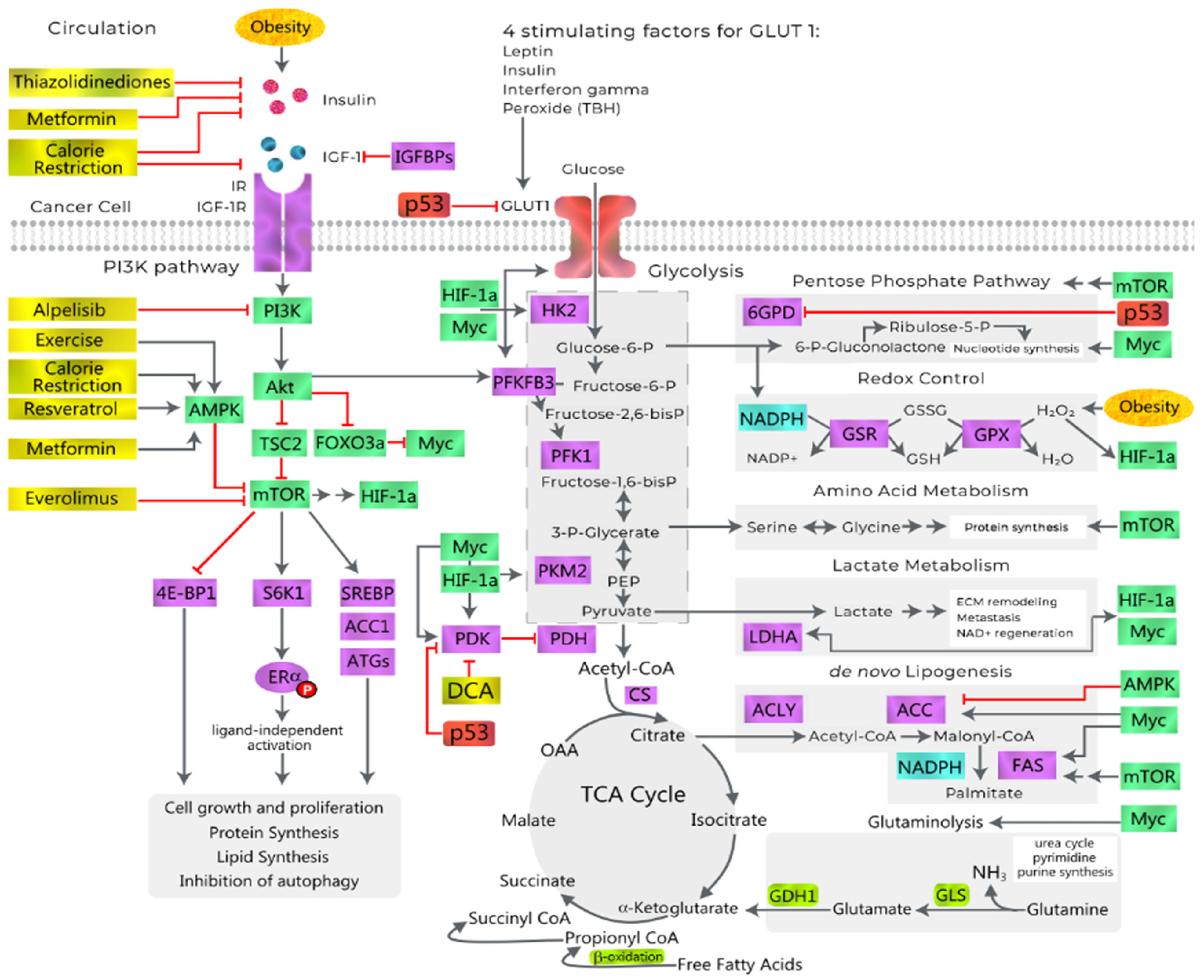
7.2. The Warburg Effect

7.3. Oscillatory HIF-1α and the Hypoxia Response Element (HRE)
- ○
- Glucose transporters (GLUT-1 and -3);
- ○
- Glycolytic enzymes (HK-1, HK-2, PFKFB3, PGK-1, PKM-2, GAPDH, LDHA);
- ○
- Metabolic reprogramming of mitochondria (PDHK);
- ○
- Erythropoeisis (EPO);
- ○
- Angiogenesis (leptin; nitric oxide synthase (NOS); vascular endothelial growth factor (VEGF); LDL-receptor-related protein 1 (LRP1); ADM, adrenomedullin (ADM); transforming growth factor-β3 (TGF-β3));
- ○
- Chromatin methylation (TET2, jumonji family histone demethylases JMJD1A and JMJD2B);
- ○
- Cellular detachment, EMT and migration (E-cadherin repression, Hedgehog, TGF β/SMAD3, ILK, Wnt/b-catenin, dynamic actin reorganization, EMT transcription factors);
- ○
- Cell survival and resistance to anoikis (ADM, EPO, NOS-2, insulin-like growth factor (IGF-2), IGF-factor-binding protein 2 (IGF-BP2), transforming growth factor α (TGF-α));
- ○
7.4. HIF and Metabolism
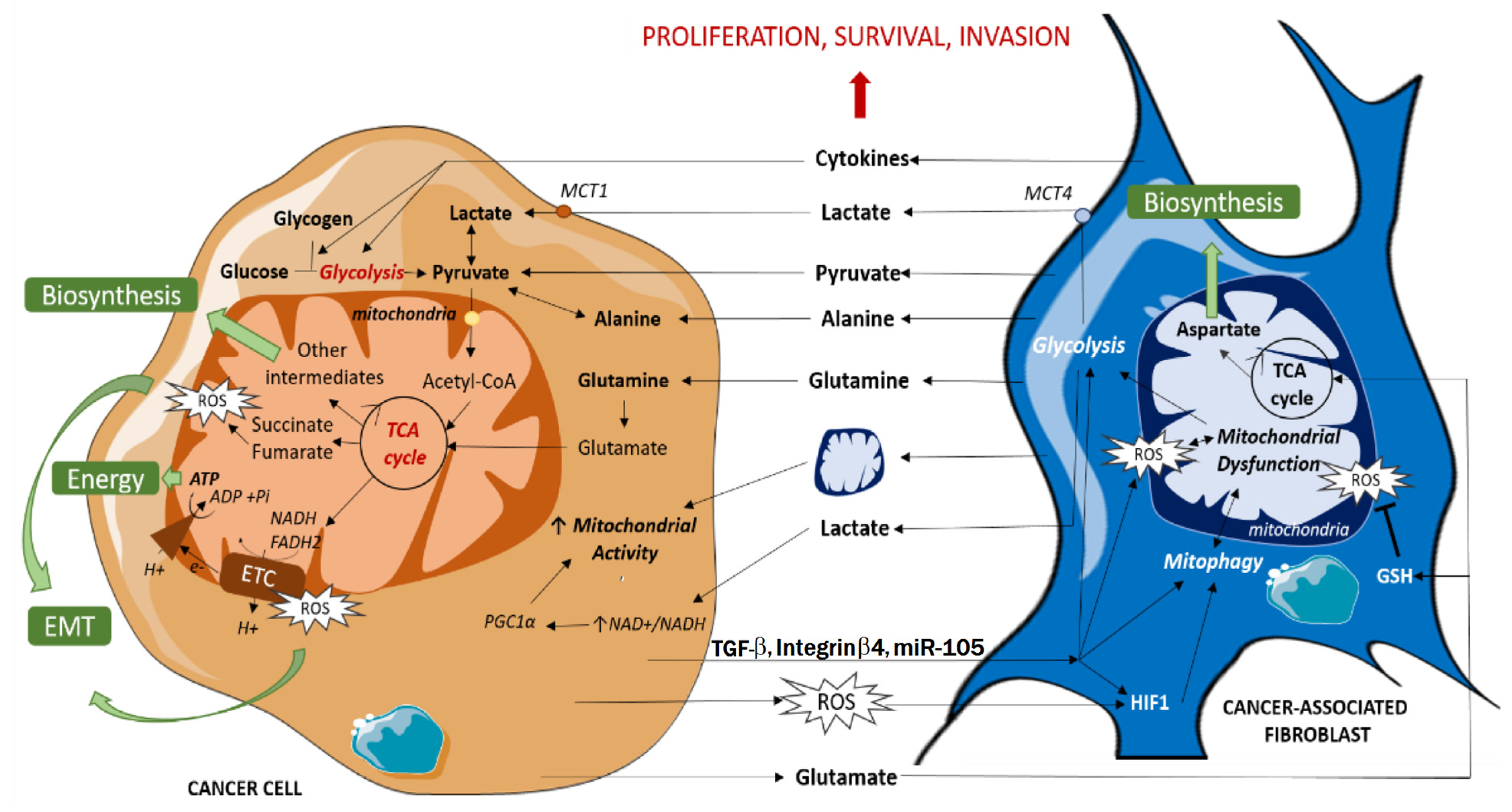
7.5. Reverse Warburg Effect
7.6. Metabolic Reprogramming and Cancer
7.7. Regulation of Cellular Anabolism vs. Catabolism
8. WAT and Hepatocyte Extracellular Matrix
CLS and NASH
9. Epigenetic Pathways
9.1. Differentially Methylated Regions
9.2. Reversal of DNA Methylation
9.3. Butyrate and Epigenetic Suppression of CRC
10. Gut, Diet and Intestinal Microbiome
10.1. Dietary Fibre
- Non-starch polysaccharides (NSP) from fruit and vegetables (cellulose, hemicellulose, pectin);
- Non-digestible oligosaccharides (galactoligosaccharides, fructoligosaccharides, inulin);
- Resistant starch from wholegrain cereals, root vegetables, nuts, seeds and legumes;
- Lignins (non-carbohydrate phenolic polymers) [163].
10.2. Short-Chain Fatty Acids, Obesity and Cancer
10.3. Obesity and Intestinal Estrolobiome
10.4. High-Fat Diet, Bile Acids and Cancer
11. Weight loss and Prevention of Cancer
12. Summary and Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviation
| 17β-HSD | 17β-hydroxysteroid dehydrogenase |
| AdipoR1/AdipoR2 | adiponectin membrane receptors |
| ADM | adrenomedullin |
| A-FABP | adipose fatty acid binding protein |
| AgRP | agouti-related peptide |
| AHI | apnoea-hypopnoea index |
| AKT | Protein kinase B (PKB) |
| AMPK | AMP-activated protein kinase |
| ANGPTL4 | angiopoietin-like 4 |
| APC | adenomatous polyposis coli regulator of WNT signaling pathway gene |
| ATM | adipocyte tissue macrophages |
| ATP | adenosine triphosphate |
| β-HB | beta-hydroxy butyrate |
| BAT | brown adipose tissue |
| BCL-2 | B cell lymphoma 2 |
| BMI | body mass index |
| BMS | bariatric metabolic surgery |
| BRAF | v-raf murine sarcoma viral oncogene homolog B1 |
| CAA | cancer-associated adipocytes |
| CAF | cancer-associated fibroblasts |
| Cav-1 | caveolin-1 structural protein |
| CCAAT | cytosine-cytosine-adenosine-adenosine-thymidine |
| CCK | cholecystokinin |
| CCL-2 | Chemokine (C-C motif) ligand 2, MCP-1 |
| CD36 | Cluster of differentiation 36 |
| CDKN1a | Cyclin Dependent Kinase Inhibitor 1A |
| CI | confidence interval |
| CIMP | CpG dinucleotide island methylator phenotype |
| CLS | crown-like structures |
| CMP-1 | chemoattractant monocyte protein 1 |
| CMS | consensus molecular subtype |
| c-Myc | cellular myelocytomatosis |
| COX-2 | cyclooxygenase |
| CpG | cytosine and guanine separated by a phosphate |
| CRC | colorectal cancer |
| CRP | C-reactive protein |
| c-Src | cellular analogue of Rous sarcoma virus |
| CXCL-1 | C-X-C Motif Chemokine Ligand 1 |
| CYP19A1 | cytochrome P450 19A1 |
| DAMPS | damage-associated molecular patterns |
| db/db | Homozygous mutation in murine leptin receptor gene |
| DDR | DNA damage response |
| DKK-3 | Dickkopf 3 protein |
| DMR | Differentially methylated regions |
| E1 | oestrone |
| E2 | 17-β-oestradiol |
| EBPα | enhancer-binding proteins α |
| ECM | extracellular matrix |
| EGFR | epidermal growth factor receptor |
| EMT | epithelial to mesenchymal transition |
| END | endurance |
| EPO | erythropoietin |
| ER-a | oestrogen receptor alpha |
| ERK | Extracellular Signal-Regulated Kinases |
| ESCRT | endosomal sorting complexes required for transport |
| FAO | Fatty acid oxidase |
| FasL | FS-7-associated surface antigen ligand |
| FFA | free fatty acid |
| G-6-PD | glucose 6-phosphate dehydrogenase |
| GAPDH | glyceraldehyde-3-phosphate dehydrogenase |
| G-CSF | granulocyte-colony stimulating factor |
| GIP | glucose dependent insulin peptide |
| GLP-1 | glucagon like peptide |
| GLUT-1 and -3 and -4 | glucose transporters |
| GPCR | G-protein coupled receptor |
| GPER | G-protein-coupled oestrogen receptor |
| GSH | glutathione |
| GUS | glucuronidase enzymes |
| HCC | hepatocellular carcinoma |
| HDACi | histone deacetylase inhibitor |
| HDL | high-density lipoprotein |
| HER2 | human epidermal growth factor receptor |
| HFD | high-fat diet |
| HGF | hepatocyte growth factor |
| HIF | hypoxia-inducible factor |
| HIF-1α | hypoxia-inducible factor-1α |
| HIIT | high intensity interval training |
| HIST1H3D | Histone linker 1 with Histone H3.D |
| HIST1H3I | Histone linker 1 with Histone H3.1 |
| HK-1 | hexokinase 1 |
| HK2 | hexokinase 2 |
| HOMA-IR | Homeostatic Model Assessment of Insulin Resistance |
| HOXB8 | homeobox B8 |
| HRE | hypoxia-response element |
| HSP | heat shock protein |
| HSP-90 | heat shock protein 90 |
| ICAM | intercellular adhesion molecule |
| ID2 | DNA-binding protein inhibitor 2 |
| IGF-1 | insulin-like growth factor 1 |
| IGF-1R | intracellular insulin receptor substrate 1 |
| IGF-2 | insulin-like growth factor 2 |
| IGF-BP | IGF binding proteins |
| IL-1 | Interleukin 1 |
| IL-10 | Interleukin 10 |
| IL-1β | Interleukin 1β |
| IL-6 | interleukin 6 |
| IL-8 | interleukin 8 |
| ILK | Integrin-linked kinase |
| INF-γ | interferon gamma |
| iNOS | inducible nitric oxide synthase |
| IRS | insulin receptor substrate |
| JAK2 | Janus kinase 2 |
| JMJD1A/JMJD2B | jumonji family histone demethylase |
| KRAS | Kirsten rat sarcoma viral oncogene homolog |
| LDHA | lactate dehydrogenase A |
| LDL | low density lipoprotein |
| LEPR | Long isoform of neural leptin receptor |
| LPA | lysophosphatidic acid |
| LSG | sleeve gastrectomy |
| LPS | lipopolysaccharide |
| MAC-2 | macrophage-2 binding protein, galectin-3 |
| MAPK | mitogen-activated protein kinase |
| MC4R | melanocortin 4 receptor |
| MCP-1 | monocyte chemoattractant protein 1 |
| MCT-1/MCT-4 | mono-carboxylate transporter |
| Mdm2 | mouse double minute 2 homolog |
| MetS | metabolic syndrome |
| MHO | metabolically healthy obesity |
| MIP-2 | macrophage inflammatory protein, CXCL2 |
| MMe | metabolically activated macrophage |
| MMP | mixed metalloproteases |
| MRAP2 | Melanocortin 2 Receptor Accessory Protein 2 |
| MSI | microsatellite instability |
| mTOR | mammalian target of rapamycin |
| MUNW | metabolically unhealthy–normal weight |
| NADPH | Reduced nicotinamide adenine dinucleotide phosphate |
| NAFLD | non-alcoholic fatty liver disease |
| NASH | non-alcoholic steatohepatitis |
| NFATC4 | nuclear factor of activated T-cells cytoplasmic 4 |
| NF-κB | nuclear factor kappa light chain enhancer of activated B cells |
| NLRC4 | nucleotide-binding domain and leucine-rich repeat receptor family CARD domain containing 4 |
| NO | nitric oxide |
| NOS | nitric oxide synthase |
| NOX | NADPH oxidase |
| NPY | neuropeptide Y |
| OAC | obesity-associated cancers |
| Ob-Rb | long form leptin receptor |
| OCT4 | octamer-binding transcription factor 4 |
| OHS | obesity hypoventilation syndrome |
| OR | odds ratio |
| OSA | obstructive sleep apnoea |
| PAI-1 | plasminogen activator inhibitor-1 |
| PCSK1 | proprotein convertase subtilisin/kexin type 1 |
| PDGF-BB | platelet-derived growth factor-BB |
| PDH | pyruvate dehydrogenase |
| PDHK | pyruvate dehydrogenase kinase |
| PFK1 | phosphofructokinase |
| PFKFB3 | 6-phosphofructo-2-kinase/fructose-2, 6-bisphosphatase 3 |
| PGE2 | Prostaglandin E2 |
| PGK-1 | phosphoglycerate kinase 1 |
| PHD2 | prolyl hydroxylase domain containing 2 |
| PHIP98 | Pleckstrin Homology Domain Interacting Protein 98 |
| PI3K | phosphoinositide 3-kinase |
| PLCγ-PKC | phospholipase C (PLC)γ-protein kinase C (PKC) |
| PKM2 | pyruvate kinase muscle 2 |
| POMC | pro-opiomelanocortin |
| PPARγ | peroxisome proliferator-activated receptor γ |
| PPP | pentose phosphate pathway |
| pVHL-VBC | von Hippel-Lindau protein (pVHL)-elonginB-elonginC (VBC) complex |
| Rab | Ras-associated binding proteins |
| RAF | rapidly accelerated fibrosarcoma |
| RAS | rat sarcoma |
| RhoA-ROCK | Ras homology gene family member A—Rho-associated kinases |
| RNA: lncRNA | long non-coding RNA |
| RNA: miRNA | micro-RNA |
| RNA: mRNA | functional RNA |
| RNA: snRNA | small nuclear RNA |
| RNA: tRNA | transfer RNA |
| ROS | reactive oxygen species |
| RYGB | Roux-en-Y gastric bypass |
| SASP | senescence-associated secretory phenotype |
| SAT | subcutaneous adipose tissue |
| SCFA | short-chain fatty acids |
| SFRP5 | secreted frizzled-related protein 5 |
| SH2B1 | Src-homology 2B adaptor protein 1 gene |
| SHBG | sex hormone-binding globulin |
| SIM1 | Single-minded 1 |
| SMAD3 | mothers against decapentaplegic homolog 3 |
| SOX2 | SRY-Box Transcription Factor 2 |
| SRGAP2C | Slit-Robo Rho GTPase activating protein 2C |
| STAT3 | Signal transducer and activator of transcription 3 |
| T2DM | Type II diabetes mellitus |
| TBW | total body weight |
| TCA | tricarboxylic acid |
| TET | ten–eleven translocation methylcytosine dioxygenase |
| TfR | transferrin receptor |
| TGF-α | transforming growth factor α |
| TGF-β | transforming growth factor β |
| TIMP | tissue inhibitor of metalloproteinase |
| TKR | tyrosine kinase receptor |
| TLR | Toll-like receptor |
| TNBC | triple negative breast cancer |
| TNF-α | tumor necrosis factor alpha |
| TP53 | tumor suppressor protein 53 |
| TPI1 | triosephosphate isomerase |
| TRAIL | tumor necrosis factor-alpha-related apoptosis inducing ligand |
| UCP-1 | uncoupling protein 1 |
| VAT | visceral adipose tissue |
| VEGF | vascular endothelial growth factor |
| WAT | white adipose tissue |
| Wnt | wingless-related integration site |
| XOX | xanthine oxidase |
| ZBTB46 | zinc finger and born-to-bind domain containing 46 |
References
- Australian Institute of Health and Welfare. Cancer Data in Australia. Cat. No. CAN 122. Available online: https://www.aihw.gov.au/reports/cancer/cancer-data-in-australia/data (accessed on 29 November 2022).
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLO-BOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef] [PubMed]
- Sung, H.; Siegel, R.L.; Torre, L.A.; Pearson-Stuttard, J.; Islami, F.; Fedewa, S.A.; Sauer, A.G.; Shuval, K.; Gapstur, S.M.; Jacobs, E.J. Global patterns in excess body weight and the associated cancer burden. CA Cancer J. Clin. 2019, 69, 88–112. [Google Scholar] [CrossRef] [PubMed]
- Murray, C.J.; Aravkin, A.Y.; Zheng, P.; Abbafati, C.; Abbas, K.M.; Abbasi-Kangevari, M.; Abd-Allah, F.; Abdelalim, A.; Abdollahi, M.; Abdollahpour, I. Global burden of 87 risk factors in 204 countries and territories, 1990–2019: A systematic analysis for the Global Burden of Disease Study. Lancet 2019, 396, 1223–1249. [Google Scholar] [CrossRef] [PubMed]
- Friedenreich, C.M.; Ryder-Burbidge, C.; McNeil, J. Physical activity, obesity and sedentary behavior in cancer etiology: Epidemio-logic evidence and biologic mechanisms. Mol. Oncol. 2021, 15, 790–800. [Google Scholar] [CrossRef] [PubMed]
- Fang, Z.; Song, M.; Lee, D.H.; Giovannucci, E.L. The role of mendelian randomization studies in deciphering the effect of obesity on cancer. J. Natl. Cancer Inst. 2022, 114, 361–371. [Google Scholar] [CrossRef]
- Younes, S.; Ibrahim, A.; Al-Jurf, R.; Zayed, H. Genetic polymorphisms associated with obesity in the Arab world: A systematic review. Int. J. Obes. 2021, 45, 1899–1913. [Google Scholar] [CrossRef]
- Centers for Disease Control and Prevention. Adult Obesity Facts. Available online: https://www.cdc.gov/obesity/data/adult.html (accessed on 13 April 2023).
- Bhaskaran, K.; Douglas, I.; Forbes, H. Body-mass index and risk of 22 specific cancers: A population-based cohort study of 5.24 million UK adults. Lancet 2014, 384, 755–765. [Google Scholar] [CrossRef]
- Vourdoumpa, A.; Paltoglou, G.; Charmandari, E. The Genetic Basis of Childhood Obesity: A Systematic Review. Nutrients 2023, 15, 1416. [Google Scholar] [CrossRef]
- Loos, R.J.F.; Yeo, G.S. The genetics of obesity: From discovery to biology. Nat. Rev. Genet. 2022, 23, 120–133. [Google Scholar] [CrossRef]
- King, S.E.; Skinner, M.K. Epigenetic Transgenerational Inheritance of Obesity Susceptibility. Trends Endocrinol. Metab. 2020, 31, 478–494. [Google Scholar] [CrossRef]
- Mohajer, N.; Joloya, E.M.; Seo, J.; Shioda, T.; Blumberg, B. Epigenetic Transgenerational Inheritance of the Effects of Obesogen Exposure. Front. Endocrinol. 2021, 12, 787580. [Google Scholar] [CrossRef] [PubMed]
- Lauby-Secretan, B.; Scoccianti, C.; Loomis, D. Body Fatness and Cancer–Viewpoint of the IARC Working Group. N. Engl. J. Med. 2016, 375, 794–802. [Google Scholar] [CrossRef] [PubMed]
- Feletto, E.; Kohar, A.; Mizrahi, D.; Grogan, P.; Steinberg, J.; Hughes, C.; Watson, W.L.; Canfell, K.; Yu, X.Q. An ecological study of obesity-related cancer incidence trends in Australia from 1983 to 2017. Lancet Reg. Health West. Pac. 2022, 29, 100575. [Google Scholar] [CrossRef]
- Steele, C.B.; Thomas, C.C.; Henley, S.J. Vital Signs: Trends in Incidence of Cancers Associated with Overweight and Obesity-United States. MMWR Morb. Mortal Wkly. Rep. 2005, 66, 1052–1058. [Google Scholar] [CrossRef] [PubMed]
- Pontzer, H. Constrained Total Energy Expenditure and Metabolic Adaptation to Physical Activity in Adult Humans. Curr. Biol. 2016, 26, 410–417. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Pederson, L.; Halberg, N. Cellular mechanisms linking cancers to obesity. Cell Stress 2021, 5, 55–72. [Google Scholar] [CrossRef] [PubMed]
- Goncalves, M.D.; Lu, C.; Tutnauer, J.; Hartman, T.E.; Hwang, S.K.; Murphy, C.J.; Pauli, C.; Morris, R.; Taylor, S.; Bosch, K.; et al. High-fructose corn syrup enhances intestinal tumor growth in mice. Science 2019, 363, 1345–1349. [Google Scholar] [CrossRef]
- Valenzuela, P.L.; Carrera-Bastos, P.; Castillo-García, A.; Lieberman, D.E.; Santos-Lozano, A.; Lucia, A. Obesity and the risk of cardiometabolic diseases. Nat. Rev. Cardiol, 2023; ahead of print. [Google Scholar] [CrossRef]
- Dvorak, H.F. Tumors: Wounds that do not heal-redux. Cancer Immunol. Res. 2015, 3, 1–11. [Google Scholar] [CrossRef]
- Balkwill, F.; Mantovani, A. Inflammation and cancer: Back to Virchow? Lancet 2001, 357, 539–545. [Google Scholar] [CrossRef]
- Rabhi, N.; Desevin, K.; Belkina, A.C.; Tilston-Lunel, A.; Varelas, X.; Layne, M.D.; Farmer, S.R. Obesity-induced senescent macrophages activate a fibrotic transcriptional program in adipocyte progenitors. Life Sci. Alliance 2022, 5, e202101286. [Google Scholar] [CrossRef]
- Seyfried, T.N. Cancer as a mitochondrial metabolic disease. Front. Cell Dev. Biol. 2015, 3, 43. [Google Scholar] [CrossRef] [PubMed]
- Wilson, R.B.; Archid, R.; Reymond, M.A. Reprogramming of Mesothelial-Mesenchymal Transition in Chronic Peritoneal Diseases by Oestrogen Receptor Modulation and TGF-β1 Inhibition. Int. J. Mol. Sci. 2020, 21, 4158. [Google Scholar] [CrossRef] [PubMed]
- Hanahan, D. Hallmarks of Cancer: New Dimensions. Cancer Discov. 2022, 12, 31–46. [Google Scholar] [CrossRef]
- Karra, P.; Winn, M.; Pauleck, S. Metabolic dysfunction and obesity related cancer: Beyond obesity and metabolic syndrome. Obes. J. 2022, 30, 1323–1334. [Google Scholar] [CrossRef]
- Murphy, N.; Cross, A.J.; Abubakar, M. A Nested Case-Control Study of Metabolically Defined Body Size Phenotypes and Risk of Colorectal Cancer in the European Prospective Investigation into Cancer and Nutrition (EPIC). PLoS Med. 2016, 13, e1001988. [Google Scholar] [CrossRef]
- Bonsignore, M.R.; Esquinas, C.; Barceló, A. Metabolic syndrome, insulin resistance and sleepiness in real-life obstructive sleep apnoea. Eur. Respir. J. 2012, 39, 1136–1143. [Google Scholar] [CrossRef] [PubMed]
- Vicks, W.S.; Lo, J.C.; Guo, L. Prevalence of prediabetes and diabetes vary by ethnicity among U.S. Asian adults at healthy weight, overweight, and obesity ranges: An electronic health record study. BMC Public Health 2022, 22, 1954. [Google Scholar] [CrossRef]
- Spaander, M.; Zauber, A.; Syngal, S.; Blaser, M.J.; Sung, J.J.; You, Y.N.; Kuipers, E.J. Young-onset colorectal cancer. Nat. Rev. Dis. Prim. 2023, 9, 21. [Google Scholar] [CrossRef]
- Spiegel, R.L.; Wagle, N.S.; Cercek, A.; Smith, R.A.; Jemal, A. Colorectal cancer statistics, 2023. CA Cancer J. Clin. 2023, 73, 233–254. [Google Scholar] [CrossRef]
- Greene, M.W.; Abraham, P.T.; Kuhlers, P.C.; Lipke, E.A.; Heslin, M.J.; Wijaya, S.T.; Odeniyi, I. Consensus molecular subtype differences linking colon adenocarcinoma and obesity revealed by a cohort transcriptomic analysis. PLoS ONE 2022, 17, e0268436. [Google Scholar] [CrossRef]
- Rebane-Klemm, E.; Truu, L.; Reinsalu, L.; Puurand, M.; Shevchuk, I.; Chekulayev, V.; Timohhina, N.; Tepp, K.; Bo-Govskaja, J.; Afanasjev, V.; et al. Mitochondrial Respiration in KRAS and BRAF Mutated Colorectal Tumors and Polyps. Cancers 2020, 12, 815. [Google Scholar] [CrossRef] [PubMed]
- Murphy, N.; Newton, C.C.; Song, M. Body mass index and molecular subtypes of colorectal cancer. J. Natl. Cancer Inst. 2022, 115, 165–173. [Google Scholar] [CrossRef] [PubMed]
- Burkitt, D.P. Related disease—Related cause? Lancet 1969, 2, 1229–1231. [Google Scholar] [CrossRef] [PubMed]
- Burkitt, D.P.; Walker, A.R.; Painter, N.S. Effect of dietary fibre on stools and the transit-times, and its role in the causation of disease. Lancet 1972, 2, 1408–1412. [Google Scholar] [CrossRef] [PubMed]
- Burkitt, D.P. Some diseases characteristic of modern Western civilization. Br. Med. J. 1973, 1, 274–278. [Google Scholar] [CrossRef] [PubMed]
- Burkitt, D.P.; Trowell, H.C. Western Diseases, Their Emergence and Prevention; Harvard University Press: Cambridge, UK, 1981; ISBN 978-0-674-95020-7. [Google Scholar]
- O’Keefe, S.J. The association between dietary fibre deficiency and high-income lifestyle-associated diseases: Burkitt’s hypothesis revisited. Lancet Gastroenterol. Hepatol. 2019, 4, 984–996. [Google Scholar] [CrossRef]
- Morkel, M.; Riemer, P.; Bläker, H.; Sers, C. Similar but different: Distinct roles for KRAS and BRAF oncogenes in colorectal cancer development and therapy resistance. Oncotarget 2015, 6, 20785–20800. [Google Scholar] [CrossRef]
- Veettil, S.K.; Wong, T.Y.; Loo, Y.S. Role of Diet in Colorectal Cancer Incidence: Umbrella Review of Meta-analyses of Prospective Observational Studies. JAMA Netw. Open 2021, 4, e2037341. [Google Scholar] [CrossRef]
- Aune, D.; Chan, D.S.; Lau, R. Dietary fibre, whole grains, and risk of colorectal cancer: Systematic review and dose-response meta-analysis of prospective studies. BMJ 2011, 43, d6617. [Google Scholar] [CrossRef]
- Bouvard, V.; Loomis, D.; Guyton, K.Z. Carcinogenicity of consumption of red and processed meat. Lancet Oncol. 2015, 16, 1599–1600. [Google Scholar] [CrossRef]
- Gianfredi, V.; Salvatori, T.; Villarini, M.; Moretti, M.; Nucci, D.; Realdon, S. Is dietary fibre truly protective against colon cancer? A systematic review and meta-analysis. Int. J. Food Sci. Nutr. 2018, 69, 904–915. [Google Scholar] [CrossRef] [PubMed]
- Gianfredi, V.; Nucci, D.; Salvatori, T.; Dallagiacoma, G.; Fatigoni, C.; Moretti, M.; Realdon, S. Rectal Cancer: 20% Risk Reduction Thanks to Dietary Fibre Intake. Systematic Review and Meta-Analysis. Nutrients 2019, 11, 1579. [Google Scholar] [CrossRef] [PubMed]
- Nucci, D.; Fatigoni, C.; Salvatori, T.; Nardi, M.; Realdon, S.; Gianfredi, V. Association between Dietary Fibre Intake and Colorectal Adenoma: A Systematic Review and Meta-Analysis. Int. J. Environ. Res. Public Health 2021, 18, 4168. [Google Scholar] [CrossRef] [PubMed]
- Cummings, J.H.; Engineer, A. Denis Burkitt and the origins of the dietary fibre hypothesis. Nutr. Res. Rev. 2017, 31, 1–15. [Google Scholar] [CrossRef]
- Guerrini, V.; Gennaro, M.L. Foam Cells: One Size Doesn’t Fit All. Trends Immunol. 2019, 40, 1163–1179. [Google Scholar] [CrossRef]
- Ramírez Molina, V.R.; Masa Jiménez, J.F.; Gómez de Terreros Caro, F.J.; Corral Peñafiel, J. Effectiveness of different treatments in obesity hypoventilation syndrome. Pulmonology 2020, 26, 370–377. [Google Scholar] [CrossRef]
- Zunica, E.R.M.; Heintz, E.C.; Axelrod, C.L.; Kirwan, J.P. Obesity Management in the Primary Prevention of Hepatocellular Carcinoma. Cancers 2022, 14, 4051. [Google Scholar] [CrossRef]
- Zelicha, H.; Kloting, N.; Kaplan, A. The effect of high-polyphenol Mediterranean diet on visceral adiposity: The DIRECT PLUS randomized controlled trial. BMC Med. 2022, 20, 327. [Google Scholar] [CrossRef]
- Teodoro, J.S.; Varela, A.T.; Rolo, A.P.; Palmeira, C.M. High-fat and obesogenic diets: Current and future strategies to fight obesity and diabetes. Genes. Nutr. 2014, 9, 406. [Google Scholar] [CrossRef]
- Kawai, T.; Autieri, M.V.; Scalia, R. Adipose tissue inflammation and metabolic dysfunction in obesity. Am. J. Physiol. Cell Physiol. 2021, 320, 375–391. [Google Scholar] [CrossRef]
- Gjermeni, E.; Kirstein, A.S.; Kolbig, F.; Kirchhof, M.; Bundalian, L.; Katzmann, J.L.; Laufs, U.; Blüher, M.; Garten, A.; Le Duc, D. Obesity—An Update on the Basic Pathophysiology and Review of Recent Therapeutic Advances. Biomolecules 2021, 11, 1426. [Google Scholar] [CrossRef] [PubMed]
- Tran, L.T.; Park, S.; Kim, S.K. Hypothalamic control of energy expenditure and thermogenesis. Exp. Mol. Med. 2022, 54, 358–369. [Google Scholar] [CrossRef] [PubMed]
- Lin, X.; Li, H. Obesity: Epidemiology, Pathophysiology, and Therapeutics. Front. Endocrinol. 2021, 12, 706978. [Google Scholar] [CrossRef] [PubMed]
- Lagarde, D.; Jeanson, Y.; Portais, J.C. Lactate Fluxes and Plasticity of Adipose Tissues: A Redox Perspective. Front. Physiol. 2021, 12, 689747. [Google Scholar] [CrossRef] [PubMed]
- Engelking, L.R. Saturated and unsaturated fatty acids. In Textbook of Veterinary Physiological Chemistry; Academic Press/Elsevier: Burlington, MA, USA, 2015; Chapter 54; pp. 345–350. ISBN 9780123919090. [Google Scholar] [CrossRef]
- Stenkula, K.G.; Erlanson-Albertsson, C. Adipose cell size: Importance in health and disease. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2018, 315, 284–295. [Google Scholar] [CrossRef]
- Palomäki, V.A.; Lehenkari, P.; Meriläinen, S.; Karttunen, T.J.; Koivukangas, V. Dynamics of adipose tissue macrophage populations after gastric bypass surgery. Obesity 2023, 31, 184–191. [Google Scholar] [CrossRef]
- Wong, C.; Kaneda, T.; Morita, H. Plant natural products as an anti-lipid droplets accumulation agent. J. Nat. Med. 2014, 68, 253–266. [Google Scholar] [CrossRef]
- Smith, U.; Li, Q.; Rydén, M.; Spalding, K.L. Cellular senescence and its role in white adipose tissue. Int. J. Obes. 2021, 45, 934–943. [Google Scholar] [CrossRef]
- Heyn, G.S.; Corrêa, L.H.; Magalhães, K.G. The Impact of Adipose Tissue-Derived miRNAs in Metabolic Syndrome, Obesity, and Cancer. Front. Endocrinol. 2020, 11, 563816. [Google Scholar] [CrossRef]
- Li, Q.; Hagberg, C.E.; Cascales, H.S.; Lang, S.; Hyvönen, M.T.; Salehzadeh, F.; Chen, P.; Alexandersson, I.; Terezaki, E.; Harms, M.J.; et al. Obesity and hyperinsulinemia drive adipocytes to activate a cell cycle program and senesce. Nat. Med. 2021, 27, 1941–1953. [Google Scholar] [CrossRef]
- Gasek, N.S.; Kuchel, G.A.; Kirkland, J.L. Strategies for targeting senescent cells in human disease. Nat. Aging 2021, 1, 870–879. [Google Scholar] [CrossRef] [PubMed]
- Berger, N.A. Crown-like Structures in Breast Adipose Tissue from Normal Weight Women: Important Impact. Cancer Prev. Res. 2017, 10, 223–225. [Google Scholar] [CrossRef] [PubMed]
- Ohtani, N. The roles and mechanisms of senescence-associated secretory phenotype (SASP): Can it be controlled by senolysis. Inflamm. Regen. 2022, 42, 11. [Google Scholar] [CrossRef] [PubMed]
- Rubinstein, M.M.; Brown, K.A.; Iyengar, N.M. Targeting obesity-related dysfunction in hormonally driven cancers. Br. J. Cancer 2021, 125, 495–509. [Google Scholar] [CrossRef] [PubMed]
- Chen, K.; Zhang, J.; Beeraka, N.M.; Tang, C.; Babayeva, Y.V.; Sinelnikov, M.Y.; Zhang, X.; Zhang, J.; Liu, J.; Reshetov, I.V.; et al. Advances in the Prevention and Treatment of Obesity-Driven Effects in Breast Cancers. Front. Oncol. 2022, 12, 820968. [Google Scholar] [CrossRef]
- Pini, M.; Czibik, G.; Sawaki, D.; Mezdari, Z.; Braud, L.; Delmont, T.; Mercedes, R.; Martel, C.; Buron, N.; Marcelin, G.; et al. Adipose tissue senescence is mediated by increased ATP content after a short-term high-fat diet exposure. Aging Cell 2021, 20, e13421. [Google Scholar] [CrossRef]
- Maharjan, B.R.; Martinez-Huenchullan, S.F.; Mclennan, S.V.; Twigg, S.M.; Williams, P.F. Exercise induces favorable metabolic changes in white adipose tissue preventing high-fat diet obesity. Physiol. Rep. 2021, 9, e14929. [Google Scholar] [CrossRef]
- Crespi, E.; Bottai, G.; Santarpia, L. Role of inflammation in obesity-related breast cancer. Curr. Opin. Pharmacol. 2016, 31, 114–122. [Google Scholar] [CrossRef]
- Quan, M.; Kuang, S. Exosomal Secretion of Adipose Tissue during Various Physiological States. Pharm. Res. 2020, 37, 221. [Google Scholar] [CrossRef]
- Zhao, C.; Wu, M.; Zeng, N. Cancer-associated adipocytes: Emerging supporters in breast cancer. J. Exp. Clin. Cancer Res. 2020, 39, 156. [Google Scholar] [CrossRef]
- Żbikowski, A.; Błachnio-Zabielska, A.; Galli, M.; Zabielski, P. Adipose-Derived Exosomes as Possible Players in the Development of Insulin Resistance. Int. J. Mol. Sci. 2021, 22, 7427. [Google Scholar] [CrossRef] [PubMed]
- Obradovic, M.; Sudar-Milovanovic, E.; Soskic, S.; Essack, M.; Arya, S.; Stewart, A.J.; Gojobori, T.; Isenovic, E.R. Leptin and Obesity: Role and Clinical Implication. Front. Endocrinol. 2021, 12, 585887. [Google Scholar] [CrossRef] [PubMed]
- Kwan, H.Y.; Chen, M.; Xu, K.; Chen, B. The impact of obesity on adipocyte-derived extracellular vesicles. Cell Mol. Life Sci. 2021, 78, 7275–7288. [Google Scholar] [CrossRef]
- Honecker, J.; Ruschke, S.; Seeliger, C.; Laber, S.; Strobel, S.; Pröll, P.; Nellaker, C.; Lindgren, C.M.; Kulozik, U.; Ecker, J.; et al. Transcriptome and fatty-acid signatures of adipocyte hypertrophy and its non-invasive MR-based characterization in human adipose tissue. eBioMedicine 2022, 79, 104020. [Google Scholar] [CrossRef] [PubMed]
- Jiménez-Cortegana, C.; García-Galey, A.; Pino, T.M.D.; Carmona, P.; López, I.; Alba, S.; Sánchez-Margalet, V. Role of Leptin in Non-Alcoholic Fatty Liver Disease. Biomedicines 2021, 9, 762. [Google Scholar] [CrossRef]
- Martins, F.F.; Santos-Reis, T.; Marinho, T.S.; Aguila, M.B.; Mandarim-De-Lacerda, C.A. Hypothalamic anorexigenic signaling pathways (leptin, amylin, and proopiomelanocortin) are semaglutide (GLP-1 analog) targets in obesity control in mice. Life Sci. 2022, 313, 121268. [Google Scholar] [CrossRef]
- Feldman, D.E.; Chen, C.; Punj, V.; Tsukamoto, H.; Machida, K. Pluripotency factor-mediated expression of the leptin receptor (OB-R) links obesity to oncogenesis through tumor-initiating stem cells. Proc. Natl. Acad. Sci. USA 2012, 109, 829–834. [Google Scholar] [CrossRef]
- Sánchez-Jiménez, F.; Pérez-Pérez, A.; Cruz-Merino, L.D.L.; Sánchez-Margalet, V. Obesity and Breast Cancer: Role of Leptin. Front. Oncol. 2019, 9, 596. [Google Scholar] [CrossRef]
- Zahid, H.; Subbaramaiah, K.; Iyengar, N.M.; Zhou, X.K.; Chen, I.C.; Bhardwaj, P.; Gucalp, A.; Morrow, M.; Hudis, C.A.; Dannenberg, A.J.; et al. Leptin regulation of the p53-HIF1α/PKM2-aromatase axis in breast adipose stromal cells: A novel mechanism for the obesity-breast cancer link. Int. J. Obes. 2017, 42, 711–720. [Google Scholar] [CrossRef]
- Olea-Flores, M.; Juárez-Cruz, J.C.; Zuñiga-Eulogio, M.D.; Acosta, E.; García-Rodríguez, E.; Zacapala-Gomez, A.E.; Mendoza-Catalán, M.A.; Ortiz-Ortiz, J.; Ortuño-Pineda, C.; Navarro-Tito, N. New Actors Driving the Epithelial-Mesenchymal Transition in Cancer: The Role of Leptin. Biomolecules 2020, 10, 1676. [Google Scholar] [CrossRef]
- Silva, C.; Andrade, N.; Guimarães, J.T.; Patrício, E.; Martel, F. The in vitro effect of the diabetes-associated markers insulin, leptin and oxidative stress on cellular characteristics promoting breast cancer progression is GLUT1-dependent. Eur. J. Pharmacol. 2021, 898, 173980. [Google Scholar] [CrossRef] [PubMed]
- Cleary, M.P.; Grossmann, M.E. Minireview: Obesity and breast cancer: The oestrogen connection. Endocrinology 2009, 150, 2537–2542. [Google Scholar] [CrossRef] [PubMed]
- Mei, R.; Qin, W.; Zheng, Y.; Wan, Z.; Liu, L. Role of Adipose Tissue Derived Exosomes in Metabolic Disease. Front. Endocrinol. 2022, 13, 873865. [Google Scholar] [CrossRef] [PubMed]
- Kawano, J.; Arora, R. The role of adiponectin in obesity, diabetes, and cardiovascular disease. J. Cardiometab. Syndr. 2009, 4, 44–49. [Google Scholar] [CrossRef]
- Mengie Ayele, T.; Muche, T.Z.; Teklemariam, B.A.; Kassie, B.A.; Chekol Abebe, E. Role of JAK2/STAT3 Signaling Pathway in the Tumorigenesis, Chemotherapy Resistance, and Treatment of Solid Tumors: A Systemic Review. J. Inflamm. Res. 2022, 15, 1349–1364. [Google Scholar] [CrossRef]
- Castagneto-Gissey, L.; Casella-Mariolo, J.; Casella, G.; Mingrone, G. Obesity Surgery and Cancer: What Are the Unanswered Questions. Front. Endocrinol. 2020, 11, 213. [Google Scholar] [CrossRef]
- Miyoshi, Y.; Funahashi, T.; Kihara, S.; Taguchi, T.; Tamaki, Y.; Matsuzawa, Y. Association of serum adiponectin levels with breast cancer risk. Clin. Cancer Res. 2003, 9, 5699–5704. [Google Scholar]
- Bao, Y.; Giovannucci, E.L.; Kraft, P.; Stampfer, M.J.; Ogino, S.; Ma, J. A prospective study of plasma adiponectin and pancreatic cancer risk in five US cohorts. J. Natl. Cancer Inst. 2013, 105, 95–103. [Google Scholar] [CrossRef]
- Xu, G.; Song, M. Recent advances in the mechanisms underlying the beneficial effects of bariatric and metabolic surgery. Surg. Obes. Relat. Dis. 2021, 17, 231–238. [Google Scholar] [CrossRef]
- Rakib, A.; Kiran, S.; Mandal, M.; Singh, U.P. MicroRNAs: A crossroad that connects obesity to immunity and aging. Immun. Ageing 2022, 19, 64. [Google Scholar] [CrossRef]
- van Eijk, M.; Aerts, J.M.P.G. The Unique Phenotype of Lipid-Laden Macrophages. Int. J. Mol. Sci. 2021, 22, 4039. [Google Scholar] [CrossRef] [PubMed]
- Jafari, N.; Llevenes, P.; Denis, G.V. Exosomes as novel biomarkers in metabolic disease and obesity-related cancers. Nat. Rev. Endocrinol. 2022, 18, 327–328. [Google Scholar] [CrossRef]
- Bonsignore, M.R. Obesity and Obstructive Sleep Apnea. Handb. Exp. Pharmacol. 2022, 274, 181–201. [Google Scholar]
- García-Estévez, L.; Cortés, J.; Pérez, S.; Calvo, I.; Gallegos, I.; Moreno-Bueno, G. Obesity and Breast Cancer: A Paradoxical and Controversial Relationship Influenced by Menopausal Status. Front. Oncol. 2021, 11, 705911. [Google Scholar] [CrossRef] [PubMed]
- Stone, T.W.; Mcpherson, M.; Gail Darlington, L. Obesity and Cancer: Existing and New Hypotheses for a Causal Connection. eBioMedicine 2018, 30, 14–28. [Google Scholar] [CrossRef]
- LeVee, A.; Mortimer, J. The Challenges of Treating Patients with Breast Cancer and Obesity. Cancers 2023, 15, 2526. [Google Scholar] [CrossRef] [PubMed]
- Vernieri, C.; Corti, F.; Nichetti, F.; Ligorio, F.; Manglaviti, S.; Zattarin, E.; Rea, C.G.; Capri, G.; Bianchi, G.V.; de Braud, F. Everolimus versus alpelisib in advanced hormone receptor-positive HER2-negative breast cancer: Targeting different nodes of the PI3K/AKT/mTORC1 pathway with different clinical implications. Breast Cancer Res. 2020, 22, 33. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, T.; Jung, S.; Nguyen, H.A. The regulation of insulin receptor/insulin-like growth factor 1 receptor ratio, an important factor for breast cancer prognosis, by TRIP-Br1. J. Hematol. Oncol. 2022, 15, 82. [Google Scholar] [CrossRef]
- Wang, L.; Zhao, R.P.; Song, X.Y. Targeting ERβ in Macrophage Reduces Crown-like Structures in Adipose Tissue by Inhibiting Osteopontin and HIF-1α. Sci. Rep. 2019, 9, 15762. [Google Scholar] [CrossRef]
- Iyengar, N.M.; Brown, K.A.; Zhou, X.K.; Gucalp, A.; Subbaramaiah, K.; Giri, D.D.; Zahid, H.; Bhardwaj, P.; Wendel, N.K.; Falcone, D.J.; et al. Metabolic Obesity, Adipose Inflammation and Elevated Breast Aromatase in Women with Normal Body Mass Index. Cancer Prev. Res. 2017, 10, 235–243. [Google Scholar] [CrossRef]
- Maliniak, M.L.; Miller-Kleinhenz, J.; Cronin-Fenton, D.P.; Lash, T.L.; Gogineni, K.; Janssen, E.A.M.; McCullough, L.E. Crown-like Structures in Breast Adipose Tissue: Early Evidence and Current Issues in Breast Cancer. Cancers 2021, 13, 2222. [Google Scholar] [CrossRef] [PubMed]
- Birts, C.N.; Savva, C.; Laversin, S.A. Prognostic significance of crown-like structures to trastuzumab response in patients with primary invasive HER2 + breast carcinoma. Sci. Rep. 2022, 12, 7802. [Google Scholar] [CrossRef] [PubMed]
- Purohit, A.; Newman, S.P.; Reed, M.J. The role of cytokines in regulating oestrogen synthesis: Implications for the etiology of breast cancer. Breast Cancer Res. 2002, 4, 65–69. [Google Scholar] [CrossRef]
- Pérez-Hernández, A.I.; Catalán, V.; Gómez-Ambrosi, J.; Rodríguez, A.; Frühbeck, G. Mechanisms linking excess adiposity and carcinogenesis promotion. Front. Endocrinol. 2014, 5, 65. [Google Scholar]
- Kundel, V.; Lehane, D.; Ramachandran, S. Measuring Visceral Adipose Tissue Metabolic Activity in Sleep Apnea Utilizing Hybrid 18F-FDG PET/MRI: A Pilot Study. Nat. Sci. Sleep 2021, 13, 1943–1953. [Google Scholar] [CrossRef]
- Singh, D.; Arora, R.; Kaur, P. Overexpression of hypoxia-inducible factor and metabolic pathways: Possible targets of cancer. Cell Biosci. 2017, 7, 62. [Google Scholar] [CrossRef] [PubMed]
- Cinti, S.; Mitchell, G.; Barbatelli, G. Adipocyte death defines macrophage localization and function in adipose tissue of obese mice and humans. J. Lipid Res. 2005, 46, 2347–2355. [Google Scholar] [CrossRef]
- Pasquarelli-Do-Nascimento, G.; Machado, S.A.; Carvalho, J.D.; Magalhães, K.G. Obesity and adipose tissue impact on T-cell response and cancer immune checkpoint blockade therapy. Immunother. Adv. 2022, 2, ltac015. [Google Scholar] [CrossRef]
- Olona, A.; Mukhopadhyay, S.; Hateley, C.; Martinez, F.O.; Gordon, S.; Behmoaras, J. Adipoclast: A multinucleated fat-eating macrophage. BMC Biol. 2021, 19, 246. [Google Scholar] [CrossRef]
- Lefere, S.; Tacke, F. Macrophages in obesity and non-alcoholic fatty liver disease: Crosstalk with metabolism. JHEP Rep. 2019, 1, 30–43. [Google Scholar] [CrossRef]
- Caslin, H.L.; Cottam, M.A.; Piñon, J.M.; Boney, L.Y.; Hasty, A.H. Weight cycling induces innate immune memory in adipose tissue macrophages. Front. Immunol. 2023, 13, 984859. [Google Scholar] [CrossRef] [PubMed]
- Liao, L.; Dang, W.; Lin, T. A potent PGK1 antagonist reveals PGK1 regulates the production of IL-1β and IL-6. Acta Pharm. Sin. B 2022, 12, 4180–4192. [Google Scholar] [CrossRef]
- Li, Q.; Spalding, K.L. The regulation of adipocyte growth in white adipose tissue. Front. Cell Dev. Biol. 2022, 10, 1003219. [Google Scholar] [CrossRef] [PubMed]
- Tam, S.Y.; Wu, V.W.C.; Law, H.K.W. Hypoxia-Induced Epithelial-Mesenchymal Transition in Cancers: HIF-1α and Beyond. Front. Oncol. 2020, 10, 486. [Google Scholar] [CrossRef]
- Poblete, J.; Ballinger, M.N.; Bao, S.; Alghothani, M.; Nevado, J.B.; Eubank, T.D.; Christman, J.W.; Magalang, U.J. Macrophage HIF-1α mediates obesity-related adipose tissue dysfunction via interleukin-1 receptor-associated kinase M. Am. J. Physiol. Endocrinol. Metab. 2020, 318, E689–E700. [Google Scholar] [CrossRef] [PubMed]
- Aird, R.; Wills, J.; Roby, K.F. Hypoxia-driven metabolic reprogramming of adipocytes fuels cancer cell proliferation. Front. Endocrinol. 2022, 13, 989523. [Google Scholar] [CrossRef] [PubMed]
- Masoud, G.N.; Li, W. HIF-1α pathway: Role, regulation and intervention for cancer therapy. Acta Pharm. Sin. B 2015, 5, 378–389. [Google Scholar] [CrossRef]
- Lee, S.H.; Golinska, M.; Griffiths, J.R. HIF-1-Independent Mechanisms Regulating Metabolic Adaptation in Hypoxic Cancer Cells. Cells 2021, 10, 2371. [Google Scholar] [CrossRef]
- Kshitiz, A.J.; Suhail, Y.; Chang, H.; Hubbi, M.E.; Hamidzadeh, A.; Goyal, R.; Liu, Y.; Sun, P.; Nicoli, S.; Dang, C.V.; et al. Lactate-dependent chaperone-mediated autophagy induces oscillatory HIF-1α activity promoting proliferation of hypoxic cells. Cell Syst. 2022, 13, 1048–1064.e7. [Google Scholar] [CrossRef]
- Li, X.; Jiang, Y.; Meisenhelder, J.; Yang, W.; Hawke, D.H.; Zheng, Y.; Xia, Y.; Aldape, K.; He, J.; Hunter, T.; et al. Mitochondria-Translocated PGK1 Functions as a Protein Kinase to Coordinate Glycolysis and the TCA Cycle in Tumorigenesis. Mol. Cell 2016, 61, 705–719. [Google Scholar] [CrossRef]
- Jiang, Y.; Li, X.; Yang, W.; Hawke, D.H.; Zheng, Y.; Xia, Y. PKM2 regulates chromosome segregation and mitosis progression of tumor cells. Mol. Cell 2014, 53, 75–87. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Y.; Wang, Y.; Wang, T.; Hawke, D.H.; Zheng, Y. PKM2 phosphorylates MLC2 and regulates cytokinesis of tumour cells. Nat. Commun. 2014, 5, 5566. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Qian, X.; Jiang, H.; Xia, Y.; Zheng, Y.; Li, J. Nuclear PGK1 alleviates ADP-dependent inhibition of CDC7 to promote DNA replication. Mol. Cell 2018, 72, 650–660. [Google Scholar] [CrossRef]
- Pellegata, N.S.; Diaz, B.; Rohm, M.; Herzig, S. Obesity and cancer-extracellular matrix, angiogenesis, and adrenergic signaling as unusual suspects linking the two diseases. Cancer Metastasis Rev. 2022, 41, 517–547. [Google Scholar] [CrossRef] [PubMed]
- Sharma, M.; Boytard, L.; Hadi, T.; Koelwyn, G.; Simon, R.; Ouimet, M.; Seifert, L.; Spiro, W.; Yan, B.; Hutchison, S.; et al. Enhanced glycolysis and HIF-1α activation in adipose tissue macrophages sustains local and systemic interleukin-1β production in obesity. Sci. Rep. 2020, 10, 5555. [Google Scholar] [CrossRef]
- Lavie, L. Intermittent Hypoxia and Obstructive Sleep Apnea: Mechanisms, Interindividual Responses and Clinical Insights, in Atherosclerosis, Arteriosclerosis and Arteriolosclerosis; IntechOpen: London, UK, 2019; Available online: https://www.intechopen.com/chapters/66924 (accessed on 22 November 2022).
- Luo, W.; Hu, H.; Chang, R.; Zhong, J.; Knabel, M.; O’Meally, R.; Cole, R.N.; Pandey, A.; Semenza, G.L. Pyruvate kinase M2 is a PHD3-stimulated coactivator for hypoxia-inducible factor 1. Cell 2011, 145, 732–744. [Google Scholar] [CrossRef]
- Nocquet, L.; Juin, P.P.; Souazé, F. Mitochondria at Center of Exchanges between Cancer Cells and Cancer-Associated Fibroblasts during Tumor Progression. Cancers 2020, 12, 3017. [Google Scholar] [CrossRef]
- Yoo, H.; Sung, Y.; Han, J. Glutamine reliance in cell metabolism. Exp. Mol. Med. 2020, 52, 1496–1516. [Google Scholar] [CrossRef]
- Li, X.; Zhu, H.; Sun, W.; Yang, X.; Nie, Q.; Fang, X. Role of glutamine and its metabolite ammonia in crosstalk of cancer-associated fibroblasts and cancer cells. Cancer Cell Int. 2021, 21, 479. [Google Scholar] [CrossRef]
- Moldogazieva, N.T.; Mokhosoev, I.M.; Terentiev, A.A. Metabolic Heterogeneity of Cancer Cells: An Interplay between HIF-1, GLUTs, and AMPK. Cancers 2020, 12, 862. [Google Scholar] [CrossRef] [PubMed]
- Doerstling, S.S.; O’Flanagan, C.H.; Hursting, S.D. Obesity and Cancer Metabolism: A Perspective on Interacting Tumor-Intrinsic and Extrinsic Factors. Front. Oncol. 2017, 7, 216. [Google Scholar] [CrossRef]
- Ma, H.-H.; Zhang, J.; Zhou, L.; Wen, S.; Tang, H.-Y.; Jiang, B.; Zhang, F.; Suleman, M.; Sun, D.; Chen, A.; et al. c-Src Promotes Tumorigenesis and Tumor Progression by Activating PFKFB3. Cell Rep. 2020, 30, 4235–4249.e6. [Google Scholar] [CrossRef] [PubMed]
- Dumas, J.F.; Brisson, L. Interaction between adipose tissue and cancer cells: Role for cancer progression. Cancer Metastasis Rev. 2020, 40, 31–46. [Google Scholar] [CrossRef] [PubMed]
- Grohmann, M.; Wiede, F.; Dodd, G.T. Obesity Drives STAT-1-Dependent NASH and STAT-3-Dependent HCC. Cell 2018, 175, 1289–1306. [Google Scholar] [CrossRef]
- Liu, Y.; Wang, C.; Wei, M.; Yang, G.; Yuan, L. Multifaceted Roles of Adipose Tissue-Derived Exosomes in Physiological and Pathological Conditions. Front. Physiol. 2021, 12, 669429. [Google Scholar] [CrossRef]
- Itoh, M.; Kato, H.; Suganami, T.; Konuma, K.; Marumoto, Y.; Terai, S.; Sakugawa, H.; Kanai, S.; Hamaguchi, M.; Fukaishi, T.; et al. Hepatic crown-like structure: A unique histological feature in non-alcoholic steatohepatitis in mice and humans. PLoS ONE 2013, 8, e82163. [Google Scholar] [CrossRef]
- Huber, Y.; Pfirrmann, D.; Gebhardt, I. Improvement of non-invasive markers of NAFLD from an individualised, web-based exercise program. Aliment. Pharm. Ther. 2019, 50, 930–939. [Google Scholar] [CrossRef]
- Kessoku, T.; Kobayashi, T.; Imajo, K.; Tanaka, K.; Yamamoto, A.; Takahashi, K.; Kasai, Y.; Ozaki, A.; Iwaki, M.; Nogami, A.; et al. Endotoxins and Non-Alcoholic Fatty Liver Disease. Front. Endocrinol. 2021, 12, 770986. [Google Scholar] [CrossRef]
- Xiong, Z.; Li, X.; Yang, L. Integrative Analysis of Gene Expression and DNA Methylation Depicting the Impact of Obesity on Breast Cancer. Front. Cell Dev. Biol. 2022, 10, 818082. [Google Scholar] [CrossRef] [PubMed]
- Milner, J.J.; Chen, Z.F.; Grayson, J.; Shiao, S.Y.P. Obesity-Associated Differentially Methylated Regions in Colon Cancer. J. Pers. Med. 2022, 12, 660. [Google Scholar] [CrossRef]
- Rigamonti, A.E.; Bollati, V.; Favero, C.; Albetti, B.; Caroli, D.; De Col, A.; Cella, S.G.; Sartorio, A. Changes in DNA Methylation of Clock Genes in Obese Adolescents after a Short-Term Body Weight Reduction Program: A Possible Metabolic and Endocrine Chrono-Resynchronization. Int. J. Environ. Res. Public Health 2022, 19, 15492. [Google Scholar] [CrossRef] [PubMed]
- Wu, F.Y.; Yin, R.X. Recent progress in epigenetics of obesity. Diabetol. Metab. Syndr. 2022, 14, 171. [Google Scholar] [CrossRef]
- Frederick, A.L.; Racicot, R.; Liu, Z. Paradoxical effects of obesity on pre- vs. post-menopausal breast cancer: The epigenetic mechanisms (Review). Int. J. Epigenetics 2021, 1, 4. [Google Scholar] [CrossRef]
- Ehrlich, M. DNA hypermethylation in disease: Mechanisms and clinical relevance. Epigenetics 2019, 14, 1141–1163. [Google Scholar] [CrossRef] [PubMed]
- Gu, Y.; Zhang, C.W.; Wang, L. Association Analysis between Body Mass Index and Genomic DNA Methylation across 15 Major Cancer Types. J. Cancer 2018, 9, 2532–2542. [Google Scholar] [CrossRef]
- Dong, L.; Ma, L.; Ma, G.H. Genome-wide Analysis Reveals DNA Methylation Alterations in Obesity Associated with High Risk of Colorectal Cancer. Sci. Rep. 2019, 9, 5100. [Google Scholar] [CrossRef]
- Shen, J.; Song, R.; Ye, Y.; Wu, X.; Chow, W.H.; Zhao, H. HIF3A DNA methylation, obesity and weight gain, and breast cancer risk among Mexican American women. Obes. Res. Clin. Pract. 2020, 14, 548–553. [Google Scholar] [CrossRef]
- Pant, D.; Mutnuru, S.A.; Shukla, S. Epigenetic regulation during hypoxia and its implications in cancer. In Metabolism and Epigenetic Regulation: Implications in Cancer; Springer International Publishing: Cham, Switzerland, 2022; Volume 100, pp. 361–390. [Google Scholar]
- Elgendy, K.; Malcomson, F.C.; Bradburn, D.M.; Mathers, J.C. Effects of bariatric surgery on DNA methylation in adults: A systematic review and meta-analysis. Surg. Obes. Relat. Dis. 2020, 16, 128–136. [Google Scholar] [CrossRef]
- Faenza, M.; Benincasa, G.; Docimo, L.; Nicoletti, G.F.; Napoli, C. Clinical epigenetics and restoring of metabolic health in severely obese patients undergoing batriatric and metabolic surgery. Updates Surg. 2022, 74, 431–438. [Google Scholar] [CrossRef] [PubMed]
- Guz, M.; Jeleniewicz, W.; Malm, A.; Korona-Glowniak, I. A Crosstalk between Diet, Microbiome and microRNA in Epigenetic Regulation of Colorectal Cancer. Nutrients 2021, 13, 2428. [Google Scholar] [CrossRef]
- Luo, J.; Yu, Z.; Tovar, J.; Nilsson, A.; Xu, B. Critical review on anti-obesity effects of phytochemicals through Wnt/β-catenin signaling pathway. Pharmacol. Res. 2022, 184, 106461. [Google Scholar] [CrossRef] [PubMed]
- World Cancer Research Fund/American Institute for Cancer Research. Continuous Update Project Expert Report: Diet, Nutrition, Physical Activity and Colorectal Cancer; World Cancer Research Fund/American Institute for Cancer Research: London, UK, 2018. [Google Scholar]
- González Hernández, M.A.; Canfora, E.E.; Jocken, J.W.E.; Blaak, E.E. The Short-Chain Fatty Acid Acetate in Body Weight Control and Insulin Sensitivity. Nutrients 1943, 11, 1943. [Google Scholar] [CrossRef] [PubMed]
- Verde, L.; Dalamaga, M.; Capó, X.; Annunziata, G.; Hassapidou, M.; Docimo, A.; Savastano, S.; Colao, A.; Muscogiuri, G.; Barrea, L. The Antioxidant Potential of the Mediterranean Diet as a Predictor of Weight Loss after a Very Low-Calorie Ketogenic Diet (VLCKD) in Women with Overweight and Obesity. Antioxidants 2023, 12, 18. [Google Scholar] [CrossRef]
- Hullings, A.G.; Sinha, R.; Liao, L.M.; Freedman, N.D.; Graubard, B.I.; Loftfield, E. Whole grain and dietary fiber intake and risk of colorectal cancer in the NIH-AARP Diet and Health Study cohort. Am. J. Clin. Nutr. 2020, 112, 603–612. [Google Scholar] [CrossRef]
- Biswas, V.; Praveen, A.; Marisetti, A.L.; Sharma, A.; Kumar, V.; Sahu, S.K.; Tewari, D. A Mechanistic Overview on Impact of Dietary Fibres on Gut Microbiota and Its Association with Colon Cancer. Dietetics 2022, 1, 182–202. [Google Scholar] [CrossRef]
- Alpuim Costa, D.; Nobre, J.G.; Batista, M.V.; Ribeiro, C.; Calle, C.; Cortes, A.; Marhold, M.; Negreiros, I.; Borralho, P.; Brito, M.; et al. Human Microbiota and Breast Cancer-Is There Any Relevant Link? —A Literature Review and New Horizons Toward Personalised Medicine. Front. Microbiol. 2021, 12, 584332. [Google Scholar] [CrossRef] [PubMed]
- Gomes, S.; Rodrigues, A.C.; Pazienza, V.; Preto, A. Modulation of the Tumor Microenvironment by Microbiota-Derived Short-Chain Fatty Acids: Impact in Colorectal Cancer Therapy. Int. J. Mol. Sci. 2023, 24, 5069. [Google Scholar] [CrossRef]
- Bordonaro, M. Oncogenic and Receptor-Mediated Wnt Signaling Influence the Sensitivity of Colonic Cells to Butyrate. J. Cancer 2023, 14, 446–453. [Google Scholar] [CrossRef]
- Geng, H.W.; Yin, F.Y.; Zhang, Z.F.; Gong, X.; Yang, Y. Butyrate Suppresses Glucose Metabolism of Colorectal Cancer Cells via GPR109a-AKT Signaling Pathway and Enhances Chemotherapy. Front. Mol. Biosci. 2021, 8, 634874. [Google Scholar] [CrossRef]
- Bose, S.; Ramesh, V.; Locasale, J. Acetate Metabolism in Physiology, Cancer, and Beyond. Trends Cell Biol. 2019, 29, 695–703. [Google Scholar] [CrossRef]
- Peled, S.; Livney, Y.D. The role of dietary proteins and carbohydrates in gut microbiome composition and activity: A review. Food Hydrocoll. 2021, 120, 106911. [Google Scholar] [CrossRef]
- Li, Z.; Zhou, E.; Liu, C.; Wicks, H.; Yildiz, S.; Razack, F.; Ying, Z.; Kooijman, S.; Koonen, D.P.; Heijink, M.; et al. Dietary butyrate ameliorates metabolic health associated with selective proliferation of gut Lachnospiraceae bacterium 28-4. JCI Insight 2023, 8, e166655. [Google Scholar] [CrossRef] [PubMed]
- Silva, L.V.; Abdalla, P.P.; Bohn, L.; de Araújo, R.G.; Batalhão, D.D.F.; Venturini, A.C.R.; Carvalho, A.D.S.; Duncan, M.; Mota, J.; Machado, D.R.L. Association of minimally processed and ultra-processed food daily consumption with obesity in overweight adults: A cross-sectional study. Nutr. Hosp. 2023, 4270. [Google Scholar] [CrossRef] [PubMed]
- Silva, Y.P.; Bernardi, A.; Frozza, R.L. The Role of Short-Chain Fatty Acids from Gut Microbiota in Gut-Brain Communication. Front. Endocrinol. 2020, 11, 25. [Google Scholar] [CrossRef] [PubMed]
- Wolf, P.G.; Byrd, D.A.; Cares, K. Bile Acids, Gut Microbes, and the Neighborhood Food Environment-a Potential Driver of Colorectal Cancer Health Disparities. mSystems 2022, 7, e0117421. [Google Scholar] [CrossRef]
- Crafts, T.D.; Tonneson, J.E.; Wolfe, B.M.; Stroud, A.M. Obesity and breast cancer: Preventive and therapeutic possibilities for bariatric surgery. Obesity 2022, 30, 587–598. [Google Scholar] [CrossRef] [PubMed]
- Ruo, S.W.; Alkayyali, T.; Win, M. Role of Gut Microbiota Dysbiosis in Breast Cancer and Novel Approaches in Prevention, Diagnosis, and Treatment. Cureus 2021, 13, e17472. [Google Scholar] [CrossRef]
- Sui, Y.; Wu, J.; Chen, J. The Role of Gut Microbial β-Glucuronidase in Oestrogen Reactivation and Breast Cancer. Front. Cell Dev. Biol. 2021, 9, 631552. [Google Scholar] [CrossRef]
- Viggiani, M.T.; Polimeno, L.; Leo, D.; Barone, M. Phytoestrogens: Dietary Intake, Bioavailability, and Protective Mechanisms against Colorectal Neoproliferative Lesions. Nutrients 2019, 11, 1709. [Google Scholar] [CrossRef]
- Goedert, J.J.; Jones, G.; Hua, X.; Xu, X.; Yu, G.; Flores, R.; Falk, R.T.; Gail, M.H.; Shi, J.; Ravel, J.; et al. Investigation of the association between the fecal microbiota and breast cancer in postmenopausal women: A population-based case-control pilot study. J. Natl. Cancer Inst. 2015, 107, djv147. [Google Scholar] [CrossRef]
- Liu, Y.; Zhang, S.; Zhou, W.; Hu, D.; Xu, H.; Ji, G. Secondary Bile Acids and Tumorigenesis in Colorectal Cancer. Front. Oncol. 2022, 12, 813745. [Google Scholar] [CrossRef]
- Bernstein, H.; Bernstein, C.; Payne, C.M.; Dvorakova, K.; Garewal, H. Bile acids as carcinogens in human gastrointestinal cancers. Mutat. Res. 2005, 589, 47–65. [Google Scholar] [CrossRef] [PubMed]
- Režen, T.; Rozman, D.; Kovács, T. The role of bile acids in carcinogenesis. Cell Mol. Life Sci. 2022, 79, 243. [Google Scholar] [CrossRef] [PubMed]
- Wang, N.; Yang, J.; Han, W.; Han, M.; Liu, X.; Jiang, L.; Cao, H.; Jing, M.; Sun, T.; Xu, J. Identifying distinctive tissue and fecal microbial signatures and the tumor-promoting effects of deoxycholic acid on breast cancer. Front. Cell Infect. Microbiol. 2022, 12, 1029905. [Google Scholar] [CrossRef] [PubMed]
- Gualano, B.; Kirwan, J.P.; Roschel, H. Exercise Is Key to Sustaining Metabolic Gains After Bariatric Surgery. Exerc. Sport Sci. Rev. 2021, 49, 197–204. [Google Scholar] [CrossRef]
- Mingrone, G.; Panunzi, S.; De Gaetano, A.; Guidone, C.; Iaconelli, A.; Capristo, E.; Chamseddine, G.; Bornstein, S.R.; Rubino, F. Metabolic surgery versus conventional medical therapy in patients with type 2 diabetes: 10-year follow-up of an open-label, single-centre, randomised controlled trial. Lancet 2021, 397, 293–304. [Google Scholar] [CrossRef]
- Schauer, D.P.; Feigelson, H.S.; Koebnick, C. Association Between Weight Loss and the Risk of Cancer after Bariatric Surgery. Obesity 2017, 25, 52–57. [Google Scholar] [CrossRef]
- Pardo, F.; Villalobos-Labra, R.; Sobrevia, B.; Toledo, F.; Sobrevia, L. Extracellular vesicles in obesity and diabetes mellitus. Mol. Asp. Med. 2018, 60, 81–91. [Google Scholar] [CrossRef]
- Athanasiadis, D.I.; Martin, A.; Kapsampelis, P.; Monfared, S.; Stefanidis, D. Factors associated with weight regain post-bariatric surgery: A systematic review. Surg. Endosc. 2021, 35, 4069–4084. [Google Scholar] [CrossRef]
- Kozarzewski, L.; Maurer, L.; Mähler, A.; Spranger, J.; Weygandt, M. Computational approaches to predicting treatment response to obesity using neuroimaging. Rev. Endocr. Metab. Disord. 2022, 23, 773–805. [Google Scholar] [CrossRef]
- Clapp, B.; Portela, R.; Sharma, I. Risk of non-hormonal cancer after bariatric surgery: Meta-analysis of retrospective observational studies. Br. J. Surg. 2022, 110, 24–33. [Google Scholar] [CrossRef] [PubMed]
- Zhang, K.; Luo, Y.; Dai, H.; Deng, Z. Effects of Bariatric Surgery on Cancer Risk: Evidence from Meta-analysis. Obes. Surg. 2020, 30, 1265–1272. [Google Scholar] [CrossRef] [PubMed]
- Sjöholm, K.; Carlsson, L.; Svensson, P.A. Association of Bariatric Surgery with Cancer Incidence in Patients with Obesity and Diabetes: Long-term Results from the Swedish Obese Subjects Study. Diabetes Care 2022, 45, 444–450. [Google Scholar] [CrossRef] [PubMed]
- Atoum, M.F.; Alzoughool, F.; Al-Hourani, H. Linkage between Obesity, Leptin and Breast Cancer. Breast Cancer 2020, 10, 1178223419898458. [Google Scholar] [CrossRef] [PubMed]
- Patterson, R.; McNamara, E.; Tainio, M. Sedentary behaviour and risk of all-cause, cardiovascular and cancer mortality, and incident type 2 diabetes: A systematic review and dose response meta-analysis. Eur. J. Epidemiol. 2018, 33, 811–829. [Google Scholar] [CrossRef]
- Maclean, P.S.; Higgins, J.A.; Giles, E.D.; Sherk, V.D.; Jackman, M.R. The role for adipose tissue in weight regain after weight loss. Obes. Rev. 2015, 16 (Suppl. 1), 45–54. [Google Scholar] [CrossRef]
- Sjöström, L. Review of the key results from the Swedish Obese Subjects (SOS) trial—A prospective controlled intervention study of bariatric surgery. J. Intern. Med. 2013, 273, 219–234. [Google Scholar] [CrossRef]
- Villarreal-Calderon, J.R.; Cuellar-Tamez, R.; Castillo, E.C.; Luna-Ceron, E.; García-Rivas, G.; Elizondo-Montemayor, L. Metabolic shift precedes the resolution of inflammation in a cohort of patients undergoing bariatric and metabolic surgery. Sci. Rep. 2021, 11, 12127. [Google Scholar] [CrossRef]
- Wilson, R.B.; Lathigara, D.; Kaushal, D. Systematic Review and Meta-Analysis of the Impact of Bariatric Surgery on Future Cancer Risk. Int. J. Mol. Sci. 2023, 24, 6192. [Google Scholar] [CrossRef]
- Cancello, R.; Henegar, C.; Viguerie, N.; Taleb, S.; Poitou, C.; Rouault, C.; Coupaye, M.; Pelloux, V.; Hugol, D.; Bouillot, J.L.; et al. Reduction of macrophage infiltration and chemoattractant gene expression changes in white adipose tissue of morbidly obese subjects after surgery-induced weight loss. Diabetes 2005, 54, 2277–2286. [Google Scholar] [CrossRef]
- Alemán, J.O.; Iyengar, N.M.; Walker, J.M.; Milne, G.L.; Rosa, D.; Liang, J.C.; Giri, Y.; Zhou, D.D.; Pollak, X.K.; Hudis, M.N.; et al. Effects of Rapid Weight Loss on Systemic and Adipose Tissue Inflammation and Metabolism in Obese Postmenopausal Women. J. Endocr. Soc. 2017, 1, 625–637. [Google Scholar] [CrossRef] [PubMed]
- Loomis, A.K.; Kabadi, S.; Preiss, D. Body Mass Index and Risk of Nonalcoholic Fatty Liver Disease: Two Electronic Health Record Prospective Studies. J. Clin. Endocrinol. Metab. 2016, 101, 945–952. [Google Scholar] [CrossRef] [PubMed]
- Alexander, M.; Loomis, A.K.; Lei, J.V.D.; Duarte-Salles, T.; Prieto-Alhambra, D.; Pasqua, A.D.; Lapi, A.; Rijnbeek, F.; Mosseveld, P.; Waterworth, M.; et al. Risks and clinical predictors of cirrhosis and hepatocellular carcinoma diagnoses in adults with diagnosed NAFLD: Real-world study of 18 million patients in four European cohorts. BMC Med. 2019, 17, 95. [Google Scholar] [CrossRef] [PubMed]
- Pais, R.; Fartoux, L.; Goumard, C.; Scatton, O.; Wendum, D.; Rosmorduc, O.; Ratziu, V. Temporal trends, clinical patterns and outcomes of NAFLD-related HCC in patients undergoing liver resection over a 20-year period. Aliment. Pharmacol. Ther. 2017, 46, 856–863. [Google Scholar] [CrossRef] [PubMed]
- Ramai, D.; Singh, J.; Lester, J. Systematic review with meta-analysis: Bariatric surgery reduces the incidence of hepatocellular carcinoma. Aliment Pharmacol. Ther. 2021, 53, 977–984. [Google Scholar]
- Xiang, Q.; Tian, F.; Xu, J.; Du, X.; Zhang, S.; Liu, L. New insight into dyslipidemia-induced cellular senescence in atherosclerosis. Biol. Rev. 2022, 97, 1844–1867. [Google Scholar] [CrossRef]
- Pham, D.V.; Park, P.H. Tumor Metabolic Reprogramming by Adipokines as a Critical Driver of Obesity-Associated Cancer Progression. Int. J. Mol. Sci. 2021, 22, 1444. [Google Scholar] [CrossRef]
- Munhoz, A.C.; Serna, J.D.C.; Vilas-Boas, E.A.; Silva, C.C.C.D.; Santos, T.G.; Mosele, F.C.; Felisbino, S.L.; Martins, V.R.; Kowaltowski, A.J. Adiponectin reverses β-Cell damage and impaired insulin secretion induced by obesity. Aging Cell 2023, 00, e13827. [Google Scholar] [CrossRef]
- Šebunova, N.; Štšepetova, J.; Kullisaar, T. Changes in adipokine levels and metabolic profiles following bariatric surgery. BMC Endocr. Disord. 2022, 22, 33. [Google Scholar] [CrossRef]
- Sima, E.; Webb, D.L.; Hellström, P.M. Non-responders after Gastric Bypass Surgery for Morbid Obesity: Peptide Hormones and Glucose Homeostasis. Obes. Surg. 2019, 29, 4008–4017. [Google Scholar] [CrossRef]
- Wiggins, T.; Antonowicz, S.S.; Markar, S.R. Cancer Risk Following Bariatric Surgery-Systematic Review and Meta-analysis of National Population-Based Cohort Studies. Obes. Surg. 2019, 29, 1031–1039. [Google Scholar] [CrossRef] [PubMed]
- Molina-Montes, E.; Salamanca-Fernández, E.; Garcia-Villanova, B.; Sánchez, M.J. The Impact of Plant-Based Dietary Patterns on Cancer-Related Outcomes: A Rapid Review and Meta-Analysis. Nutrients 2020, 12, 2010. [Google Scholar] [CrossRef] [PubMed]
- Mahmod, A.I.; Haif, S.K.; Kamal, A.; Al-Ataby, I.A.; Talib, W.H. Chemoprevention effect of the Mediterranean diet on colorectal cancer: Current studies and future prospects. Front. Nutr. 2022, 9, 1772. [Google Scholar] [CrossRef] [PubMed]
- Van Blarigan, E.L.; Fuchs, C.S.; Niedzwiecki, D.; Zhang, S.; Saltz, L.B.; Mayer, R.J. Association of Survival with Adherence to the American Cancer Society Nutrition and Physical Activity Guidelines for Cancer Survivors After Colon Cancer Diagnosis: The CALGB 89803/Alliance Trial. JAMA Oncol. 2018, 4, 783–790. [Google Scholar] [CrossRef] [PubMed]
- Collado, M.; Castillo, M.; Mier, M.D.; Pinta, G.J.D.L.; Peña, C. The Diet as a Modulator of Tumor Microenvironment in Colorectal Cancer Patients. Int. J. Mol. Sci. 2023, 24, 7317. [Google Scholar] [CrossRef]
- Cramer, T. Impact of dietary carbohydrate restriction on the pathobiology of Hepatocellular Carcinoma: The gut-liver axis and beyond. Semin. Immunol. 2023, 66, 101736. [Google Scholar] [CrossRef]
- Chang, K.; Gunter, M.J.; Rauber, F. Ultra-processed food consumption, cancer risk and cancer mortality: A large-scale prospective analysis within the UK Biobank. eClinicalMedicine 2023, 56, 101840. [Google Scholar] [CrossRef]
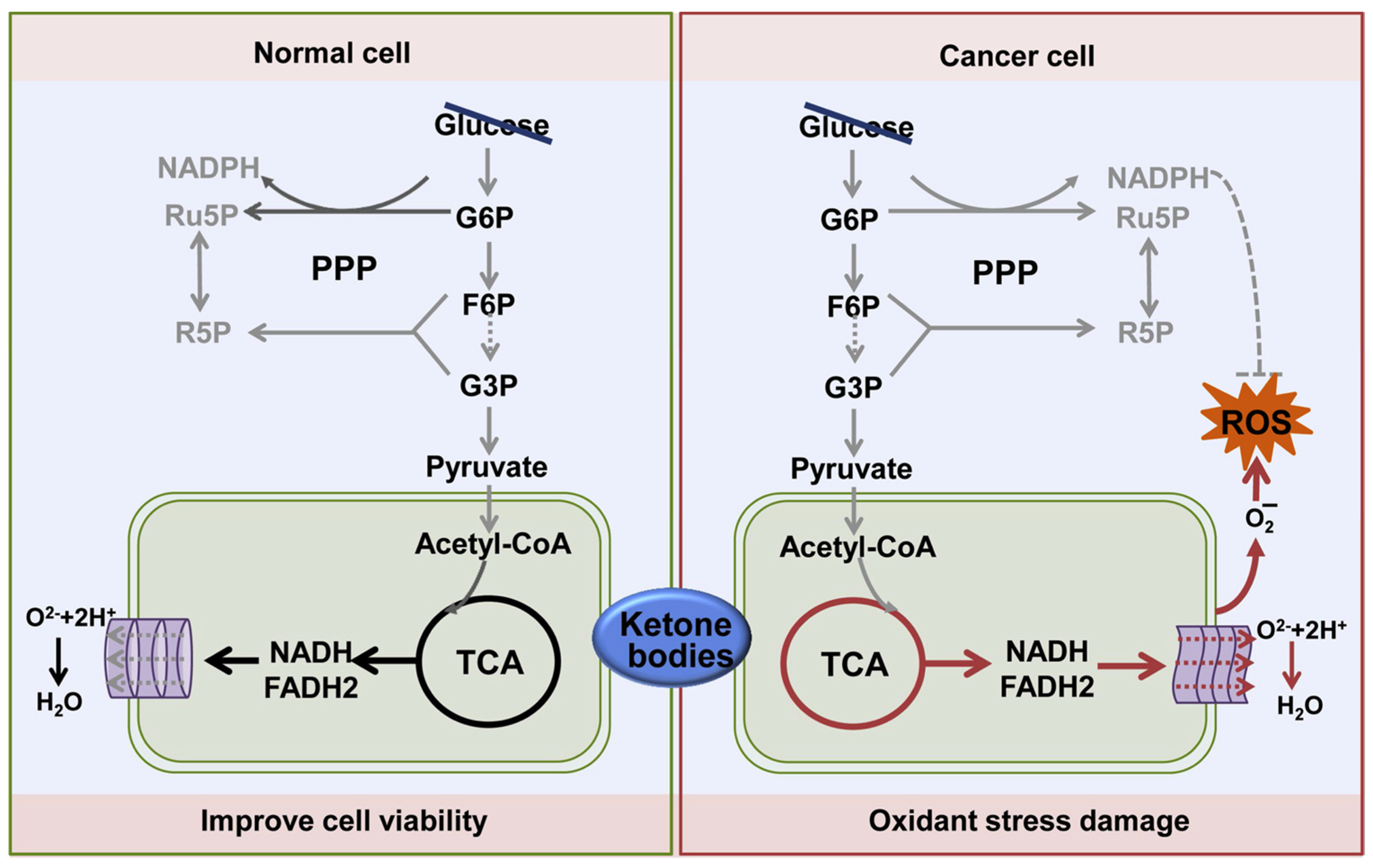
- Li, J.; Zhang, H.; Dai, Z. Cancer Treatment with the Ketogenic Diet: A Systematic Review and Meta-analysis of Animal Studies. Front. Nutr. 2021, 8, 594408. [Google Scholar] [CrossRef]
- Feng, S.; Wang, H.; Liu, J.; Aa, J.; Zhou, F.; Wang, G. Multi-dimensional roles of ketone bodies in cancer biology: Opportunities for cancer therapy. Pharmacol. Res. 2019, 150, 104500. [Google Scholar] [CrossRef]
- Yang, Y.F.; Mattamel, P.B.; Joseph, T.; Huang, J.; Chen, Q.; Akinwunmi, B.O.; Zhang, C.J.P.; Ming, W.K. Efficacy of Low-Carbohydrate Ketogenic Diet as an Adjuvant Cancer Therapy: A Systematic Review and Meta-Analysis of Randomized Controlled Trials. Nutrients. 2021, 13, 1388. [Google Scholar] [CrossRef]
- Römer, M.; Dörfler, J.; Huebner, J. The use of ketogenic diets in cancer patients: A systematic review. Clin. Exp. Med. 2021, 21, 501–536. [Google Scholar] [CrossRef] [PubMed]
- Ferreri, C.; Sansone, A.; Chatgilialoglu, C.; Ferreri, R.; Amézaga, J.; Burgos, M.C.; Arranz, S.; Tueros, I. Critical Review on Fatty Acid-Based Food and Nutraceuticals as Supporting Therapy in Cancer. Int. J. Mol. Sci. 2022, 23, 6030. [Google Scholar] [CrossRef] [PubMed]
- Hu, J.X.; Zhao, C.F.; Chen, W.B.; Liu, Q.C.; Li, Q.W.; Lin, Y.Y.; Gao, F. Pancreatic cancer: A review of epidemiology, trend, and risk factors. World J. Gastroenterol. 2021, 27, 4298–4321. [Google Scholar] [CrossRef] [PubMed]
- National Cancer Institute, Obesity and Cancer, United States, National Cancer Institute, 2022. Available online: https://www.cancer.gov/about-cancer/causes-prevention/risk/obesity/obesity-fact-sheet (accessed on 18 December 2022).
- Feigelson, H.S.; Bodelon, C.; Powers, J.D. Body mass index and risk of second cancer among women with breast cancer. J. Natl. Cancer Inst. 2021, 113, 1156–1160. [Google Scholar] [CrossRef] [PubMed]
- Sung, H.; Hyun, N.; Leach, C.R.; Yabroff, K.R.; Jemal, A. Association of first primary cancer with risk of subsequent primary cancer among survivors of adult-onset cancers in the United States. JAMA 2020, 324, 2521–2535. [Google Scholar] [CrossRef]
- Fu, J.; Jiang, J.; Liu, K. Metabolic syndrome and survival of patients with hepatocellular carcinoma: A meta-analysis. Front. Oncol. 2023, 13, 1117846. [Google Scholar] [CrossRef]
- Sjöström, L.; Narbro, K.; Sjöström, C.D. Effects of bariatric surgery on mortality in Swedish obese subjects. N. Engl. J. Med. 2007, 357, 741–752. [Google Scholar] [CrossRef]
- Schauer, D.P. What is currently known about the association between bariatric surgery and cancer. Surg. Obes. Relat. Dis. 2023, 19, 530–533. [Google Scholar] [CrossRef]
- Aminian, A.; Wilson, R.; Al-Kurd, A.; Tu, C.; Milinovich, A.; Kroh, M.; Rosenthal, R.J.; Brethauer, S.A.; Schauer, P.R.; Kattan, M.W.; et al. Association of Bariatric Surgery with Cancer Risk and Mortality in Adults with Obesity. JAMA 2022, 327, 2423–2433. [Google Scholar] [CrossRef]






Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lathigara, D.; Kaushal, D.; Wilson, R.B. Molecular Mechanisms of Western Diet-Induced Obesity and Obesity-Related Carcinogenesis—A Narrative Review. Metabolites 2023, 13, 675. https://doi.org/10.3390/metabo13050675
Lathigara D, Kaushal D, Wilson RB. Molecular Mechanisms of Western Diet-Induced Obesity and Obesity-Related Carcinogenesis—A Narrative Review. Metabolites. 2023; 13(5):675. https://doi.org/10.3390/metabo13050675
Chicago/Turabian StyleLathigara, Dhruvi, Devesh Kaushal, and Robert Beaumont Wilson. 2023. "Molecular Mechanisms of Western Diet-Induced Obesity and Obesity-Related Carcinogenesis—A Narrative Review" Metabolites 13, no. 5: 675. https://doi.org/10.3390/metabo13050675